
Tumor Molecular and Microenvironment Characteristics in EBV-Associated Malignancies as Potential Therapeutic Targets: Focus on Gastric Cancer
 , , ,
, , ,
Abstract
:1. Introduction
2. Viruses as Carcinogens
3. Direct Carcinogenesis: EBV Infectious Cycle and Related Oncogenic Alterations
3.1. EBV Cell-Cycle Dysregulation
3.2. EBV-Induced Epigenetic Mutations
3.2.1. DNA Methylation
3.2.2. Histone Acetylation
3.3. EBV miRNAs and Their Role in Immunosuppression
4. Indirect Carcinogenesis: Tumor Microenvironment (TME) in EBV-Associated Malignancies
5. EBV-Associated Gastric Cancer
EBVaGC TME
6. EBV Induces IDO1, a Potent Immunosuppressor That Potentiates Malignant Transformation
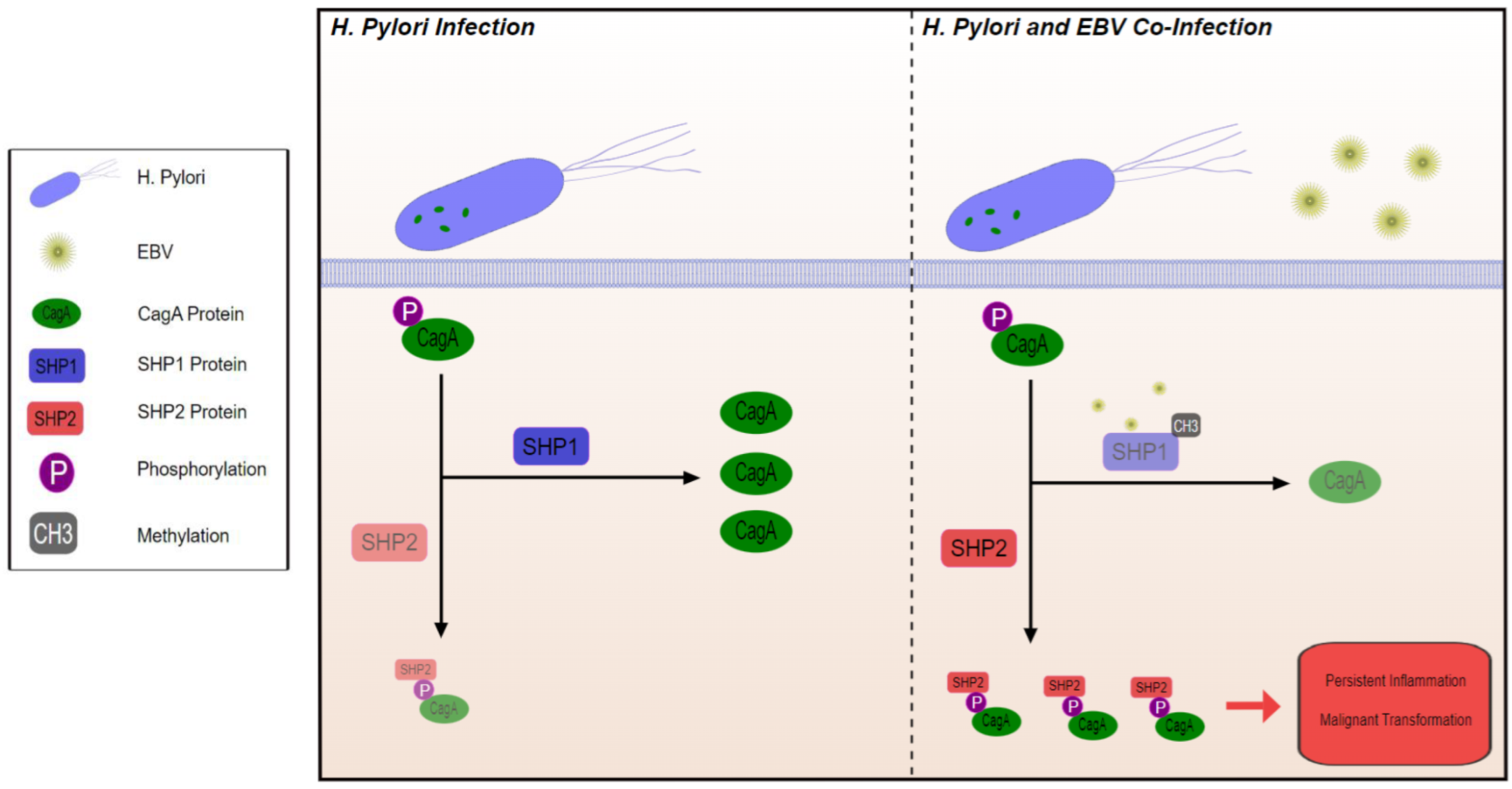
7. Microbial Community as Part of the TME: EBV and H. pylori Co-Infection
8. Tumor-Associated Macrophages (TAMS), M1 to M2 Switch Mediated by TME
9. Perspectives and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 1-methyl-tryptophan | (1MT) |
| Alternatively activated macrophages | (M2) |
| Burkitt Lymphoma | (BL) |
| Carcinoma with Crohn’s disease-like lymphoid reaction | (CLR) |
| Chimeric antigen receptor | (CAR) |
| Classically activated macrophages | (M1) |
| Conventional adenocarcinoma | (CA) |
| EBV Related Breast Cancer | (EBVrBCa) |
| EBV-associated gastric cancer | (EBVaGC) |
| Epstein–Barr Virus | (EBV) |
| Hepatitis B virus | (HBV) |
| Hepatitis C virus | (HCV) |
| High risk human papillomavirus | (HPV high-risk types) |
| Histone deacetylase inhibitors | (HDACi) |
| Hodgkin’s Lymphoma | (HL) |
| Human herpesvirus type 8 | (HHV-8) |
| Human T-cell lymphotropic virus type 1 | (HTLV) |
| IFN-regulatory factors | (IRFs) |
| IFN-stimulated genes | (ISGs) |
| Indoleamine 2,3-dioxygenase | (IDO1) |
| Infectious mononucleosis | (IM) |
| International Agency for Research on Cancer | (IARC) |
| Kynurenine | (Kyn) |
| Lymphoepithelioma-like carcinoma | (LELC) |
| Nasopharyngeal Carcinoma | (NPC) |
| NK/T Cell Lymphoma | (NKTCL) |
| The Cancer Genome Atlas | (TCGA) |
| Tryptophan | (Trp) |
| Tumor microenvironment | (TME) |
| Vascular endothelial growth factor | (VEGF) |
References
- Wong, Y.; Meehan, M.T.; Burrows, S.R.; Doolan, D.L.; Miles, J.J. Estimating the Global Burden of Epstein–Barr Virus-Related Cancers. J. Cancer Res. Clin. Oncol. 2022, 148, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Snapper, C.M. Epstein Barr Virus: Development of Vaccines and Immune Cell Therapy for EBV-Associated Diseases. Front. Immunol. 2021, 12, 734471. [Google Scholar] [CrossRef] [PubMed]
- Umakanthan, S.; Bukelo, M.M. Molecular Genetics in Epstein–Barr Virus-Associated Malignancies. Life 2021, 11, 593. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Kang, M.-S.; Kim, K.-M. Epstein-Barr Virus-Associated Gastric Carcinoma and Specific Features of the Accompanying Immune Response. J. Gastric Cancer 2016, 16, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatano, Y.; Ideta, T.; Hirata, A.; Hatano, K.; Tomita, H.; Okada, H.; Shimizu, M.; Tanaka, T.; Hara, A. Virus-Driven Carcinogenesis. Cancers 2021, 13, 2625. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Chan, Y.-T.; Tan, H.-Y.; Li, S.; Wang, N.; Feng, Y. Epigenetic Regulation in Human Cancer: The Potential Role of Epi-Drug in Cancer Therapy. Mol. Cancer 2020, 19, 79. [Google Scholar] [CrossRef]
- Lunn, R.M.; Jahnke, G.D.; Rabkin, C.S. Tumour Virus Epidemiology. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372. [Google Scholar] [CrossRef] [Green Version]
- Cancer and Infections. Available online: https://gco.iarc.fr/infections/home (accessed on 5 June 2022).
- Chen, X.; Kost, J.; Sulovari, A.; Wong, N.; Liang, W.S.; Cao, J.; Li, D. A Virome-Wide Clonal Integration Analysis Platform for Discovering Cancer Viral Etiology. Genome Res. 2019, 29, 819–830. [Google Scholar] [CrossRef] [Green Version]
- Mesri, E.A.; Feitelson, M.A.; Munger, K. Human Viral Oncogenesis: A Cancer Hallmarks Analysis. Cell Host Microbe 2014, 15, 266–282. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Longnecker, R. Epithelial Cell Infection by Epstein–Barr Virus. FEMS Microbiol. Rev. 2019, 43, 674–683. [Google Scholar] [CrossRef]
- Odumade, O.A.; Hogquist, K.A.; Balfour, H.H., Jr. Progress and Problems in Understanding and Managing Primary Epstein-Barr Virus Infections. Clin. Microbiol. Rev. 2011, 24, 193–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kempkes, B.; Robertson, E.S. Epstein-Barr Virus Latency: Current and Future Perspectives. Curr. Opin. Virol. 2015, 14, 138–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frappier, L. Epstein-Barr Virus: Current Questions and Challenges. Tumour Virus Res. 2021, 12, 200218. [Google Scholar] [CrossRef]
- Bauer, M.; Jasinski-Bergner, S.; Mandelboim, O.; Wickenhauser, C.; Seliger, B. Epstein–Barr Virus—Associated Malignancies and Immune Escape: The Role of the Tumor Microenvironment and Tumor Cell Evasion Strategies. Cancers 2021, 13, 5189. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Liu, Y.; Wang, C.; Gan, R. Signaling Pathways of EBV-Induced Oncogenesis. Cancer Cell Int. 2021, 21, 93. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Pang, P.S.; Tsang, C.M.; Hau, P.M.; Yip, Y.L.; Cheung, A.L.M.; Tsao, S.W. Epstein-Barr Virus-Encoded Latent Membrane Protein 1 Impairs G2 Checkpoint in Human Nasopharyngeal Epithelial Cells through Defective Chk1 Activation. PLoS ONE 2012, 7, e39095. [Google Scholar] [CrossRef] [Green Version]
- Manet, E.; Polvèche, H.; Mure, F.; Mrozek-Gorska, P.; Roisné-Hamelin, F.; Hammerschmidt, W.; Auboeuf, D.; Gruffat, H. Modulation of Alternative Splicing during Early Infection of Human Primary B Lymphocytes with Epstein-Barr Virus (EBV): A Novel Function for the Viral EBNA-LP Protein. Nucleic Acids Res. 2021, 49, 10657–10676. [Google Scholar] [CrossRef]
- Zhao, J.; Liang, Q.; Cheung, K.-F.; Kang, W.; Lung, R.W.M.; Tong, J.H.M.; To, K.F.; Sung, J.J.Y.; Yu, J. Genome-Wide Identification of Epstein-Barr Virus-Driven Promoter Methylation Profiles of Human Genes in Gastric Cancer Cells. Cancer 2013, 119, 304–312. [Google Scholar] [CrossRef]
- Böger, C.; Krüger, S.; Behrens, H.M.; Bock, S.; Haag, J.; Kalthoff, H.; Röcken, C. Epstein–Barr Virus-Associated Gastric Cancer Reveals Intratumoral Heterogeneity of PIK3CA Mutations. Ann. Oncol. 2017, 28, 1005–1014. [Google Scholar] [CrossRef]
- Wang, K.; Kan, J.; Yuen, S.T.; Shi, S.T.; Chu, K.M.; Law, S.; Chan, T.L.; Kan, Z.; Chan, A.S.Y.; Tsui, W.Y.; et al. Exome Sequencing Identifies Frequent Mutation of ARID1A in Molecular Subtypes of Gastric Cancer. Nat. Genet. 2011, 43, 1219–1223. [Google Scholar] [CrossRef]
- Sun, K.; Jia, K.; Lv, H.; Wang, S.-Q.; Wu, Y.; Lei, H.; Chen, X. EBV-Positive Gastric Cancer: Current Knowledge and Future Perspectives. Front. Oncol. 2020, 10, 583463. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, P.M.; Hardwick, J.M.; Sample, J.; Hayward, G.S.; Hayward, S.D. The Zta Transactivator Involved in Induction of Lytic Cycle Gene Expression in Epstein-Barr Virus-Infected Lymphocytes Binds to Both AP-1 and ZRE Sites in Target Promoter and Enhancer Regions. J. Virol. 1990, 64, 1143–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woellmer, A.; Hammerschmidt, W. Epstein-Barr Virus and Host Cell Methylation: Regulation of Latency, Replication and Virus Reactivation. Curr. Opin. Virol. 2013, 3, 260–265. [Google Scholar] [CrossRef] [Green Version]
- Countryman, J.K.; Gradoville, L.; Miller, G. Histone Hyperacetylation Occurs on Promoters of Lytic Cycle Regulatory Genes in Epstein-Barr Virus-Infected Cell Lines Which Are Refractory to Disruption of Latency by Histone Deacetylase Inhibitors. J. Virol. 2008, 82, 4706–4719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassani, A.; Khan, G. Epstein-Barr Virus and miRNAs: Partners in Crime in the Pathogenesis of Multiple Sclerosis? Front. Immunol. 2019, 10, 695. [Google Scholar] [CrossRef] [Green Version]
- Abusalah, M.A.H.; Irekeola, A.A.; Hanim Shueb, R.; Jarrar, M.; Yean Yean, C. Prognostic Epstein-Barr Virus (EBV) miRNA Biomarkers for Survival Outcome in EBV-Associated Epithelial Malignancies: Systematic Review and Meta-Analysis. PLoS ONE 2022, 17, e0266893. [Google Scholar] [CrossRef]
- Zheng, X.; Huang, Y.; Li, K.; Luo, R.; Cai, M.; Yun, J. Immunosuppressive Tumor Microenvironment and Immunotherapy of Epstein-Barr Virus-Associated Malignancies. Viruses 2022, 14, 1017. [Google Scholar] [CrossRef]
- Tan, G.; Visser, L.; Tan, L.; Berg, A.; Diepstra, A. The Microenvironment in Epstein–Barr Virus-Associated Malignancies. Pathogens 2018, 7, 40. [Google Scholar] [CrossRef] [Green Version]
- Young, L.S.; Yap, L.F.; Murray, P.G. Epstein-Barr virus: More than 50 years old and still providing surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef]
- Scott, D.W.; Gascoyne, R.D. The tumour microenvironment in B cell lymphomas. Nat. Rev. Cancer 2014, 14, 517–534. [Google Scholar] [CrossRef]
- van Beek, J.; zur Hausen, A.; Snel, S.N.; Berkhof, J.; Kranenbarg, E.K.; van de Velde, C.J.; van den Brule, A.J.; Middeldorp, J.M.; Meijer, C.J.; Bloemena, E. Morphological evidence of an activated cytotoxic T-cell infiltrate in EBV-positive gastric carcinoma preventing lymph node metastases. Am. J. Surg. Pathol. 2006, 30, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Chapel, F.; Fabiani, B.; Davi, F.; Raphael, M.; Tepper, M.; Champault, G.; Guettier, C. Epstein-Barr virus and gastric carcinoma in Western patients: Comparison of pathological parameters and p53 expression in EBV-positive and negative tumours. Histopathology 2000, 36, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.F.; Zhang, L.H.; Shan, L.H.; Sun, W.G.; Chai, C.C.; Wu, H.M.; Ibla, J.C.; Wang, L.F.; Liu, J.R. Effects of the fibroblast activation protein on the invasion and migration of gastric cancer. Exp. Mol. Pathol. 2013, 95, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chang, E.W.; Wong, S.C.; Ong, S.M.; Chong, D.Q.; Ling, K.L. Increased myeloid-derived suppressor cells in gastric cancer correlate with cancer stage and plasma S100A8/A9 proinflammatory proteins. J. Immunol. (Baltim. Md. 1950) 2013, 190, 794–804. [Google Scholar] [CrossRef]
- Evrard, C.; Louvet, C.; Hajbi, F.E.; Fiore, F.D.; Malicot, K.L.; Aparicio, T.; Bouché, O.; Laurent-Puig, P.; Bibeau, F.; Lecomte, T.; et al. PRODIGE 59-DURIGAST trial: A randomised phase II study evaluating FOLFIRI + Durvalumab ± Tremelimumab in second-line of patients with advanced gastric cancer. Dig. Liver Dis. Off. J. Ital. Soc. Gastroenterol. Ital. Assoc. Study Liver 2021, 53, 420–426. [Google Scholar] [CrossRef]
- Evrard, C.; Aparicio, T.; Soularue, E.; Le Malicot, K.; Desramé, J.; Botsen, D.; El Hajbi, F.; Gonzalez, D.; Lepage, C.; Bouché, O.; et al. Safety of FOLFIRI + Durvalumab +/- Tremelimumab in Second Line of Patients with Advanced Gastric Cancer: A Safety Run-In from the Randomized Phase II Study DURIGAST PRODIGE 59. Biomedicines 2022, 10, 1211. [Google Scholar] [CrossRef]
- Kwon, M.; Kim, G.; Kim, R.; Kim, K.T.; Kim, S.T.; Smith, S.; Mortimer, P.; Hong, J.Y.; Loembé, A.B.; Irurzun-Arana, I.; et al. Phase II study of ceralasertib (AZD6738) in combination with durvalumab in patients with advanced gastric cancer. J. Immunother. Cancer 2022, 10, e005041. [Google Scholar] [CrossRef]
- Murfin, K. 3 Things to Know about the Tumor Microenvironment. Available online: https://www.mdanderson.org/cancerwise/what-is-the-tumor-microenvironment-3-things-to-know.h00-159460056.html (accessed on 1 June 2022).
- Cancer Today. Available online: http://gco.iarc.fr/today/home (accessed on 21 June 2022).
- Wang, Q.; Liu, G.; Hu, C. Molecular Classification of Gastric Adenocarcinoma. Gastroenterol. Res. Pract. 2019, 12, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.Y.; Cheong, J.-H. Beyond Precision Surgery: Molecularly Motivated Precision Care for Gastric Cancer. Eur. J. Surg. Oncol. 2017, 43, 856–864. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas—Gastric Adenocarcinoma Study. Available online: https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga/studied-cancers/stomach (accessed on 21 June 2022).
- Yang, J.; Liu, Z.; Zeng, B.; Hu, G.; Gan, R. Epstein-Barr Virus-Associated Gastric Cancer: A Distinct Subtype. Cancer Lett. 2020, 495, 191–199. [Google Scholar] [CrossRef]
- Song, H.-J.; Kim, K.-M. Pathology of Epstein-Barr Virus-Associated Gastric Carcinoma and Its Relationship to Prognosis. Gut Liver 2011, 5, 143–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Wang, L.J.; Lombardo, K.; Kwak, Y.; Kim, W.H.; Resnick, M.B. Expression of Indoleamine 2, 3-Dioxygenase 1 (IDO1) and Tryptophanyl-tRNA Synthetase (WARS) in Gastric Cancer Molecular Subtypes. Appl. Immunohistochem. Mol. Morphol. 2020, 28, 360–368. [Google Scholar] [CrossRef] [PubMed]
- IDO1 Indoleamine 2,3-Dioxygenase 1 [Homo Sapiens (human)]—Gene—NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene/3620 (accessed on 21 June 2022).
- Hwu, P.; Du, M.X.; Lapointe, R.; Do, M.; Taylor, M.W.; Young, H.A. Indoleamine 2,3-Dioxygenase Production by Human Dendritic Cells Results in the Inhibition of T Cell Proliferation. J. Immunol. 2000, 164, 3596–3599. [Google Scholar] [CrossRef] [Green Version]
- Puccetti, P. On Watching the Watchers: IDO and Type I/II IFN. Eur. J. Immunol. 2007, 37, 876–879. [Google Scholar] [CrossRef] [PubMed]
- Strong, M.J.; Xu, G.; Coco, J.; Baribault, C.; Vinay, D.S.; Lacey, M.R.; Strong, A.L.; Lehman, T.A.; Seddon, M.B.; Lin, Z.; et al. Differences in Gastric Carcinoma Microenvironment Stratify according to EBV Infection Intensity: Implications for Possible Immune Adjuvant Therapy. PLoS Pathog. 2013, 9, e1003341. [Google Scholar] [CrossRef]
- Panda, A.; Ganesan, S. Genomic and Immunologic Correlates of Indoleamine 2,3-Dioxygenase Pathway Expression in Cancer. Front. Genet. 2021, 12, 706435. [Google Scholar] [CrossRef]
- Hou, D.-Y.; Muller, A.J.; Sharma, M.D.; DuHadaway, J.; Banerjee, T.; Johnson, M.; Mellor, A.L.; Prendergast, G.C.; Munn, D.H. Inhibition of Indoleamine 2,3-Dioxygenase in Dendritic Cells by Stereoisomers of 1-Methyl-Tryptophan Correlates with Antitumor Responses. Cancer Res. 2007, 67, 792–801. [Google Scholar] [CrossRef] [Green Version]
- Incyte and Merck Provide Update on Phase 3 Study of Epacadostat in Combination with KEYTRUDA® (pembrolizumab) in Patients with Unresectable or Metastatic Melanoma. Available online: https://www.businesswire.com/news/home/20180406005141/en/Incyte-and-Merck-Provide-Update-on-Phase-3-Study-of-Epacadostat-in-Combination-with-KEYTRUDA%C2%AE-pembrolizumab-in-Patients-with-Unresectable-or-Metastatic-Melanoma (accessed on 1 June 2022).
- Muller, A.J.; Manfredi, M.G.; Zakharia, Y.; Prendergast, G.C. Inhibiting IDO pathways to treat cancer: Lessons from the ECHO-301 trial and beyond. Semin. Immunopathol. 2019, 41, 41–48. [Google Scholar] [CrossRef]
- Du, Q.; Feng, X.; Wang, Y.; Xu, X.; Zhang, Y.; Qu, X.; Li, Z.; Bian, J. Discovery of phosphonamidate IDO1 inhibitors for the treatment of non-small cell lung cancer. Eur. J. Med. Chem. 2019, 182, 111629. [Google Scholar] [CrossRef]
- Hooi, J.K.Y.; Lai, W.Y.; Ng, W.K.; Suen, M.M.Y.; Underwood, F.E.; Tanyingoh, D.; Malfertheiner, P.; Graham, D.Y.; Wong, V.W.S.; Wu, J.C.Y.; et al. Global Prevalence of Helicobacter Pylori Infection: Systematic Review and Meta-Analysis. Gastroenterology 2017, 153, 420–429. [Google Scholar] [CrossRef]
- Díaz, P.; Valenzuela Valderrama, M.; Bravo, J.; Quest, A.F.G. And Gastric Cancer: Adaptive Cellular Mechanisms Involved in Disease Progression. Front. Microbiol. 2018, 9, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, H.J.; Lee, D.S. Helicobacter Pylori in Gastric Carcinogenesis. World J. Gastrointest. Oncol. 2015, 7, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Rihane, F.E.; Erguibi, D.; Elyamine, O.; Abumsimir, B.; Ennaji, M.M.; Chehab, F. Helicobacter Pylori Co-Infection with Epstein-Barr Virus and the Risk of Developing Gastric Adenocarcinoma at an Early Age: Observational Study Infectious Agents and Cancer. Ann. Med. Surg. 2021, 68, 102651. [Google Scholar] [CrossRef] [PubMed]
- Saju, P.; Murata-Kamiya, N.; Hayashi, T.; Senda, Y.; Nagase, L.; Noda, S.; Matsusaka, K.; Funata, S.; Kunita, A.; Urabe, M.; et al. Host SHP1 Phosphatase Antagonizes Helicobacter Pylori CagA and Can Be Downregulated by Epstein-Barr Virus. Nat. Microbiol. 2016, 1, 16026. [Google Scholar] [CrossRef] [PubMed]
- Takahashi-Kanemitsu, A.; Knight, C.T.; Hatakeyama, M. Molecular Anatomy and Pathogenic Actions of Helicobacter Pylori CagA That Underpin Gastric Carcinogenesis. Cell. Mol. Immunol. 2020, 17, 50–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Désage, A.L.; Léonce, C.; Swalduz, A.; Ortiz-Cuaran, S. Targeting KRAS Mutant in Non-Small Cell Lung Cancer: Novel Insights Into Therapeutic Strategies. Front. Oncol. 2022, 12, 796832. [Google Scholar] [CrossRef]
- Watson, H.A.; Wehenkel, S.; Matthews, J.; Ager, A. SHP-1: The next Checkpoint Target for Cancer Immunotherapy? Biochem. Soc. Trans. 2016, 44, 356–362. [Google Scholar] [CrossRef] [Green Version]
- Prockop, S.; Doubrovina, E.; Suser, S.; Heller, G.; Barker, J.; Dahi, P.; Perales, M.A.; Papadopoulos, E.; Sauter, C.; Castro-Malaspina, H.; et al. Off-the-shelf EBV-specific T cell immunotherapy for rituximab-refractory EBV-associated lymphoma following transplantation. J. Clin. Investig. 2020, 130, 733–747. [Google Scholar] [CrossRef] [Green Version]
- Gambardella, V.; Castillo, J.; Tarazona, N.; Gimeno-Valiente, F.; Martínez-Ciarpaglini, C.; Cabeza-Segura, M.; Roselló, S.; Roda, D.; Huerta, M.; Cervantes, A.; et al. The Role of Tumor-Associated Macrophages in Gastric Cancer Development and Their Potential as a Therapeutic Target. Cancer Treat. Rev. 2020, 86, 102015. [Google Scholar] [CrossRef] [Green Version]
- Moyano, A.; Ferressini Gerpe, N.M.; De Matteo, E.; Preciado, M.V.; Chabay, P. M1 Macrophage Polarization Prevails in Epstein-Barr Virus-Infected Children in an Immunoregulatory Environment. J. Virol. 2022, 96, e0143421. [Google Scholar] [CrossRef]
- Shen, Z.; Kauttu, T.; Seppänen, H.; Vainionpää, S.; Ye, Y.; Wang, S.; Mustonen, H.; Puolakkainen, P. Both Macrophages and Hypoxia Play Critical Role in Regulating Invasion of Gastric Cancer in Vitro. Acta Oncol. 2013, 52, 852–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.T.; Sa, J.K.; Oh, S.Y.; Kim, K.; Hong, J.Y.; Kang, W.K.; Kim, K.-M.; Lee, J. Comprehensive Molecular Characterization of Gastric Cancer Patients from Phase II Second-Line Ramucirumab plus Paclitaxel Therapy Trial. Genome Med. 2021, 13, 11. [Google Scholar] [CrossRef] [PubMed]
- Boutilier, A.J.; Elsawa, S.F. Macrophage Polarization States in the Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 6995. [Google Scholar] [CrossRef] [PubMed]
- Deci, M.B.; Ferguson, S.W.; Scatigno, S.L.; Nguyen, J. Modulating Macrophage Polarization through CCR2 Inhibition and Multivalent Engagement. Mol. Pharm. 2018, 15, 2721–2731. [Google Scholar] [CrossRef]
- Myers, K.V.; Amend, S.R.; Pienta, K.J. Targeting Tyro3, Axl and MerTK (TAM Receptors): Implications for Macrophages in the Tumor Microenvironment. Mol. Cancer 2019, 18, 94. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Roca, C.A.; Italiano, A.; Le Tourneau, C.; Cassier, P.A.; Toulmonde, M.; D’Angelo, S.P.; Campone, M.; Weber, K.L.; Loirat, D.; Cannarile, M.A.; et al. Phase I Study of Emactuzumab Single Agent or in Combination with Paclitaxel in Patients with Advanced/metastatic Solid Tumors Reveals Depletion of Immunosuppressive M2-like Macrophages. Ann. Oncol. 2019, 30, 1381–1392. [Google Scholar] [CrossRef]


{kind=link}
| Trial Name | Identification | Phase | Status |
|---|---|---|---|
| Indoleamine 2,3-dioxygenase (IDO) Activity in Patients with Chronic Lymphocytic Leukemia (CLL) | NCT01397916 | 2 | Completed |
| Indoleamine 2,3-Dioxygenase (IDO) Inhibitor in Advanced Solid Tumors | NCT02048709 | 1 | Completed |
| NLG802 Indoleamine 2,3-Dioxygenase (IDO) Inhibitor in Advanced Solid Tumors | NCT03164603 | 1 | Completed |
| Intraperitoneal Natural Killer Cells and INCB024360 for Recurrent Ovarian, Fallopian Tube, and Primary Peritoneal Cancer | NCT02118285 | 1 | Completed |
| Epacadostat and Vaccine Therapy in Treating Patients with Stage III-IV Melanoma | NCT01961115 | 2 | Completed |
| Pembrolizumab in Combination with Epacadostat or Placebo in Cisplatin-ineligible Urothelial Carcinoma (KEYNOTE-672/ECHO-307) | NCT03361865 | 3 | Completed |
| Pembrolizumab + Epacadostat vs. Pembrolizumab + Placebo in Recurrent or Progressive Metastatic Urothelial Carcinoma | NCT03374488 | 3 | Completed |
| Pembrolizumab Plus Epacadostat, Pembrolizumab Monotherapy, and the EXTREME Regimen in Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma (KEYNOTE-669/ECHO-304) | NCT03358472 | 3 | Active |
| Pembrolizumab Plus Epacadostat vs. Pembrolizumab Plus Placebo in Metastatic Non-Small Cell Lung Cancer (KEYNOTE-654-05/ECHO-305-05) | NCT03322540 | 2 | Completed |
| Pembrolizumab (MK-3475) Plus Epacadostat vs. Standard of Care in mRCC (KEYNOTE-679/ECHO-302) | NCT03260894 | 3 | Active |
| A Study of Pembrolizumab Plus Epacadostat with Platinum-based Chemotherapy versus Pembrolizumab Plus Platinum-based Chemotherapy Plus Placebo in Metastatic Non-Small Cell Lung Cancer (KEYNOTE-715-06/ECHO-306-06) | NCT03322566 | 2 | Completed |
| Chemo-immunotherapy Using Ibrutinib Plus Indoximod for Patients with Pediatric Brain Cancer | NCT05106296 | 1 | Recruiting |
| Pediatric Trial of Indoximod with Chemotherapy and Radiation for Relapsed Brain Tumors or Newly Diagnosed DIPG | NCT04049669 | 2 | Recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atri-Schuller, A.; Abushukair, H.; Cavalcante, L.; Hentzen, S.; Saeed, A.; Saeed, A. Tumor Molecular and Microenvironment Characteristics in EBV-Associated Malignancies as Potential Therapeutic Targets: Focus on Gastric Cancer. Curr. Issues Mol. Biol. 2022, 44, 5756-5767. https://doi.org/10.3390/cimb44110390
Atri-Schuller A, Abushukair H, Cavalcante L, Hentzen S, Saeed A, Saeed A. Tumor Molecular and Microenvironment Characteristics in EBV-Associated Malignancies as Potential Therapeutic Targets: Focus on Gastric Cancer. Current Issues in Molecular Biology. 2022; 44(11):5756-5767. https://doi.org/10.3390/cimb44110390
Chicago/Turabian StyleAtri-Schuller, Aviva, Hassan Abushukair, Ludimila Cavalcante, Stijn Hentzen, Azhar Saeed, and Anwaar Saeed. 2022. "Tumor Molecular and Microenvironment Characteristics in EBV-Associated Malignancies as Potential Therapeutic Targets: Focus on Gastric Cancer" Current Issues in Molecular Biology 44, no. 11: 5756-5767. https://doi.org/10.3390/cimb44110390
APA StyleAtri-Schuller, A., Abushukair, H., Cavalcante, L., Hentzen, S., Saeed, A., & Saeed, A. (2022). Tumor Molecular and Microenvironment Characteristics in EBV-Associated Malignancies as Potential Therapeutic Targets: Focus on Gastric Cancer. Current Issues in Molecular Biology, 44(11), 5756-5767. https://doi.org/10.3390/cimb44110390