
The Type I IFN-Induced miRNA, miR-21



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. MiR-21 Expression and Biological Functions
2.1. IFN-Induced miR-21 Expression
2.2. Signaling Pathways in IFN-Induced miR-21 Expression
2.3. The Role of miR-21 in Cancer
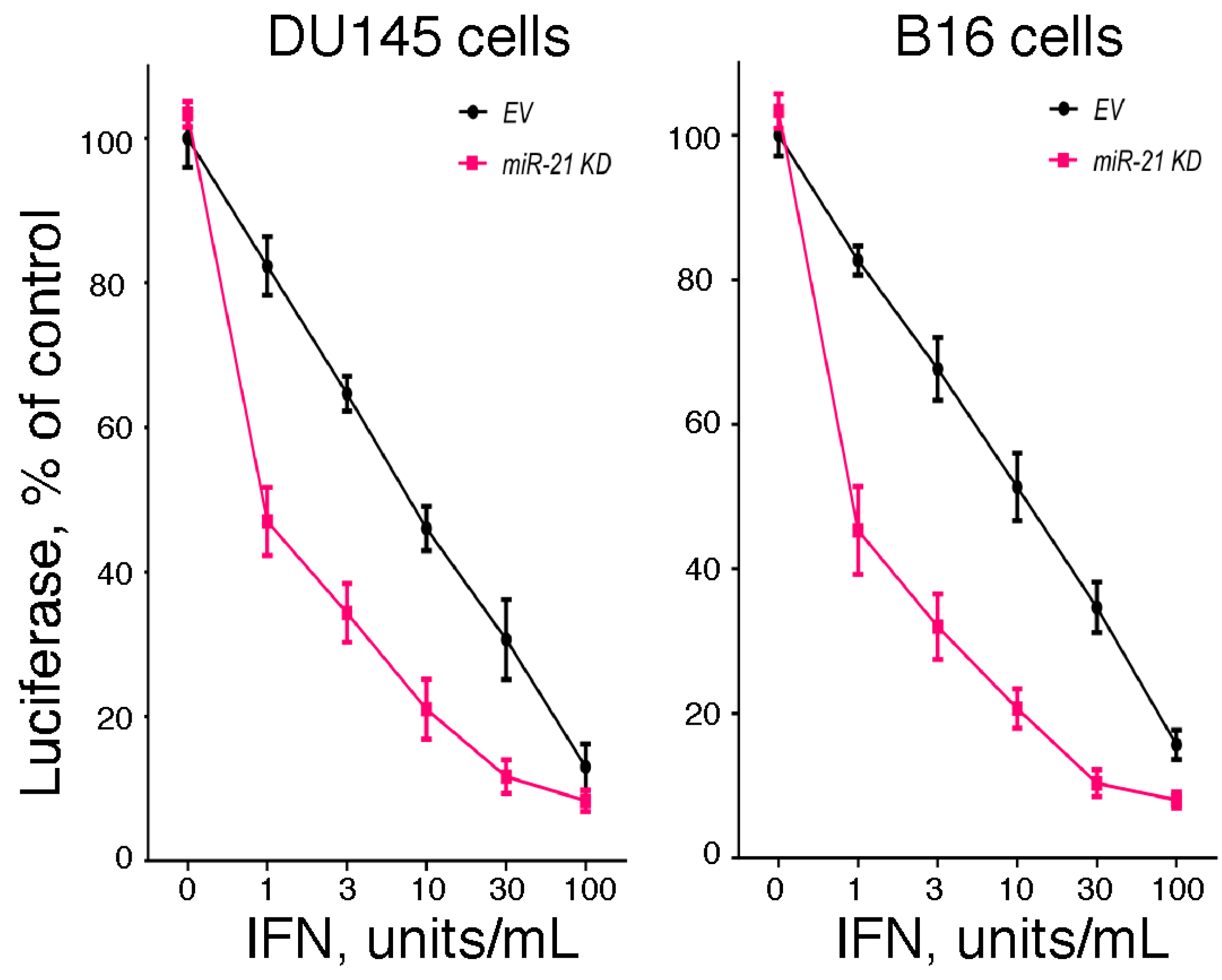
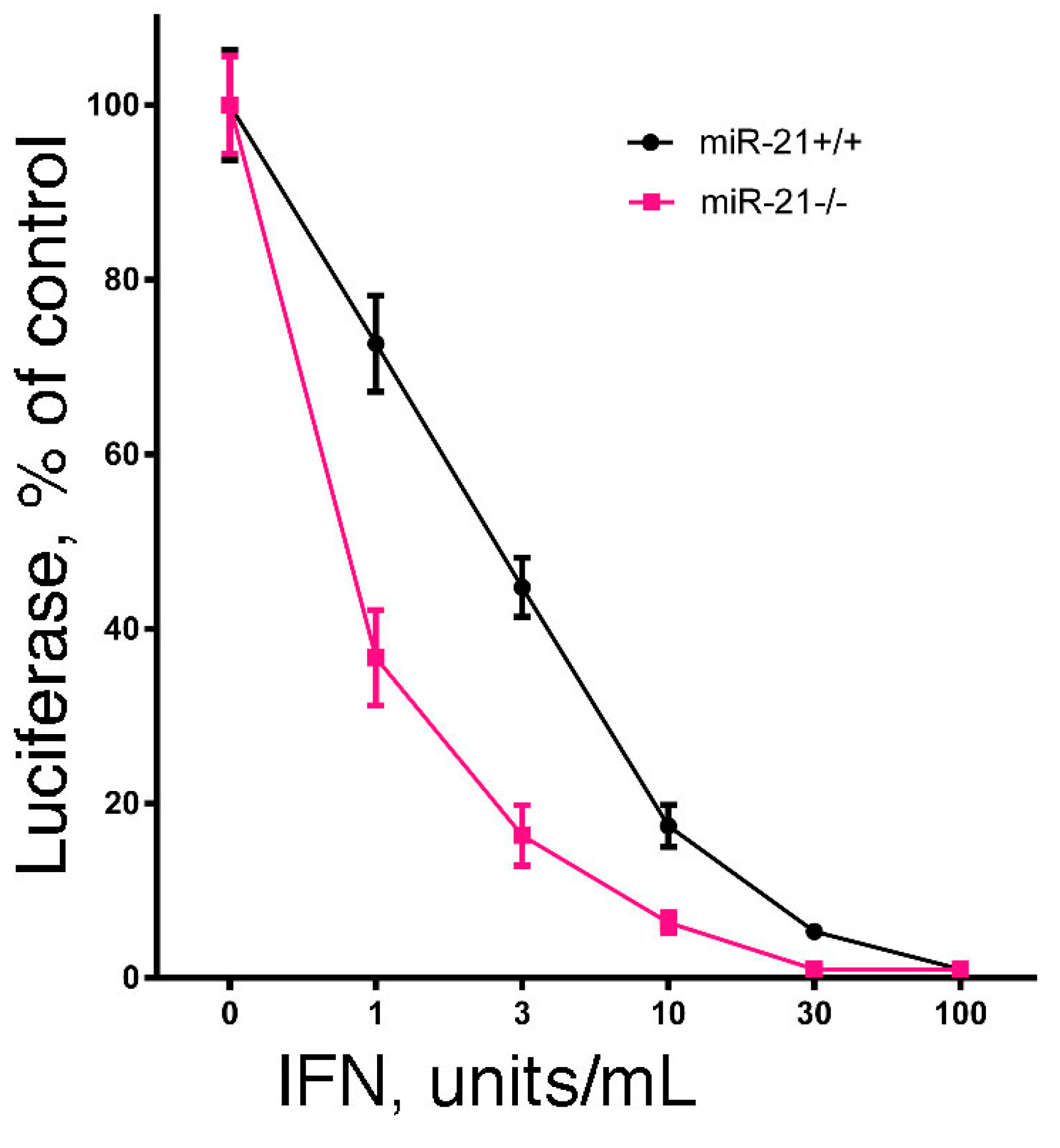
2.4. The Role of miR-21 in Regulating IFN-Mediated Antiviral Action


2.5. The Role of miR-21 in Regulating Immune Mechanisms Leading to IFN Expression
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pfeffer, L.M.; Dinarello, C.A.; Herberman, R.B.; Williams, B.R.; Borden, E.C.; Bordens, R.; Walter, M.R.; Nagabhushan, T.L.; Trotta, P.P.; Pestka, S. Biological properties of recombinant α-interferons: 40th Anniversary of the discovery of interferons. Cancer Res. 1998, 58, 2489–2499. [Google Scholar] [PubMed]
- Tanabe, T.; Kominsky, S.L.; Subramaniam, P.S.; Johnson, H.M.; Torres, B.A. Inhibition of the glioblastoma cell cycle by type I IFNs occurs at both the G1 and S phases and correlates with the upregulation of P21(WAF1/CIP1). J. Neuro-Oncol. 2000, 48, 225–232. [Google Scholar] [CrossRef]
- Chawla-Sarkar, M.; Lindner, D.J.; Liu, Y.F.; Williams, B.R.; Sen, G.C.; Silverman, R.H.; Borden, E.C. Apoptosis and interferons: Role of interferon-stimulated genes as mediators of apoptosis. Apoptosis 2003, 8, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, J.M.; Richards, T.; Zarour, H.M.; Sosman, J.; Ernstoff, M.; Whiteside, T.L.; Ibrahim, J.; Blum, R.; Wieand, S.; Mascari, R. Immunomodulatory effects of high-dose and low-dose interferon α2b in patients with high-risk resected melanoma: The e2690 laboratory corollary of intergroup adjuvant trial e1690. Cancer 2002, 95, 1101–1112. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F.; Gresser, I. Microvascular injury in pathogenesis of interferon-induced necrosis of subcutaneous tumors in mice. J. Natl. Cancer Inst. 1989, 81, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.W.; Swann, J.B.; Koebel, C.M.; Schreiber, R.D.; Smyth, M.J. Immune-mediated dormancy: An equilibrium with cancer. J. Leukoc. Biol. 2008, 84, 988–993. [Google Scholar] [CrossRef] [PubMed]
- Larner, A.C.; Jonak, G.; Cheng, Y.-S.E.; Korant, B.; Knight, E.; Darnell, J.E.J. Transcriptional induction of two genes in human cells by β interferon. Proc. Natl. Acad. Sci. USA 1984, 81, 6733–6737. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.L.; Manly, S.P.; McMahon, M.; Kerr, I.M.; Stark, G.R. Transcriptional and post-transcriptional regulation of interferon-induced gene expression in human cells. Cell 1984, 38, 745–755. [Google Scholar] [CrossRef]
- Darnell, J.E.J.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.L.; Stark, G.R. α-Interferon-induced transcription of HLA and metallothionein genes containing homologous upstream sequences. Nature 1985, 314, 637–639. [Google Scholar] [CrossRef] [PubMed]
- Larner, A.C.; Chaudhuri, A.; Darnell, J.E.J. Transcriptional induction by interferon: New protein(s) determine the length and extent of induction. J. Biol.Chem. 1986, 261, 453–459. [Google Scholar] [PubMed]
- Pfeffer, L.M.; Mullersman, J.E.; Pfeffer, S.R.; Murti, A.; Shi, W.; Yang, C.H. STAT3 as an adapter to couple phosphatidylinositol-3 kinase to the IFNAR-1 chain of the type I IFN receptor. Science 1997, 276, 1418–1420. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Murti, A.; Baker, S.J.; Frangou-Lazaridis, M.; Vartapetian, A.B.; Murti, K.G.; Pfeffer, L.M. Interferon induces the interaction of prothymosin-α with STAT3 and results in the nuclear translocation of the complex. Exp. Cell Res. 2004, 298, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Murti, A.; Pfeffer, L.M. STAT3 complements defects in an interferon-resistant cell line: Evidence for an essential role for STAT3 in interferon signaling and biological activities. Proc. Natl. Acad. Sci. USA 1998, 95, 5568–5572. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Shi, W.; Basu, L.; Murti, A.; Constantinescu, S.N.; Blatt, L.; Croze, E.; Mullersman, J.E.; Pfeffer, L.M. Direct association of STAT3 with the IFNAR1 signal transducing chain of the type I IFN receptor. J. Biol. Chem 1996, 271, 8057–8061. [Google Scholar] [PubMed]
- Pedersen, I.M.; Cheng, G.; Wieland, S.; Volinia, S.; Croce, C.M.; Chisari, F.V.; David, M. Interferon modulation of cellular micrornas as an antiviral mechanism. Nature 2007, 449, 919–922. [Google Scholar] [CrossRef] [PubMed]
- Sarasin-Filipowicz, M.; Krol, J.; Markiewicz, I.; Heim, M.H.; Filipowicz, W. Decreased levels of microRNA miR-122 in individuals with hepatitis C responding poorly to interferon therapy. Nat. Med. 2009, 15, 31–33. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Yue, J.; Fan, M.; Pfeffer, L.M. IFN induces miR-21 through a signal transducer and activator of transcription 3-dependent pathway as a suppressive negative feedback on IFN-induced apoptosis. Cancer Res. 2010, 70, 8108–8116. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Yue, J.; Pfeffer, S.R.; Handorf, C.R.; Pfeffer, L.M. Microrna miR-21 regulates the metastatic behavior of B16 melanoma cells. J. Biol. Chem 2011, 286, 39172–39178. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. Micrornas: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Lim, L.P.; Lau, N.C.; Garrett-Engele, P.; Grimson, A.; Schelter, J.M.; Castle, J.; Bartel, D.P.; Linsley, P.S.; Johnson, J.M. Microarray analysis shows that some micrornas downregulate large numbers of target mrnas. Nature 2005, 433, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Scagnolari, C.; Zingariello, P.; Vecchiet, J.; Selvaggi, C.; Racciatti, D.; Taliani, G.; Riva, E.; Pizzigallo, E.; Antonelli, G. Differential expression of interferon-induced micrornas in patients with chronic hepatitis C virus infection treated with pegylated interferon α. Virol. J. 2010, 7, 311. [Google Scholar] [CrossRef] [PubMed]
- tenOever, B.R. RNA viruses and the host microRNA machinery. Nat. Rev. Microbiol. 2013, 11, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Folini, M.; Gandellini, P.; Longoni, N.; Profumo, V.; Callari, M.; Pennati, M.; Colecchia, M.; Supino, R.; Veneroni, S.; Salvioni, R.; et al. MiR-21: An oncomir on strike in prostate cancer. Mol. Cancer 2010, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Loffler, D.; Brocke-Heidrich, K.; Pfeifer, G.; Stocsits, C.; Hackermuller, J.; Kretzschmar, A.K.; Burger, R.; Gramatzki, M.; Blumert, C.; Bauer, K.; et al. Interleukin-6 dependent survival of multiple myeloma cells involves the STAT3-mediated induction of microRNA-21 through a highly conserved enhancer. Blood 2007, 110, 1330–1333. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.; Edwards, S.; Feber, A.; Flohr, P.; John, M.; Giddings, I.; Crossland, S.; Stratton, M.R.; Wooster, R.; Campbell, C.; et al. Genome-wide screening for complete genetic loss in prostate cancer by comparative hybridization onto cdna microarrays. Oncogene 2003, 22, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Nishio, Y.; Inoue, M.; Wang, X.-J.; Wei, S.; Matsusaka, T.; Yoshida, K.; Sudo, T.; Naruto, M.; Kishimoto, T. Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell 1994, 77, 63–71. [Google Scholar] [CrossRef]
- Zhong, Z.; Wen, Z.; Darnell, J.E.J. STAT3: A STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 1994, 264, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Turkson, J.; Jove, R. STAT proteins: Novel molecular targets for cancer drug discovery. Oncogene 2000, 19, 6613–6626. [Google Scholar] [CrossRef] [PubMed]
- Bowman, T.; Garcia, R.; Turkson, J.; Jove, R. STATS in oncogenesis. Oncogene 2000, 19, 2474–2488. [Google Scholar] [CrossRef] [PubMed]
- Beg, A.A.; Baltimore, D. An essential role for NF-κb in preventing TNF-α-induced cell death. Science 1996, 274, 782–784. [Google Scholar] [CrossRef] [PubMed]
- Beg, A.A.; Sha, W.C.; Bronson, R.T.; Ghosh, S.; Baltimore, D. Embryonic lethality and liver degeneration in mice lacking the rela component of NF-κb. Nature 1995, 376, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Van Antwerp, D.J.; Martin, S.J.; Kafri, T.; Green, D.R.; Verma, I.M. Suppression of TNF-α-induced apotosis by NF-κb. Science 1996, 274, 787–789. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-Y.; Mayo, M.W.; Baldwin, A.S., Jr. TNF- and cancer therapy-induced apoptosis: Potentiation by inhibition of NF-κb. Science 1996, 274, 784–787. [Google Scholar] [CrossRef] [PubMed]
- Sheedy, F.J.; Palsson-McDermott, E.; Hennessy, E.J.; Martin, C.; O’Leary, J.J.; Ruan, Q.; Johnson, D.S.; Chen, Y.; O’Neill, L.A. Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat. Immunol. 2010, 11, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Wei, L.; Pfeffer, S.R.; Du, Z.; Murti, A.; Valentine, W.J.; Zheng, Y.; Pfeffer, L.M. Identification of CXCL11 as a STAT3-dependent gene induced by IFN. J. Immunol. 2007, 178, 986–992. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Karin, M. Dangerous liaisons: STAT3 and Nf-κb collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010, 21, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Ben-Neriah, Y.; Karin, M. Inflammation meets cancer, with NF-κb as the matchmaker. Nat. Immunol. 2011, 12, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Malizos, K.N.; Oikonomou, P.; Tsezou, A. Integrative microRNA and proteomic approaches identify novel osteoarthritis genes and their collaborative metabolic and inflammatory networks. PLoS ONE 2008, 3, e3740. [Google Scholar] [CrossRef] [PubMed]
- Sonkoly, E.; Wei, T.; Janson, P.C.; Saaf, A.; Lundeberg, L.; Tengvall-Linder, M.; Norstedt, G.; Alenius, H.; Homey, B.; Scheynius, A.; et al. MicroRNAs: Novel regulators involved in the pathogenesis of psoriasis? PLoS ONE 2007, 2, e610. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zikusoka, M.; Trindade, A.; Dassopoulos, T.; Harris, M.L.; Bayless, T.M.; Brant, S.R.; Chakravarti, S.; Kwon, J.H. MicroRNAs are differentially expressed in ulcerative colitis and alter expression of macrophage inflammatory peptide-2α. Gastroenterology 2008, 135, 1624–1635. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Ji, R.; Yue, J.; Yang, J.; Liu, X.; Chen, H.; Dean, D.B.; Zhang, C. MicroRNAs are aberrantly expressed in hypertrophic heart: Do they play a role in cardiac hypertrophy? Am. J. Pathol. 2007, 170, 1831–1840. [Google Scholar] [CrossRef] [PubMed]
- Hatley, M.E.; Patrick, D.M.; Garcia, M.R.; Richardson, J.A.; Bassel-Duby, R.; van Rooij, E.; Olson, E.N. Modulation of K-Ras-dependent lung tumorigenesis by microRNA-21. Cancer Cell. 2010, 18, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Krichevsky, A.M.; Gabriely, G. miR-21: A small multi-faceted RNA. J. Cell. Mol. Med. 2009, 13, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Si, M.L.; Zhu, S.; Wu, H.; Lu, Z.; Wu, F.; Mo, Y.Y. MiR-21-mediated tumor growth. Oncogene 2007, 26, 2799–2803. [Google Scholar] [CrossRef] [PubMed]
- Volinia, S.; Calin, G.A.; Liu, C.G.; Ambs, S.; Cimmino, A.; Petrocca, F.; Visone, R.; Iorio, M.; Roldo, C.; Ferracin, M.; et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. USA 2006, 103, 2257–2261. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.A.; Krichevsky, A.M.; Kosik, K.S. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005, 65, 6029–6033. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Wu, H.; Wu, F.; Nie, D.; Sheng, S.; Mo, Y.Y. MicroRNA-21 targets tumor suppressor genes in invasion and metastasis. Cell. Res. 2008, 18, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zou, F.; Zhang, X.; Li, H.; Dulak, A.; Tomko, R.J., Jr.; Lazo, J.S.; Wang, Z.; Zhang, L.; Yu, J. MicroRNA-21 negatively regulates Cdc25a and cell cycle progression in colon cancer cells. Cancer Res. 2009, 69, 8157–8165. [Google Scholar] [CrossRef] [PubMed]
- Medina, P.P.; Nolde, M.; Slack, F.J. OncomiR addiction in an in vivo model of microRNA-21-induced pre-B-cell lymphoma. Nature 2010, 467, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Murti, A.; Basu, L.; Kim, J.G.; Pfeffer, L.M. IFNα/β promotes cell survival by activating Nf-κb. Proc. Natl. Acad. Sci. USA 2000, 97, 13631–13636. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Murti, A.; Pfeffer, S.R.; Kim, J.G.; Donner, D.B.; Pfeffer, L.M. Interferon α/β promotes cell survival by activating NF-κb through phosphatidylinositol-3 kinase and Akt. J. Biol. Chem. 2001, 276, 13756–13761. [Google Scholar] [PubMed]
- Poste, G.; Doll, J.; Brown, A.E.; Tzeng, J.; Zeidman, I. Comparison of the metastatic properties of B16 melanoma clones isolated from cultured cell lines, subcutaneous tumors, and individual lung metastases. Cancer Res. 1982, 42, 2770–2778. [Google Scholar] [PubMed]
- Nicolson, G.L.; Brunson, K.W.; Fidler, I.J. Specificity of arrest, survival, and growth of selected metastatic variant cell lines. Cancer Res. 1978, 38, 4105–4111. [Google Scholar] [PubMed]
- Nicolson, G.L. Cancer metastasis. Sci. Am. 1979, 240, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Buscaglia, L.E.; Li, Y. Apoptosis and the target genes of microRNA-21. Chin. J. Cancer 2011, 30, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.; Cermak, L.; Pagan, J.K.; Rossi, M.; Martinengo, C.; di Celle, P.F.; Chapuy, B.; Shipp, M.; Chiarle, R.; Pagano, M. FBXO11 targets BCL6 for degradation and is inactivated in diffuse large B-cell lymphomas. Nature 2012, 481, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Trojan, J.; Cloix, J.F.; Ardourel, M.Y.; Chatel, M.; Anthony, D.D. Insulin-like growth factor type I biology and targeting in malignant gliomas. Neuroscience 2007, 145, 795–811. [Google Scholar] [CrossRef] [PubMed]
- Panasiti, V.; Naspi, A.; Devirgiliis, V.; Curzio, M.; Roberti, V.; Curzio, G.; Gobbi, S.; Calvieri, S.; Londei, P. Correlation between insulin-like growth factor binding protein-3 serum level and melanoma progression. J. Am. Acad. Dermatol. 2011, 64, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Li, K.; Pfeffer, L.M. University of Tennessee Health Science Center: Memphis, TN, Unpublished work; 2015.
- Chen, Y.; Chen, J.; Wang, H.; Shi, J.; Wu, K.; Liu, S.; Liu, Y.; Wu, J. HCV-induced miR-21 contributes to evasion of host immune system by targeting MyD88 and IRAK1. PLoS Pathog. 2013, 9, e1003248. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.X.; Hartner, J.; Lim, E.J.; Fabry, V.; Mingler, M.K.; Cole, E.T.; Orkin, S.H.; Aronow, B.J.; Rothenberg, M.E. MicroRNA-21 limits in vivo immune response-mediated activation of the IL-12/IFN-γ pathway, Th1 polarization, and the severity of delayed-type hypersensitivity. J. Immunol. 2011, 187, 3362–3373. [Google Scholar] [CrossRef] [PubMed]
- Carissimi, C.; Carucci, N.; Colombo, T.; Piconese, S.; Azzalin, G.; Cipolletta, E.; Citarella, F.; Barnaba, V.; Macino, G.; Fulci, V. MiR-21 is a negative modulator of T-cell activation. Biochimie 2014, 107, 319–326. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, C.H.; Li, K.; Pfeffer, S.R.; Pfeffer, L.M. The Type I IFN-Induced miRNA, miR-21. Pharmaceuticals 2015, 8, 836-847. https://doi.org/10.3390/ph8040836
Yang CH, Li K, Pfeffer SR, Pfeffer LM. The Type I IFN-Induced miRNA, miR-21. Pharmaceuticals. 2015; 8(4):836-847. https://doi.org/10.3390/ph8040836
Chicago/Turabian StyleYang, Chuan He, Kui Li, Susan R. Pfeffer, and Lawrence M. Pfeffer. 2015. "The Type I IFN-Induced miRNA, miR-21" Pharmaceuticals 8, no. 4: 836-847. https://doi.org/10.3390/ph8040836
APA StyleYang, C. H., Li, K., Pfeffer, S. R., & Pfeffer, L. M. (2015). The Type I IFN-Induced miRNA, miR-21. Pharmaceuticals, 8(4), 836-847. https://doi.org/10.3390/ph8040836