

The Role of WRAP53 in Cell Homeostasis and Carcinogenesis Onset

 , ,
, ,  and
and 
Abstract
:1. Introduction
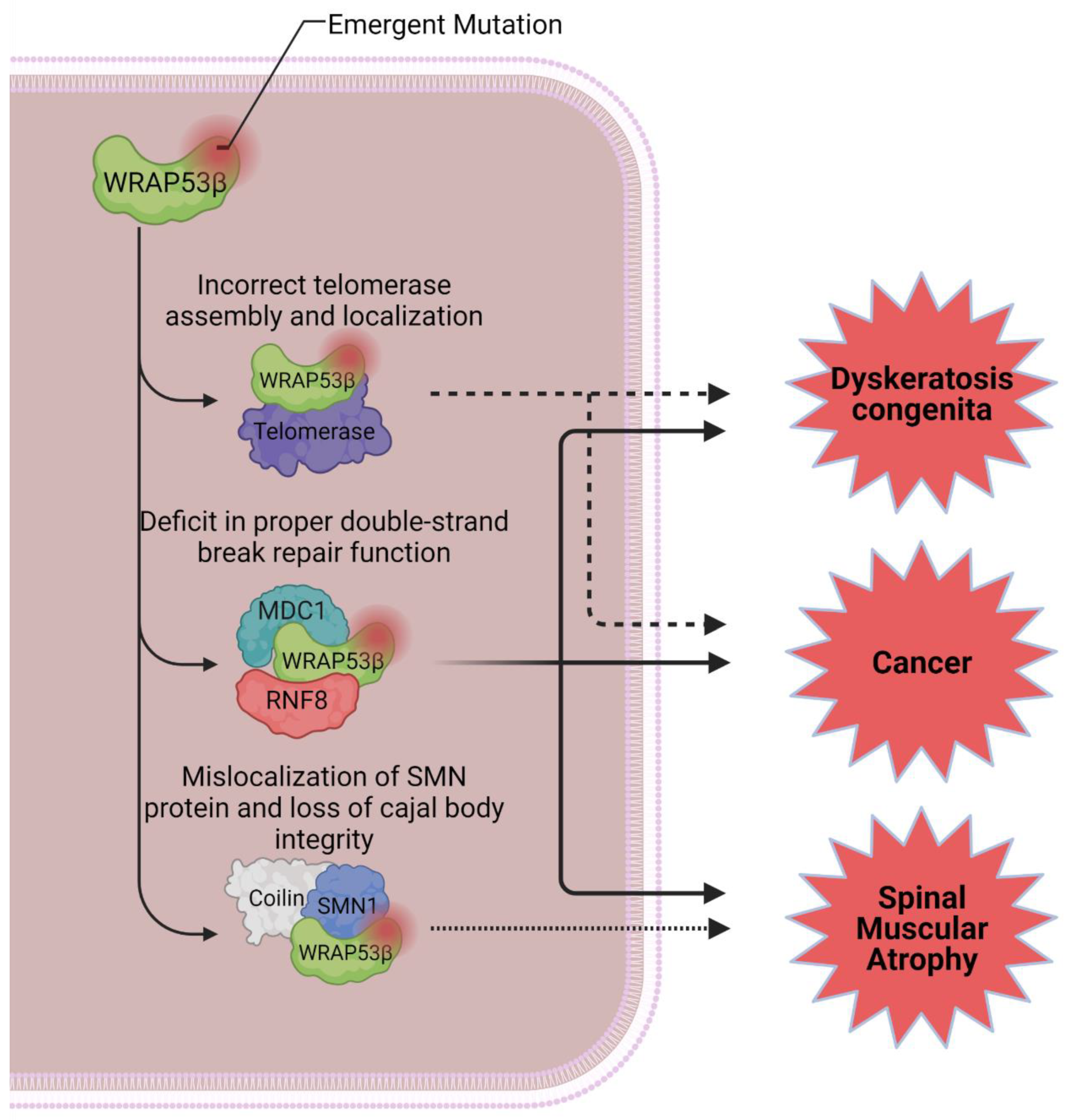
2. WRAP53 Characterization and Cellular Roles
2.1. WRAP53 and Telomerase
2.2. DNA Repair
2.3. Protein Trafficking
2.4. WRAP53 and Diseases
3. WRAP53 as a Potential Novel Biomarker for Cancer
4. WRAP53 as a Prognostic Factor in Cancer
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Basu, A. DNA Damage, Mutagenesis and Cancer. Int. J. Mol. Sci. 2018, 19, 970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopnin, B.P. Targets of Oncogenes and Tumor Suppressors: Key for Understanding Basic Mechanisms of Carcinogenesis. Biochemistry 2000, 65, 2–27. [Google Scholar]
- McNally, E.J.; Luncsford, P.J.; Armanios, M. Long Telomeres and Cancer Risk: The Price of Cellular Immortality. J. Clin. Investig. 2019, 129, 3474–3481. [Google Scholar] [CrossRef] [Green Version]
- Vogelstein, B.; Kinzler, K.W. The Path to Cancer—Three Strikes and You’re Out. N. Engl. J. Med. 2015, 373, 1895–1898. [Google Scholar] [CrossRef]
- Shay, J.W. Telomerase and Cancer. Hum. Mol. Genet. 2001, 10, 677–685. [Google Scholar] [CrossRef] [Green Version]
- De Lange, T. Telomere-Related Genome Instability in Cancer. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 197–204. [Google Scholar] [CrossRef] [Green Version]
- Jafri, M.A.; Ansari, S.A.; Alqahtani, M.H.; Shay, J.W. Roles of Telomeres and Telomerase in Cancer, and Advances in Telomerase-Targeted Therapies. Genome Med. 2016, 8, 69. [Google Scholar] [CrossRef] [Green Version]
- Kong, F.; Zheng, C.; Xu, D. Telomerase as a “Stemness” Enzyme. Sci. China Life Sci. 2014, 57, 564–570. [Google Scholar] [CrossRef]
- Palm, W.; de Lange, T. How Shelterin Protects Mammalian Telomeres. Annu. Rev. Genet. 2008, 42, 301–334. [Google Scholar] [CrossRef] [Green Version]
- Barthel, F.P.; Wei, W.; Tang, M.; Martinez-Ledesma, E.; Hu, X.; Amin, S.B.; Akdemir, K.C.; Seth, S.; Song, X.; Wang, Q.; et al. Systematic Analysis of Telomere Length and Somatic Alterations in 31 Cancer Types. Nat. Genet. 2017, 49, 349–357. [Google Scholar] [CrossRef]
- Wang, K.; Liu, T.; Liu, L.; Liu, J.; Liu, C.; Wang, C.; Ge, N.; Ren, H.; Yan, K.; Hu, S.; et al. TERT Promoter Mutations in Renal Cell Carcinomas and Upper Tract Urothelial Carcinomas. Oncotarget 2014, 5, 1829–1836. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Espín, D.; Serrano, M. Cellular Senescence: From Physiology to Pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Kastan, M.B.; Bartek, J. Cell-Cycle Checkpoints and Cancer. Nature 2004, 432, 316–323. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA Damage, Repair, and Mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [Green Version]
- Nagy, Á.; Munkácsy, G.; Győrffy, B. Pancancer Survival Analysis of Cancer Hallmark Genes. Sci. Rep. 2021, 11, 6047. [Google Scholar] [CrossRef]
- Carter, S.L.; Eklund, A.C.; Kohane, I.S.; Harris, L.N.; Szallasi, Z. A Signature of Chromosomal Instability Inferred from Gene Expression Profiles Predicts Clinical Outcome in Multiple Human Cancers. Nat. Genet. 2006, 38, 1043–1048. [Google Scholar] [CrossRef]
- Venteicher, A.S.; Abreu, E.B.; Meng, Z.; McCann, K.E.; Terns, R.M.; Veenstra, T.D.; Terns, M.P.; Artandi, S.E. A Human Telomerase Holoenzyme Protein Required for Cajal Body Localization and Telomere Synthesis. Science 2009, 323, 644–648. [Google Scholar] [CrossRef] [Green Version]
- Tycowski, K.T.; Shu, M.-D.; Kukoyi, A.; Steitz, J.A. A Conserved WD40 Protein Binds the Cajal Body Localization Signal of ScaRNP Particles. Mol. Cell 2009, 34, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, S.; Henriksson, S.; Corcoran, M.; Méndez-Vidal, C.; Wiman, K.G.; Farnebo, M. Wrap53, a Natural P53 Antisense Transcript Required for P53 Induction upon DNA Damage. Mol. Cell 2009, 33, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Stirnimann, C.U.; Petsalaki, E.; Russell, R.B.; Müller, C.W. WD40 Proteins Propel Cellular Networks. Trends Biochem. Sci. 2010, 35, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Min, J. Structure and Function of WD40 Domain Proteins. Protein Cell 2011, 2, 202–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schapira, M.; Tyers, M.; Torrent, M.; Arrowsmith, C.H. WD40 Repeat Domain Proteins: A Novel Target Class? Nat. Rev. Drug Discov. 2017, 16, 773–786. [Google Scholar] [CrossRef]
- Li, D.; Roberts, R. Human Genome and Diseases:¶WD-Repeat Proteins: Structure Characteristics, Biological Function, and Their Involvement in Human Diseases. Cell. Mol. Life Sci. 2001, 58, 2085–2097. [Google Scholar] [CrossRef]
- Suganuma, T.; Pattenden, S.G.; Workman, J.L. Diverse Functions of WD40 Repeat Proteins in Histone Recognition: Figure 1. Genes Dev. 2008, 22, 1265–1268. [Google Scholar] [CrossRef] [Green Version]
- Jain, B.P.; Pandey, S. WD40 Repeat Proteins: Signalling Scaffold with Diverse Functions. Protein J. 2018, 37, 391–406. [Google Scholar] [CrossRef]
- Xie, X.; Wang, Z.; Chen, Y. Association of LKB1 with a WD-Repeat Protein WDR6 Is Implicated in Cell Growth Arrest and P27Kip1 Induction. Mol. Cell Biochem. 2007, 301, 115–122. [Google Scholar] [CrossRef]
- Welcker, M.; Clurman, B.E. FBW7 Ubiquitin Ligase: A Tumour Suppressor at the Crossroads of Cell Division, Growth and Differentiation. Nat. Rev. Cancer 2008, 8, 83–93. [Google Scholar] [CrossRef]
- Halder, S.K.; Anumanthan, G.; Maddula, R.; Mann, J.; Chytil, A.; Gonzalez, A.L.; Washington, M.K.; Moses, H.L.; Beauchamp, R.D.; Datta, P.K. Oncogenic Function of a Novel WD-Domain Protein, STRAP, in Human Carcinogenesis. Cancer Res. 2006, 66, 6156–6166. [Google Scholar] [CrossRef]
- Honoré, B.; Baandrup, U.; Nielsen, S.; Vorum, H. Endonuclein Is a Cell Cycle Regulated WD-Repeat Protein That Is up-Regulated in Adenocarcinoma of the Pancreas. Oncogene 2002, 21, 1123–1129. [Google Scholar] [CrossRef] [Green Version]
- Adams, D.R.; Ron, D.; Kiely, P.A. RACK1, A Multifaceted Scaffolding Protein: Structure and Function. Cell Commun. Signal. 2011, 9, 22. [Google Scholar] [CrossRef] [Green Version]
- Silva, F.P.; Hamamoto, R.; Nakamura, Y.; Furukawa, Y. WDRPUH, A Novel WD-Repeat—Containing Protein, Is Highly Expressed in Human Hepatocellular Carcinoma and Involved in Cell Proliferation. Neoplasia 2005, 7, 348–355. [Google Scholar] [CrossRef] [Green Version]
- Henriksson, S.; Farnebo, M. On the Road with WRAP53Î2: Guardian of Cajal Bodies and Genome Integrity. Front. Genet. 2015, 6, 91. [Google Scholar] [CrossRef] [Green Version]
- Mahmoudi, S.; Henriksson, S.; Weibrecht, I.; Smith, S.; Söderberg, O.; Strömblad, S.; Wiman, K.G.; Farnebo, M. WRAP53 Is Essential for Cajal Body Formation and for Targeting the Survival of Motor Neuron Complex to Cajal Bodies. PLoS Biol. 2010, 8, e1000521. [Google Scholar] [CrossRef] [Green Version]
- Savage, S.A. Dyskeratosis Congenita; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; GeneReviews®, University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Bergstrand, S.; Böhm, S.; Malmgren, H.; Norberg, A.; Sundin, M.; Nordgren, A.; Farnebo, M. Biallelic Mutations in WRAP53 Result in Dysfunctional Telomeres, Cajal Bodies and DNA Repair, Thereby Causing Hoyeraal–Hreidarsson Syndrome. Cell Death Dis. 2020, 11, 238. [Google Scholar] [CrossRef] [Green Version]
- Magnusson, T.; Godby, R.C.; Bachiashvili, K.; Jamy, O. First Report of Novel Heterozygous WRAP53 p.Ala522Glyfs*8 Mutation Associated Dyskeratosis Congenita. Br. J. Haematol. 2022, 196, 27–29. [Google Scholar] [CrossRef]
- Shao, Y.; Feng, S.; Huang, J.; Huo, J.; You, Y.; Zheng, Y. A Unique Homozygous WRAP53 Arg298Trp Mutation Underlies Dyskeratosis Congenita in a Chinese Han Family. BMC Med. Genet. 2018, 19, 40. [Google Scholar] [CrossRef]
- Barkats, M. Amyotrophie Spinale Infantile. Médecine/Sci. 2020, 36, 137–140. [Google Scholar] [CrossRef] [Green Version]
- Zhong, F.; Savage, S.A.; Shkreli, M.; Giri, N.; Jessop, L.; Myers, T.; Chen, R.; Alter, B.P.; Artandi, S.E. Disruption of Telomerase Trafficking by TCAB1 Mutation Causes Dyskeratosis Congenita. Genes Dev. 2011, 25, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Dokal, I. Dyskeratosis Congenita. Hematology 2011, 2011, 480–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kansagra, A.; Dahiya, S.; Litzow, M. Continuing Challenges and Current Issues in Acute Lymphoblastic Leukemia. Leuk. Lymphoma 2018, 59, 526–541. [Google Scholar] [CrossRef] [PubMed]
- Venteicher, A.S.; Artandi, S.E. TCAB1: Driving Telomerase to Cajal Bodies. Cell Cycle 2009, 8, 1329–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farnebo, M. Wrap53, a Novel Regulator of P53. Cell Cycle 2009, 8, 2343–2346. [Google Scholar] [CrossRef]
- Gall, J.G. The Centennial of the Cajal Body. Nat. Rev. Mol. Cell Biol. 2003, 4, 975–980. [Google Scholar] [CrossRef]
- Gall, J.G. Cajal Bodies: The First 100 Years. Annu. Rev. Cell Dev. Biol. 2000, 16, 273–300. [Google Scholar] [CrossRef]
- Cristofari, G.; Adolf, E.; Reichenbach, P.; Sikora, K.; Terns, R.M.; Terns, M.P.; Lingner, J. Human Telomerase RNA Accumulation in Cajal Bodies Facilitates Telomerase Recruitment to Telomeres and Telomere Elongation. Mol. Cell 2007, 27, 882–889. [Google Scholar] [CrossRef]
- Cioce, M.; Lamond, A.I. Cajal Bodies: A Long History of Discovery. Annu. Rev. Cell Dev. Biol. 2005, 21, 105–131. [Google Scholar] [CrossRef] [Green Version]
- Lafarga, M.; Tapia, O.; Romero, A.M.; Berciano, M.T. Cajal Bodies in Neurons. RNA Biol. 2017, 14, 712–725. [Google Scholar] [CrossRef] [Green Version]
- Machyna, M.; Neugebauer, K.M.; Staněk, D. Coilin: The First 25 Years. RNA Biol. 2015, 12, 590–596. [Google Scholar] [CrossRef]
- Tucker, K.E.; Berciano, M.T.; Jacobs, E.Y.; LePage, D.F.; Shpargel, K.B.; Rossire, J.J.; Chan, E.K.L.; Lafarga, M.; Conlon, R.A.; Matera, A.G. Residual Cajal Bodies in Coilin Knockout Mice Fail to Recruit Sm SnRNPs and SMN, the Spinal Muscular Atrophy Gene Product. J. Cell Biol. 2001, 154, 293–308. [Google Scholar] [CrossRef] [Green Version]
- Kiss, T. Biogenesis of Small Nuclear RNPs. J. Cell Sci. 2004, 117, 5949–5951. [Google Scholar] [CrossRef] [Green Version]
- Praveen, K.; Wen, Y.; Gray, K.M.; Noto, J.J.; Patlolla, A.R.; van Duyne, G.D.; Matera, A.G. SMA-Causing Missense Mutations in Survival Motor Neuron (Smn) Display a Wide Range of Phenotypes When Modeled in Drosophila. PLoS Genet. 2014, 10, e1004489. [Google Scholar] [CrossRef] [Green Version]
- Deryusheva, S.; Gall, J.G. ScaRNAs and SnoRNAs: Are They Limited to Specific Classes of Substrate RNAs? RNA 2019, 25, 17–22. [Google Scholar] [CrossRef]
- Kiss, T. Small Nucleolar RNAs. Cell 2002, 109, 145–148. [Google Scholar] [CrossRef] [Green Version]
- Richard, P. A Common Sequence Motif Determines the Cajal Body-Specific Localization of Box H/ACA ScaRNAs. EMBO J. 2003, 22, 4283–4293. [Google Scholar] [CrossRef]
- Herrmann, M.; Pusceddu, I.; März, W.; Herrmann, W. Telomere Biology and Age-Related Diseases. Clin. Chem. Lab. Med. (CCLM) 2018, 56, 1210–1222. [Google Scholar] [CrossRef]
- Artandi, S.E.; DePinho, R.A. Telomeres and Telomerase in Cancer. Carcinogenesis 2010, 31, 9–18. [Google Scholar] [CrossRef]
- Jády, B.E.; Richard, P.; Bertrand, E.; Kiss, T. Cell Cycle-Dependent Recruitment of Telomerase RNA and Cajal Bodies to Human Telomeres. Mol. Biol. Cell 2006, 17, 944–954. [Google Scholar] [CrossRef] [Green Version]
- Henriksson, S.; Rassoolzadeh, H.; Hedström, E.; Coucoravas, C.; Julner, A.; Goldstein, M.; Imreh, G.; Zhivotovsky, B.; Kastan, M.B.; Helleday, T.; et al. The Scaffold Protein WRAP53β Orchestrates the Ubiquitin Response Critical for DNA Double-Strand Break Repair. Genes Dev. 2014, 28, 2726–2738. [Google Scholar] [CrossRef] [PubMed]
- Iyama, T.; Wilson, D.M. DNA Repair Mechanisms in Dividing and Non-Dividing Cells. DNA Repair 2013, 12, 620–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rassoolzadeh, H.; Coucoravas, C.; Farnebo, M. The Proximity Ligation Assay Reveals That at DNA Double-Strand Breaks WRAP53β Associates with ΓH2AX and Controls Interactions between RNF8 and MDC1. Nucleus 2015, 6, 417–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rassoolzadeh, H.; Böhm, S.; Hedström, E.; Gad, H.; Helleday, T.; Henriksson, S.; Farnebo, M. Overexpression of the Scaffold WD40 Protein WRAP53β Enhances the Repair of and Cell Survival from DNA Double-Strand Breaks. Cell Death Dis. 2016, 7, e2267. [Google Scholar] [CrossRef] [Green Version]
- Lefebvre, S.; Bürglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and Characterization of a Spinal Muscular Atrophy-Determining Gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Rochette, C.; Gilbert, N.; Simard, L. SMN Gene Duplication and the Emergence of the SMN2 Gene Occurred in Distinct Hominids: SMN2 Is Unique to Homo Sapiens. Hum. Genet. 2001, 108, 255–266. [Google Scholar] [CrossRef]
- Lefebvre, S.; Burlet, P.; Liu, Q.; Bertrandy, S.; Clermont, O.; Munnich, A.; Dreyfuss, G.; Melki, J. Correlation between Severity and SMN Protein Level in Spinal Muscular Atrophy. Nat. Genet. 1997, 16, 265–269. [Google Scholar] [CrossRef]
- Hebert, M.D.; Poole, A.R. Towards an Understanding of Regulating Cajal Body Activity by Protein Modification. RNA Biol. 2017, 14, 761–778. [Google Scholar] [CrossRef] [Green Version]
- Dokal, I. Dyskeratosis Congenita in All Its Forms. Br. J. Haematol. 2000, 110, 768–779. [Google Scholar] [CrossRef]
- Tummala, H.; Walne, A.; Collopy, L.; Cardoso, S.; de la Fuente, J.; Lawson, S.; Powell, J.; Cooper, N.; Foster, A.; Mohammed, S.; et al. Poly(A)-Specific Ribonuclease Deficiency Impacts Telomere Biology and Causes Dyskeratosis Congenita. J. Clin. Investig. 2015, 125, 2151–2160. [Google Scholar] [CrossRef]
- Heiss, N.S.; Knight, S.W.; Vulliamy, T.J.; Klauck, S.M.; Wiemann, S.; Mason, P.J.; Poustka, A.; Dokal, I. X-Linked Dyskeratosis Congenita Is Caused by Mutations in a Highly Conserved Gene with Putative Nucleolar Functions. Nat. Genet. 1998, 19, 32–38. [Google Scholar] [CrossRef]
- Vulliamy, T.; Beswick, R.; Kirwan, M.; Marrone, A.; Digweed, M.; Walne, A.; Dokal, I. Mutations in the Telomerase Component NHP2 Cause the Premature Ageing Syndrome Dyskeratosis Congenita. Proc. Natl. Acad. Sci. USA 2008, 105, 8073–8078. [Google Scholar] [CrossRef] [Green Version]
- Ballew, B.J.; Savage, S.A. Updates on the Biology and Management of Dyskeratosis Congenita and Related Telomere Biology Disorders. Expert Rev. Hematol. 2013, 6, 327–337. [Google Scholar] [CrossRef] [Green Version]
- Vasko, T.; Kaifie, A.; Stope, M.; Kraus, T.; Ziegler, P. Telomeres and Telomerase in Hematopoietic Dysfunction: Prognostic Implications and Pharmacological Interventions. Int. J. Mol. Sci. 2017, 18, 2267. [Google Scholar] [CrossRef] [Green Version]
- Albanell, J.; Bosl, G.J.; Reuter, V.E.; Engelhardt, M.; Franco, S.; Moore, M.A.S.; Dmitrovsky, E. Telomerase Activity in Germ Cell Cancers and Mature Teratomas. JNCI J. Natl. Cancer Inst. 1999, 91, 1321–1326. [Google Scholar] [CrossRef] [Green Version]
- Fiorini, E.; Santoni, A.; Colla, S. Dysfunctional Telomeres and Hematological Disorders. Differentiation 2018, 100, 1–11. [Google Scholar] [CrossRef]
- M’kacher, R.; Colicchio, B.; Borie, C.; Junker, S.; Marquet, V.; Heidingsfelder, L.; Soehnlen, K.; Najar, W.; Hempel, W.M.; Oudrhiri, N.; et al. Telomere and Centromere Staining Followed by M-FISH Improves Diagnosis of Chromosomal Instability and Its Clinical Utility. Genes 2020, 11, 475. [Google Scholar] [CrossRef]
- Mehrotra, M.; Luthra, R.; Ravandi, F.; Sargent, R.L.; Barkoh, B.A.; Abraham, R.; Mishra, B.M.; Medeiros, L.J.; Patel, K.P. Identification of Clinically Important Chromosomal Aberrations in Acute Myeloid Leukemia by Array-Based Comparative Genomic Hybridization. Leuk. Lymphoma 2014, 55, 2538–2548. [Google Scholar] [CrossRef] [Green Version]
- Terwilliger, T.; Abdul-Hay, M. Acute Lymphoblastic Leukemia: A Comprehensive Review and 2017 Update. Blood Cancer J. 2017, 7, e577. [Google Scholar] [CrossRef] [Green Version]
- Nogueira, B.M.D.; da Costa Pantoja, L.; da Silva, E.L.; Mello Júnior, F.A.R.; Teixeira, E.B.; Wanderley, A.V.; da Silva Maués, J.H.; de Moraes Filho, M.O.; de Moraes, M.E.A.; Montenegro, R.C.; et al. Telomerase (HTERT) Overexpression Reveals a Promising Prognostic Biomarker and Therapeutical Target in Different Clinical Subtypes of Pediatric Acute Lymphoblastic Leukaemia. Genes 2021, 12, 1632. [Google Scholar] [CrossRef] [PubMed]
- Lansdorp, P.M. Maintenance of Telomere Length in AML. Blood Adv. 2017, 1, 2467–2472. [Google Scholar] [CrossRef] [PubMed]
- Keller, G.; Brassat, U.; Braig, M.; Heim, D.; Wege, H.; Brümmendorf, T.H. Telomeres and Telomerase in Chronic Myeloid Leukaemia: Impact for Pathogenesis, Disease Progression and Targeted Therapy. J. Hematol. Oncol. 2009, 27, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Davison, G.M. Telomeres and Telomerase in Leukaemia and Lymphoma. Transfus. Apher. Sci. 2007, 37, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Yik, M.Y.; Azlan, A.; Rajasegaran, Y.; Rosli, A.; Yusoff, N.M.; Moses, E.J. Mechanism of Human Telomerase Reverse Transcriptase (HTERT) Regulation and Clinical Impacts in Leukemia. Genes 2021, 12, 1188. [Google Scholar] [CrossRef]
- Jebaraj, B.M.C.; Stilgenbauer, S. Telomere Dysfunction in Chronic Lymphocytic Leukemia. Front. Oncol. 2021, 10, 612665. [Google Scholar] [CrossRef]
- Mascarenhas, J.O.; Komrokji, R.S.; Cavo, M.; Martino, B.; Niederwieser, D.; Reiter, A.; Scott, B.L.; Baer, M.R.; Hoffman, R.; Odenike, O.; et al. Telomerase Activity, Telomere Length and HTERT Expression Correlate with Clinical Outcomes in Higher-Risk Myelofibrosis (MF) Relapsed/Refractory (R/R) to Janus Kinase Inhibitor Treated with Imetelstat. Hemasphere 2020, 4, 1098. [Google Scholar]
- Berardinelli, F.; Nieri, D.; Sgura, A.; Tanzarella, C.; Antoccia, A. Telomere Loss, Not Average Telomere Length, Confers Radiosensitivity to TK6-Irradiated Cells. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2012, 740, 13–20. [Google Scholar] [CrossRef]
- McCaul, J.A.; Gordon, K.E.; Minty, F.; Fleming, J.; Parkinson, E.K. Telomere Dysfunction Is Related to the Intrinsic Radio-Resistance of Human Oral Cancer Cells. Oral Oncol. 2008, 44, 261–269. [Google Scholar] [CrossRef]
- Nogueira, B.M.D.; Machado, C.B.; Montenegro, R.C.; de Moraes, M.E.A.; Moreira-Nunes, C.A. Telomere Length and Hematological Disorders: A Review. In Vivo 2020, 34, 3093–3101. [Google Scholar] [CrossRef]
- Andor, N.; Maley, C.C.; Ji, H.P. Genomic Instability in Cancer: Teetering on the Limit of Tolerance. Cancer Res. 2017, 77, 2179–2185. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, J. Ueber Chronische Spinale Muskelatrophie Im Kindesalter, Auf Familiärer Basis. Dtsch. Z. Nervenheilkd. 1893, 3, 427–470. [Google Scholar] [CrossRef]
- Werdnig, G. Zwei Frühinfantile Hereditäre Fälle von Progressiver Muskelatrophie Unter Dem Bilde Der Dystrophie, Aber Anf Neurotischer Grundlage. Arch. Psychiatr. Nervenkr. 1891, 22, 437–480. [Google Scholar] [CrossRef] [Green Version]
- Goh, A.M.; Coffill, C.R.; Lane, D.P. The Role of Mutant P53 in Human Cancer. J. Pathol. 2011, 223, 116–126. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational Landscape and Significance across 12 Major Cancer Types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, F.; Collavin, L.; del Sal, G. Mutant P53 as a Guardian of the Cancer Cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef] [Green Version]
- Leroy, B.; Anderson, M.; Soussi, T. TP53 Mutations in Human Cancer: Database Reassessment and Prospects for the Next Decade. Hum. Mutat. 2014, 35, 672–688. [Google Scholar] [CrossRef]
- Weinstein, I.B.; Joe, A. Oncogene Addiction. Cancer Res. 2008, 68, 3077–3080. [Google Scholar] [CrossRef] [Green Version]
- Mina, M.; Raynaud, F.; Tavernari, D.; Battistello, E.; Sungalee, S.; Saghafinia, S.; Laessle, T.; Sanchez-Vega, F.; Schultz, N.; Oricchio, E.; et al. Conditional Selection of Genomic Alterations Dictates Cancer Evolution and Oncogenic Dependencies. Cancer Cell 2017, 32, 155–168.e6. [Google Scholar] [CrossRef]
- Sun, Y.; Yang, C.; Chen, J.; Song, X.; Li, Z.; Duan, M.; Li, J.; Hu, X.; Wu, K.; Yan, G.; et al. Overexpression of WDR 79 in Non-small Cell Lung Cancer Is Linked to Tumour Progression. J. Cell Mol. Med. 2016, 20, 698–709. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Cao, L.; Sheng, X.; Chen, J.; Zhou, Y.; Yang, C.; Deng, T.; Ma, H.; Feng, P.; Liu, J.; et al. WDR79 Promotes the Proliferation of Non-Small Cell Lung Cancer Cells via USP7-Mediated Regulation of the Mdm2-P53 Pathway. Cell Death Dis. 2017, 8, e2743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Sheng, X.; Ma, H.; Tang, Z.; Yang, C.; Cao, L.; Sun, Y.; Deng, T.; Feng, P.; Hu, B.; et al. WDR79 Mediates the Proliferation of Non-small Cell Lung Cancer Cells by Regulating the Stability of UHRF1. J. Cell Mol. Med. 2018, 22, 2856–2864. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Zhan, Y.; Feng, J.; Fan, S.; Zang, H. Expression of WDR79 Is Associated with TP53 Mutation and Poor Prognosis in Surgically Resected Non-Small Cell Lung Cancer. J. Cancer 2019, 10, 3046–3053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, X.-S.; Cao, L.-X.; Hu, Y.-J.; Bao, F.-C.; Wang, Z.-T.; Cao, J.-L.; Yuan, P.; Lv, W.; Hu, J. Clinical, Cellular, and Bioinformatic Analyses Reveal Involvement of WRAP53 Overexpression in Carcinogenesis of Lung Adenocarcinoma. Tumor Biol. 2017, 39, 101042831769430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, X.; Huang, D.; Sui, X.; Liu, G.; Song, X.; Xie, J.; Huang, D. Overexpression of WRAP53 Is Associated with Development and Progression of Esophageal Squamous Cell Carcinoma. PLoS ONE 2014, 9, e91670. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, D.-W.; Adell, G.; Sun, X.-F. WRAP53 Is an Independent Prognostic Factor in Rectal Cancer- a Study of Swedish Clinical Trial of Preoperative Radiotherapy in Rectal Cancer Patients. BMC Cancer 2012, 12, 294. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.-J.; Ping, J.; Li, Y.; Adell, G.; Arbman, G.; Nodin, B.; Meng, W.-J.; Zhang, H.; Yu, Y.-Y.; Wang, C.; et al. The Prognostic Factors and Multiple Biomarkers in Young Patients with Colorectal Cancer. Sci. Rep. 2015, 5, 10645. [Google Scholar] [CrossRef] [Green Version]
- Wen, Y.; Zhao, S.; Holmqvist, A.; Hahn-Stromberg, V.; Adell, G.; Holmlund, B.; Pathak, S.; Peng, Z.; Sun, X.-F. Predictive Role of Biopsy Based Biomarkers for Radiotherapy Treatment in Rectal Cancer. J. Pers. Med. 2020, 10, 168. [Google Scholar] [CrossRef]
- Meng, W.-J.; Pathak, S.; Zhang, X.; Adell, G.; Jarlsfelt, I.; Holmlund, B.; Wang, Z.-Q.; Zhang, A.S.; Zhang, H.; Zhou, Z.-G.; et al. Expressions of MiR-302a, MiR-105, and MiR-888 Play Critical Roles in Pathogenesis, Radiotherapy, and Prognosis on Rectal Cancer Patients: A Study from Rectal Cancer Patients in a Swedish Rectal Cancer Trial of Preoperative Radiotherapy to Big Database Analyses. Front. Oncol. 2020, 10, 567042. [Google Scholar] [CrossRef]
- Zhu, Y.; Ding, L.; Chen, B.-F.; Song, J.-G.; Yao, Y.-S. Oncogenic Activity of Wrap53 in Human Colorectal Cancer In Vitro and in Nude Mouse Xenografts. Med. Sci. Monit. 2018, 24, 6129–6136. [Google Scholar] [CrossRef]
- Kamel, M.M.; Matboli, M.; Sallam, M.; Montasser, I.F.; Saad, A.S.; El-Tawdi, A.H.F. Investigation of Long Noncoding RNAs Expression Profile as Potential Serum Biomarkers in Patients with Hepatocellular Carcinoma. Transl. Res. 2016, 168, 134–145. [Google Scholar] [CrossRef]
- Schildkraut, J.M.; Goode, E.L.; Clyde, M.A.; Iversen, E.S.; Moorman, P.G.; Berchuck, A.; Marks, J.R.; Lissowska, J.; Brinton, L.; Peplonska, B.; et al. Single Nucleotide Polymorphisms in the TP53 Region and Susceptibility to Invasive Epithelial Ovarian Cancer. Cancer Res. 2009, 69, 2349–2357. [Google Scholar] [CrossRef]
- Hedström, E.; Pederiva, C.; Farnebo, J.; Nodin, B.; Jirström, K.; Brennan, D.J.; Farnebo, M. Downregulation of the Cancer Susceptibility Protein WRAP53β in Epithelial Ovarian Cancer Leads to Defective DNA Repair and Poor Clinical Outcome. Cell Death Dis. 2015, 6, e1892. [Google Scholar] [CrossRef] [Green Version]
- Mędrek, K.; Magnowski, P.; Masojć, B.; Chudecka-Głaz, A.; Torbe, B.; Menkiszak, J.; Spaczyński, M.; Gronwald, J.; Lubiński, J.; Górski, B. Association of Common WRAP 53 Variant with Ovarian Cancer Risk in the Polish Population. Mol. Biol. Rep. 2013, 40, 2145–2147. [Google Scholar] [CrossRef] [Green Version]
- Silwal-Pandit, L.; Russnes, H.; Borgen, E.; Skarpeteig, V.; Moen Vollan, H.K.; Schlichting, E.; Kåresen, R.; Naume, B.; Børresen-Dale, A.-L.; Farnebo, M.; et al. The Sub-Cellular Localization of WRAP53 Has Prognostic Impact in Breast Cancer. PLoS ONE 2015, 10, e0139965. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Closas, M.; Kristensen, V.; Langerød, A.; Qi, Y.; Yeager, M.; Burdett, L.; Welch, R.; Lissowska, J.; Peplonska, B.; Brinton, L.; et al. Common Genetic Variation in TP53 and Its Flanking Genes, WDR79 and ATP1B2, and Susceptibility to Breast Cancer. Int. J. Cancer 2007, 121, 2532–2538. [Google Scholar] [CrossRef]
- Pouladi, N.; Abdolahi, S.; Farajzadeh, D.; Hosseinpour Feizi, M.A. Haplotype and Linkage Disequilibrium of TP53-WRAP53 Locus in Iranian-Azeri Women with Breast Cancer. PLoS ONE 2019, 14, e0220727. [Google Scholar] [CrossRef] [Green Version]
- Mahmoudi, S.; Henriksson, S.; Farnebo, L.; Roberg, K.; Farnebo, M. WRAP53 Promotes Cancer Cell Survival and Is a Potential Target for Cancer Therapy. Cell Death Dis. 2011, 2, e114. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Luo, X.; Gou, Y.; Hu, L.; Wang, K.; Li, C.; Xiang, Z.; Zhang, P.; Kong, X.; Zhang, C.; et al. TCAB1: A Potential Target for Diagnosis and Therapy of Head and Neck Carcinomas. Mol. Cancer 2014, 13, 180. [Google Scholar] [CrossRef] [Green Version]
- Tiefenböck-Hansson, K.; Haapaniemi, A.; Farnebo, L.; Palmgren, B.; Tarkkanen, J.; Farnebo, M.; Munck-Wikland, E.; Mäkitie, A.; Garvin, S.; Roberg, K. WRAP53β, Survivin and P16INK4a Expression as Potential Predictors of Radiotherapy/Chemoradiotherapy Response in T2N0-T3N0 Glottic Laryngeal Cancer. Oncol. Rep. 2017, 38, 2062–2068. [Google Scholar] [CrossRef] [Green Version]
- Garvin, S.; Tiefenböck, K.; Farnebo, L.; Thunell, L.K.; Farnebo, M.; Roberg, K. Nuclear Expression of WRAP53β Is Associated with a Positive Response to Radiotherapy and Improved Overall Survival in Patients with Head and Neck Squamous Cell Carcinoma. Oral Oncol. 2015, 51, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Zhao, D.-Y.; Yuan, L.-M.; Zhang, G.; Xie, C.-H. Regulatory Effects of WRAP53 on Radiosensitivity of Laryngeal Squamous Cell Carcinoma Cells. Asian Pac. J. Cancer Prev. 2015, 16, 2975–2979. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Kim, P.; Jia, P.; Park, A.K.; Liang, H.; Zhao, Z. Distinct Telomere Length and Molecular Signatures in Seminoma and Non-Seminoma of Testicular Germ Cell Tumor. Brief. Bioinform. 2019, 20, 1502–1512. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Ge, Y.; Ni, C.; Cui, B.; Du, J.; Zhang, B.; Hu, X.; Chen, J.; Xiao, L.; Sun, C.; et al. Epstein-Barr Virus-Induced up-Regulation of TCAB1 Is Involved in the DNA Damage Response in Nasopharyngeal Carcinoma. Sci. Rep. 2017, 7, 3218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, R.; Mitra, A. An Overview of Effective Therapies and Recent Advances in Biomarkers for Chronic Liver Diseases and Associated Liver Cancer. Int. Immunopharmacol. 2015, 24, 335–345. [Google Scholar] [CrossRef]
- Altekruse, S.F.; McGlynn, K.A.; Reichman, M.E. Hepatocellular Carcinoma Incidence, Mortality, and Survival Trends in the United States From 1975 to 2005. J. Clin. Oncol. 2009, 27, 1485–1491. [Google Scholar] [CrossRef] [Green Version]
- Takayama, T.; Makuuchi, M.; Kojiro, M.; Lauwers, G.Y.; Adams, R.B.; Wilson, S.R.; Jang, H.-J.; Charnsangavej, C.; Taouli, B. Early Hepatocellular Carcinoma: Pathology, Imaging, and Therapy. Ann. Surg. Oncol. 2008, 15, 972–978. [Google Scholar] [CrossRef]
- Kyo, S.; Inoue, M. Complex Regulatory Mechanisms of Telomerase Activity in Normal and Cancer Cells: How Can We Apply Them for Cancer Therapy? Oncogene 2002, 21, 688–697. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.-F. Relationships of Tumor Inflammatory Infiltration and Necrosis with Microsatellite Instability in Colorectal Cancers. World J. Gastroenterol. 2005, 11, 2179. [Google Scholar] [CrossRef]
- Pollheimer, M.J.; Kornprat, P.; Lindtner, R.A.; Harbaum, L.; Schlemmer, A.; Rehak, P.; Langner, C. Tumor Necrosis Is a New Promising Prognostic Factor in Colorectal Cancer. Hum. Pathol. 2010, 41, 1749–1757. [Google Scholar] [CrossRef]
- Park, J.; Cho, J.; Song, E.J. Ubiquitin–Proteasome System (UPS) as a Target for Anticancer Treatment. Arch. Pharm. Res. 2020, 43, 1144–1161. [Google Scholar] [CrossRef]
- Zhang, X.; Linder, S.; Bazzaro, M. Drug Development Targeting the Ubiquitin–Proteasome System (UPS) for the Treatment of Human Cancers. Cancers 2020, 12, 902. [Google Scholar] [CrossRef]
- Shammas, M.A.; Koley, H.; Batchu, R.B.; Bertheau, R.C.; Protopopov, A.; Munshi, N.C.; Goyal, R.K. Telomerase Inhibition by SiRNA Causes Senescence and Apoptosis in Barrett’s Adenocarcinoma Cells: Mechanism and Therapeutic Potential. Mol. Cancer 2005, 4, 24. [Google Scholar] [CrossRef] [Green Version]
- Stewart, Z.A.; Westfall, M.D.; Pietenpol, J.A. Cell-Cycle Dysregulation and Anticancer Therapy. Trends Pharmacol. Sci. 2003, 24, 139–145. [Google Scholar] [CrossRef]
- Watkins, N.J.; Bohnsack, M.T. The Box C/D and H/ACA SnoRNPs: Key Players in the Modification, Processing and the Dynamic Folding of Ribosomal RNA. Wiley Interdiscip. Rev. RNA 2012, 3, 397–414. [Google Scholar] [CrossRef]
- Qiu, X.-B.; Lin, Y.-L.; Thome, K.C.; Pian, P.; Schlegel, B.P.; Weremowicz, S.; Parvin, J.D.; Dutta, A. An Eukaryotic RuvB-like Protein (RUVBL1) Essential for Growth. J. Biol. Chem. 1998, 273, 27786–27793. [Google Scholar] [CrossRef] [Green Version]
- Lund, A.; Knudsen, S.M.; Vissing, H.; Clark, B.; Tommerup, N. Assignment of Human Elongation Factor 1α Genes: EEF1AMaps to Chromosome 6q14 AndEEF1A2to 20q13.3. Genomics 1996, 36, 359–361. [Google Scholar] [CrossRef]
- Dayton, T.L.; Jacks, T.; vander Heiden, M.G. PKM2, Cancer Metabolism, and the Road Ahead. EMBO Rep. 2016, 17, 1721–1730. [Google Scholar] [CrossRef] [Green Version]
- Chang, M.T.; Asthana, S.; Gao, S.P.; Lee, B.H.; Chapman, J.S.; Kandoth, C.; Gao, J.; Socci, N.D.; Solit, D.B.; Olshen, A.B.; et al. Identifying Recurrent Mutations in Cancer Reveals Widespread Lineage Diversity and Mutational Specificity. Nat. Biotechnol. 2016, 34, 155–163. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S.; Fisher, K.; Snowden, J.; Danson, S.; Brown, S.; Zeidler, M. Effect of Methotrexate on JAK/STAT Pathway Activation in Myeloproliferative Neoplasms. Lancet 2015, 385, S98. [Google Scholar] [CrossRef]
- Wen, X. The PI3K AKT Pathway in the Pathogenesis of Prostate Cancer. Front. Biosci. 2016, 21, 4443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, E.G.; Kim, M.S.; Nam, H.K.; Min, C.K.; Lee, S.; Chung, Y.J.; Yoo, N.J.; Lee, S.H. Somatic Mutations of JAK1 and JAK3 in Acute Leukemias and Solid Cancers. Clin. Cancer Res. 2008, 14, 3716–3721. [Google Scholar] [CrossRef]
- Brimo, F.; Wu, C.; Zeizafoun, N.; Tanguay, S.; Aprikian, A.; Mansure, J.J.; Kassouf, W. Prognostic Factors in T1 Bladder Urothelial Carcinoma: The Value of Recording Millimetric Depth of Invasion, Diameter of Invasive Carcinoma, and Muscularis Mucosa Invasion. Hum. Pathol. 2013, 44, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Wu, J.; Wang, W.; Fei, X.; Zong, Y.; Chen, X.; Huang, O.; He, J.; Chen, W.; Li, Y.; et al. Biologic Behavior and Long-Term Outcomes of Breast Ductal Carcinoma in Situ with Microinvasion. Oncotarget 2016, 7, 64182–64190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Cao, Y.; Ding, M.; Liu, J.; Zuo, X.; Li, H.; Fan, R. Oncological and Prognostic Impact of Lymphovascular Invasion in Colorectal Cancer Patients. Int. J. Med. Sci. 2021, 18, 1721–1729. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-Y.; Yang, M.-C.; Huang, C.-H.; Wu, W.-J.; Yu, T.-J.; Lung, F.-W. Polymorphisms of TP53 Are Markers of Bladder Cancer Vulnerability and Prognosis. Urol. Oncol. Semin. Orig. Investig. 2013, 31, 1231–1241. [Google Scholar] [CrossRef]
- Ran, R.; Tu, G.; Li, H.; Wang, H.; Mou, E.; Liu, C. Genetic Variants Associated with Thyroid Cancer Risk: Comprehensive Research Synopsis, Meta-Analysis, and Cumulative Epidemiological Evidence. J. Oncol. 2021, 2021, 9967599. [Google Scholar] [CrossRef]
- Li, Q.; Liu, X.; Hua, R.-X.; Wang, F.; An, H.; Zhang, W.; Zhu, J.-H. Association of Three 8q24 Polymorphisms with Prostate Cancer Susceptibility: Evidence from a Meta-Analysis with 50,854 Subjects. Sci. Rep. 2015, 5, 12069. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Wen, X.; Liu, X.; Wang, Y.; Liang, C.; Tu, J. Association between 8q24 Rs6983267 Polymorphism and Cancer Susceptibility: A Meta-Analysis Involving 170,737 Subjects. Oncotarget 2017, 8, 57421–57439. [Google Scholar] [CrossRef] [Green Version]
- Orth, M.; Lauber, K.; Niyazi, M.; Friedl, A.A.; Li, M.; Maihöfer, C.; Schüttrumpf, L.; Ernst, A.; Niemöller, O.M.; Belka, C. Current Concepts in Clinical Radiation Oncology. Radiat. Environ. Biophys. 2014, 53, 1–29. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Li, L.L.; Yang, J.; Tao, Y.G.; Ye, M.; Shi, Y.; Tang, M.; Yi, W.; Li, X.L.; Gong, J.P.; et al. Epstein–Barr Virus Encoded Latent Membrane Protein 1 Modulates Nuclear Translocation of Telomerase Reverse Transcriptase Protein by Activating Nuclear Factor-ΚB P65 in Human Nasopharyngeal Carcinoma Cells. Int. J. Biochem. Cell Biol. 2005, 37, 1881–1889. [Google Scholar] [CrossRef]
- Mei, Y.-P.; Zhu, X.-F.; Zhou, J.-M.; Huang, H.; Deng, R.; Zeng, Y.-X. SiRNA Targeting LMP1-Induced Apoptosis in EBV-Positive Lymphoma Cells Is Associated with Inhibition of Telomerase Activity and Expression. Cancer Lett. 2006, 232, 189–198. [Google Scholar] [CrossRef]
- Terrin, L.; Dal Col, J.; Rampazzo, E.; Zancai, P.; Pedrotti, M.; Ammirabile, G.; Bergamin, S.; Rizzo, S.; Dolcetti, R.; de Rossi, A. Latent Membrane Protein 1 of Epstein-Barr Virus Activates the HTERT Promoter and Enhances Telomerase Activity in B Lymphocytes. J. Virol. 2008, 82, 10175–10187. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Deng, X.; Deng, L.; Gu, H.; Fan, W.; Cao, Y. Telomerase Activation by Epstein-Barr Virus Latent Membrane Protein 1 Is Associated with c-Myc Expression in Human Nasopharyngeal Epithelial Cells. J. Exp. Clin. Cancer Res. 2004, 23, 495–506. [Google Scholar]
- Yang, L.; Xu, Z.; Liu, L.; Luo, X.; Lu, J.; Sun, L.; Cao, Y. Targeting EBV-LMP1 DNAzyme Enhances Radiosensitivity of Nasopharyngeal Carcinoma Cells by Inhibiting Telomerase Activity. Cancer Biol. Ther. 2014, 15, 61–68. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Cancer Subtype | Biological Samples and Methodology | Molecular Mechanisms | Clinical Status | Clinical Outcome or Overall Survival (OS) | Referen-ce |
|---|---|---|---|---|---|
| NSCLC | Patient samples; A549, H1299 95-C, 95-D, HTB182, and HBE and in vivo | WRAP53β plays an important role in tumorigenesis regulating cell cycle progression and apoptosis. | N.I | N.I | [101] |
| H1299 and A549 cells | WRAP53β induced clonal proliferation through mediation of USP7-MDM2-TP53 pathway. | N.I | N.I | [102] | |
| H1299 and A549 cells | WRAP53β induced clonal proliferation through UHRF1 activity. | N.I | N.I | [103] | |
| Patient samples | WRAP53β overexpression may act as an independent biomarker to predict a poor prognosis. | Patients with primary NSCLC and not previously treated with radiotherapy | WRAP5β expression was an unfavorable prognostic factor related to low overall survival rates. | [104] | |
| Patient samples; A549 and SPC-A-1 cells | Overexpression of WRAP53β reported as a poor prognostic factor, and its downregulation reduced cell proliferation. | Patients with pathological stages I–IV | WRAP53β was present in all patients. | [105] | |
| Esophageal squamous cell carcinoma | Patient samples; KYSE150, KYSE180, EC109, and EC9706 cells | Overexpression of WRAP53β was correlated with tumor infiltration, clinical stage, and lymph node metastasis. | Patient’s selected at the first diagnosis and without the use of radiotherapy | Presence of altered expression of WRAP53β in 95.6% of cases. | [106] |
| Colorectal cancer | Patient samples | WRAP53β is overexpressed in the primary rectal tumor compared with the normal mucosa. | Patients with stages I, IIA+IIIA+IIIB, and IIIC+IV of the disease | In the group without radiotherapy or metastasis, WRAP53β expression was associated with a worse prognosis. | [107] |
| in silico analysis of clinicopathological features | WRAP53β is a biomarker of prognoses for young patients. | Patients with stages: I, II, III, IV, and stage 0 and missing cases | WRAP53β was shown to be differentially expressed between the young and elderly groups. | [108] | |
| Patient samples | Overexpression of cytoplasmic or nuclear WRAP53β is indicative of poor prognosis. | N.I | Patients with high expression of cytoplasmic WRAP53β have a low OS and DFS, while its nuclear presence impairs the radiotherapy response. | [109] | |
| Patient samples | Overexpression WRAP53β is reported for colorectal cancer. | Patients with stages I, II, III, and IV of the disease | Cancer patients in the necrotic state had a strong expression of WRAP53β. | [110] | |
| In silico analysis of TCGA database; SW480, HT-29, HCT116, and LoVo cells and in vivo | The elimination of WRAP53β reduced tumor cell proliferation and invasion. | N.I | Higher expression of WRAP53β was observed in colorectal cancer tissues than in normal tissues. | [111] | |
| Hepatocellular carcinoma | Patient samples | WRAP53α has clinical value as a promising biomarker with precision in primary screening in hepatocarcinoma and HCV. | Patients with primary HCC and patients with chronic HCV infection | Patients with positive WRAP53α RNA are related to a lower DFS. | [112] |
| Ovarian cancer | Patient samples | The rs2287498 polymorphism is associated with increased risk of invasive ovarian cancer. | Patients with invasive epithelial ovarian cancer in non-Hispanic white women | N.I | [113] |
| Patient samples; A2780 and SKOV-3 cells | Low nuclear expression of WRAP53β correlates with aggressiveness and poor prognosis of epithelial ovarian cancer. | Patients with serous, endometrioid, mucinous, and other tumors | Aggressive disease, poor prognosis, and reduced survival of the patients. | [114] | |
| Patient samples | SNPs located in WRAP53-TP53 regions rs1042522, rs2287497, and rs2287498 are more strongly associated with a risk of ovarian cancer. | N.I | N.I | [115] | |
| Breast cancer | Patient samples | WRAP53β was shown to be a potential prognostic biomarker. | Patients with a primary breast tumor | The cellular localization of WRAP53β is linked to prognosis and OS. | [116] |
| Patient samples | Polymorphisms linked to TP53 or WRAP53 rs2287499 and rs2287498 may be associated with estrogen receptor (ER)-negative breast cancer. | Patients with stage I and II disease and invasive breast cancer | N.I | [117] | |
| Patient samples and in silico analysis | SNPs rs2287499 and rs1042522 may play an important role in breast cancer susceptibility. | N.I | N.I | [118] | |
| Head and neck carcinomas | Patient samples; U2OS, HeLa, H1299, HEK293, HCT116, HDF, AG06814, and MCF10A cells | Overexpression of WRAP53β is a marker of poor prognosis. | N.I | WRAP53β expression levels were higher in patients with recurrent disease. | [119] |
| Patient samples; HSC-3, Cal-27, and CNE1 ACC2 cells and in vivo | WRAP53β is overexpressed in clinical specimens as well as carcinoma cell lines. | Human nasopharyngeal carcinoma tissue samples and nasopharyngitis tissues | N.I | [120] | |
| Patient samples | Cytoplasmic WRAP53β is a potential predictive marker for poor response to chemoradiotherapy. | Patients treated for primary stage T2N0 or T3N0 glottic laryngeal SCC | A worse DFS and a tendency for worse OS in those patients where WRAP53β was more present in the cytoplasm compared with patients with nuclear staining. | [121] | |
| Patient samples; LK0412 and LK0949 cells | WRAP53β plays an important role in radiotherapy response, and its nuclear localization may be a promising biomarker for overall survival. | Patients with stages T1,T2,T3, N0, N1, and N2 | Patients with nuclear expression of WRAP53β demonstrated greater overall survival than those with non-nuclear staining. | [122] | |
| Hep-2 cells | WRAP53β can be an ideal target for increasing radiosensitivity. | N.I | N.I | [123] | |
| Testicular germ cell tumors | In silico analysis of 168 unique telomere-related genes | Overexpression of WRAP53 can induce telomere lengthening. | Primary tumor samples | In testicular germ cell tumors of the non-seminoma, subtype WRAP53 was overexpressed. | [124] |
| Nasopharyngeal carcinoma associated with EBV | Patient samples; CNE1, CNE1-LMP1, NP69, HOK, and B95-8 cells | EBV increases the expression of WRAP53β in vitro and, consequently, overactivates the enzymatic activity of telomerase. Its downregulation reduced cell proliferation. | N.I | N.I | [125] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gadelha, R.B.; Machado, C.B.; Pessoa, F.M.C.d.P.; Pantoja, L.d.C.; Barreto, I.V.; Ribeiro, R.M.; de Moraes Filho, M.O.; de Moraes, M.E.A.; Khayat, A.S.; Moreira-Nunes, C.A. The Role of WRAP53 in Cell Homeostasis and Carcinogenesis Onset. Curr. Issues Mol. Biol. 2022, 44, 5498-5515. https://doi.org/10.3390/cimb44110372
Gadelha RB, Machado CB, Pessoa FMCdP, Pantoja LdC, Barreto IV, Ribeiro RM, de Moraes Filho MO, de Moraes MEA, Khayat AS, Moreira-Nunes CA. The Role of WRAP53 in Cell Homeostasis and Carcinogenesis Onset. Current Issues in Molecular Biology. 2022; 44(11):5498-5515. https://doi.org/10.3390/cimb44110372
Chicago/Turabian StyleGadelha, Renan Brito, Caio Bezerra Machado, Flávia Melo Cunha de Pinho Pessoa, Laudreísa da Costa Pantoja, Igor Valentim Barreto, Rodrigo Monteiro Ribeiro, Manoel Odorico de Moraes Filho, Maria Elisabete Amaral de Moraes, André Salim Khayat, and Caroline Aquino Moreira-Nunes. 2022. "The Role of WRAP53 in Cell Homeostasis and Carcinogenesis Onset" Current Issues in Molecular Biology 44, no. 11: 5498-5515. https://doi.org/10.3390/cimb44110372
APA StyleGadelha, R. B., Machado, C. B., Pessoa, F. M. C. d. P., Pantoja, L. d. C., Barreto, I. V., Ribeiro, R. M., de Moraes Filho, M. O., de Moraes, M. E. A., Khayat, A. S., & Moreira-Nunes, C. A. (2022). The Role of WRAP53 in Cell Homeostasis and Carcinogenesis Onset. Current Issues in Molecular Biology, 44(11), 5498-5515. https://doi.org/10.3390/cimb44110372