
GCEN: An Easy-to-Use Toolkit for Gene Co-Expression Network Analysis and lncRNAs Annotation

 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Normalization
2.2. Co-Expression Network Construction
2.3. Module Identification
2.4. Function Annotation
3. Results
3.1. The Main Analysis Process of GCEN
3.2. Performance Evaluation
3.3. Data Visualization
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, J.; Xuan, Z.; Liu, C. Long non-coding RNAs and complex human diseases. Int. J. Mol. Sci. 2013, 14, 18790–18808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Wei, Y. A Novel Regulatory Player in the Innate Immune System: Long Non-Coding RNAs. Int. J. Mol. Sci. 2021, 22, 9535. [Google Scholar] [CrossRef] [PubMed]
- Schonrock, N.; Harvey, R.P.; Mattick, J.S. Long noncoding RNAs in cardiac development and pathophysiology. Circ. Res. 2012, 111, 1349–1362. [Google Scholar] [CrossRef] [Green Version]
- Ulitsky, I. Evolution to the rescue: Using comparative genomics to understand long non-coding RNAs. Nat. Rev. Genet. 2016, 17, 601–614. [Google Scholar] [CrossRef]
- Liao, Q.; Liu, C.; Yuan, X.; Kang, S.; Miao, R.; Xiao, H.; Zhao, G.; Luo, H.; Bu, D.; Zhao, H.; et al. Large-scale prediction of long non-coding RNA functions in a coding-non-coding gene co-expression network. Nucleic Acids Res. 2011, 39, 3864–3878. [Google Scholar] [CrossRef]
- Van Dam, S.; Vosa, U.; van der Graaf, A.; Franke, L.; de Magalhaes, J.P. Gene co-expression analysis for functional classification and gene-disease predictions. Brief Bioinform. 2018, 19, 575–592. [Google Scholar] [CrossRef] [PubMed]
- Cogill, S.B.; Wang, L. Co-expression Network Analysis of Human lncRNAs and Cancer Genes. Cancer Inf. 2014, 13, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Zhang, X.; Li, J.; Huang, S.; Xiang, S.; Hu, X.; Liu, C. Comprehensive analysis of coding-lncRNA gene co-expression network uncovers conserved functional lncRNAs in zebrafish. BMC Genom. 2018, 19, 112. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Liao, Q.; Xiao, H.; Bu, D.; Xie, C.; Miao, R.; Luo, H.; Zhao, G.; Yu, K.; Zhao, H.; Skogerbo, G.; et al. ncFANs: A web server for functional annotation of long non-coding RNAs. Nucleic Acids Res. 2011, 39, W118–W124. [Google Scholar] [CrossRef] [Green Version]
- Ballouz, S.; Verleyen, W.; Gillis, J. Guidance for RNA-seq co-expression network construction and analysis: Safety in numbers. Bioinformatics 2015, 31, 2123–2130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, J.; Ye, X.; Feng, W.; Zhang, Q.; Han, Y.; Liu, Y.; Li, Y.; Wei, Y. Distance correlation application to gene co-expression network analysis. BMC Bioinform. 2022, 23, 81. [Google Scholar] [CrossRef] [PubMed]
- Saelens, W.; Cannoodt, R.; Saeys, Y. A comprehensive evaluation of module detection methods for gene expression data. Nat. Commun. 2018, 9, 1090. [Google Scholar] [CrossRef]
- Wang, Z.; San Lucas, F.A.; Qiu, P.; Liu, Y. Improving the sensitivity of sample clustering by leveraging gene co-expression networks in variable selection. BMC Bioinform. 2014, 15, 153. [Google Scholar] [CrossRef] [Green Version]
- Cowen, L.; Ideker, T.; Raphael, B.J.; Sharan, R. Network propagation: A universal amplifier of genetic associations. Nat. Rev. Genet. 2017, 18, 551–562. [Google Scholar] [CrossRef]
- Klopfenstein, D.V.; Zhang, L.; Pedersen, B.S.; Ramirez, F.; Warwick Vesztrocy, A.; Naldi, A.; Mungall, C.J.; Yunes, J.M.; Botvinnik, O.; Weigel, M.; et al. GOATOOLS: A Python library for Gene Ontology analyses. Sci. Rep. 2018, 8, 10872. [Google Scholar] [CrossRef]
- Liang, M.; Zhang, F.; Jin, G.; Zhu, J. FastGCN: A GPU accelerated tool for fast gene co-expression networks. PLoS ONE 2015, 10, e0116776. [Google Scholar] [CrossRef]
- Pauli, A.; Valen, E.; Lin, M.F.; Garber, M.; Vastenhouw, N.L.; Levin, J.Z.; Fan, L.; Sandelin, A.; Rinn, J.L.; Regev, A.; et al. Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Res. 2012, 22, 577–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, C.; Hardin, J.; Stoebel, D.M. Selecting between-sample RNA-Seq normalization methods from the perspective of their assumptions. Brief Bioinform. 2018, 19, 776–792. [Google Scholar] [CrossRef] [PubMed]
- Liesecke, F.; De Craene, J.O.; Besseau, S.; Courdavault, V.; Clastre, M.; Vergès, V.; Papon, N.; Giglioli-Guivarc’h, N.; Glévarec, G.; Pichon, O.; et al. Improved gene co-expression network quality through expression dataset down-sampling and network aggregation. Sci. Rep. 2019, 9, 14431. [Google Scholar] [CrossRef]
- Paci, P.; Colombo, T.; Fiscon, G.; Gurtner, A.; Pavesi, G.; Farina, L. SWIM: A computational tool to unveiling crucial nodes in complex biological networks. Sci. Rep. 2017, 7, 44797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petti, M.; Verrienti, A.; Paci, P.; Farina, L. SEaCorAl: Identifying and contrasting the regulation-correlation bias in RNA-Seq paired expression data of patient groups. Comput. Biol. Med. 2021, 135, 104567. [Google Scholar] [CrossRef]
- Russo, P.S.T.; Ferreira, G.R.; Cardozo, L.E.; Burger, M.C.; Arias-Carrasco, R.; Maruyama, S.R.; Hirata, T.D.C.; Lima, D.S.; Passos, F.M.; Fukutani, K.F.; et al. CEMiTool: A Bioconductor package for performing comprehensive modular co-expression analyses. BMC Bioinform. 2018, 19, 56. [Google Scholar] [CrossRef]
- Lemoine, G.G.; Scott-Boyer, M.P.; Ambroise, B.; Perin, O.; Droit, A. GWENA: Gene co-expression networks analysis and extended modules characterization in a single Bioconductor package. BMC Bioinform. 2021, 22, 267. [Google Scholar] [CrossRef]
- Sang, S.; Chen, W.; Zhang, D.; Zhang, X.; Yang, W.; Liu, C. Data integration and evolutionary analysis of long non-coding RNAs in 25 flowering plants. BMC Genom. 2021, 22, 739. [Google Scholar] [CrossRef]
- Mathias, C.; Groeneveld, C.S.; Trefflich, S.; Zambalde, E.P.; Lima, R.S.; Urban, C.A.; Prado, K.B.; Ribeiro, E.; Castro, M.A.A.; Gradia, D.F.; et al. Novel lncRNAs Co-Expression Networks Identifies LINC00504 with Oncogenic Role in Luminal A Breast Cancer Cells. Int. J. Mol. Sci. 2021, 22, 2420. [Google Scholar] [CrossRef]
- Bayraktar, A.; Lam, S.; Altay, O.; Li, X.; Yuan, M.; Zhang, C.; Arif, M.; Turkez, H.; Uhlén, M.; Shoaie, S.; et al. Revealing the Molecular Mechanisms of Alzheimer’s Disease Based on Network Analysis. Int. J. Mol. Sci. 2021, 22, 11556. [Google Scholar] [CrossRef]
- Paci, P.; Fiscon, G.; Conte, F.; Wang, R.S.; Farina, L.; Loscalzo, J. Gene co-expression in the interactome: Moving from correlation toward causation via an integrated approach to disease module discovery. NPJ Syst. Biol. Appl. 2021, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Fiscon, G.; Paci, P. SWIMmeR: An R-based software to unveiling crucial nodes in complex biological networks. Bioinformatics 2021, 38, 586–588. [Google Scholar] [CrossRef]




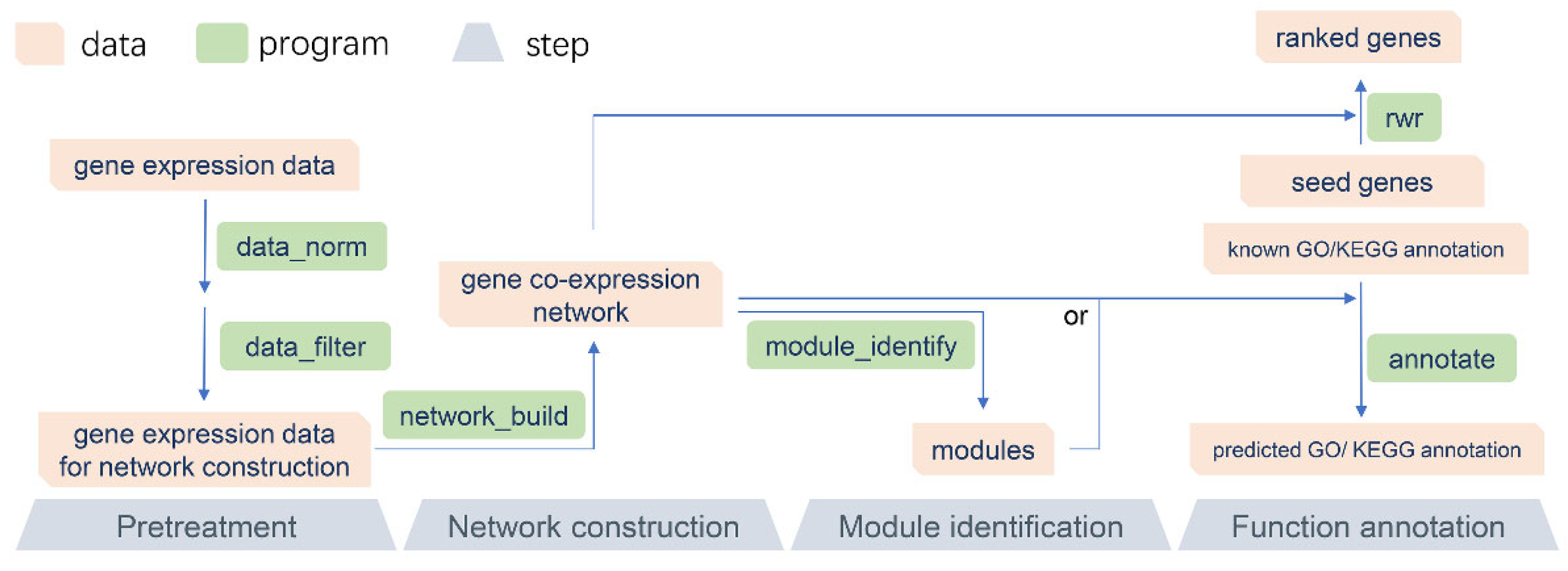{kind=link}
{kind=link}
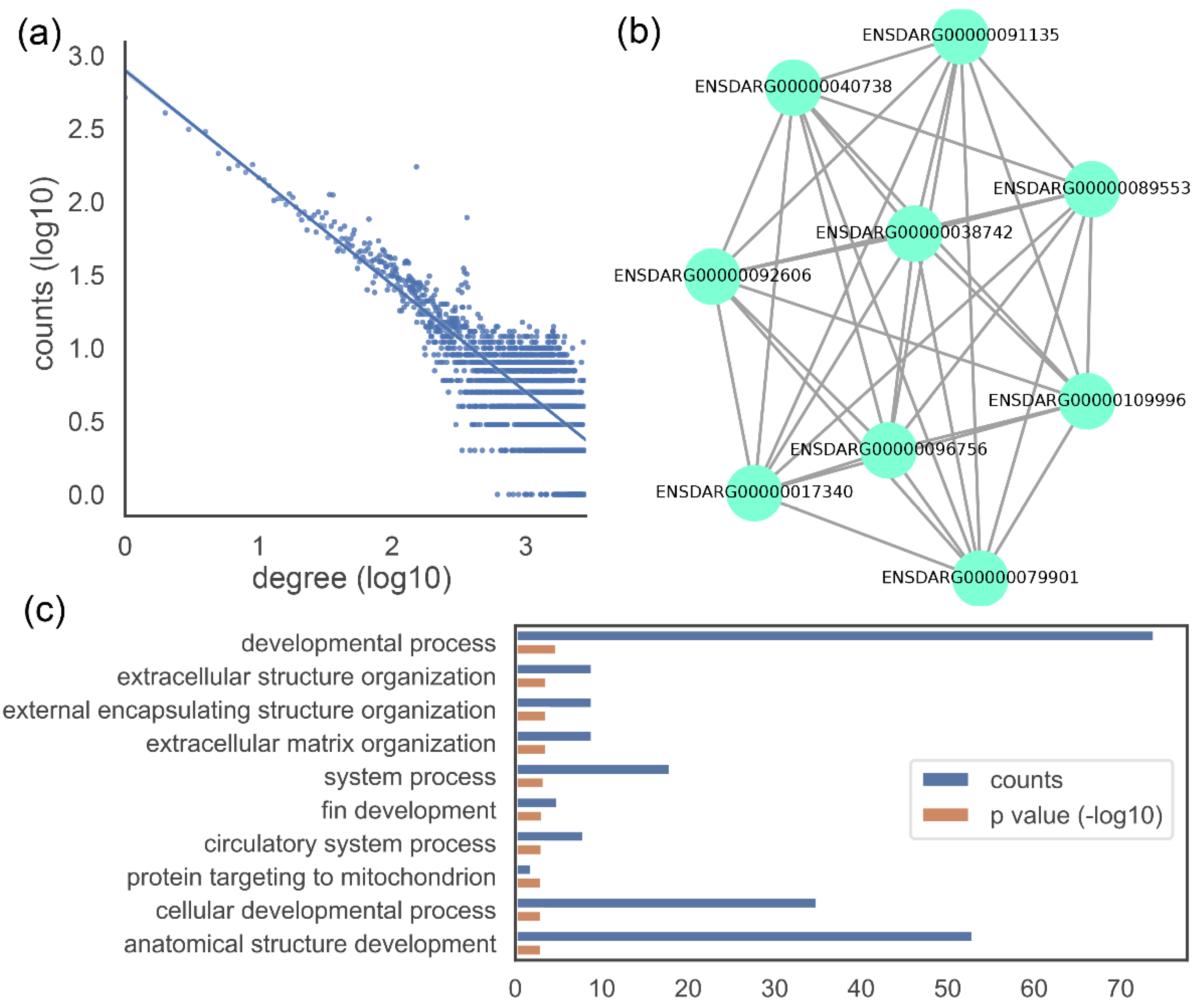{kind=link}
| Gene Number | GCEN | FastGCN | WGCNA |
|---|---|---|---|
| 10k | 9.51 s/5.93 MiB | 16.98 s/1.31 GiB | 59.36 s/1.84 GiB |
| 20k | 37.86 s/8.50 MiB | 2 m 11.59 s/5.25 GiB | 3 m 47.15 s/6.36 GiB |
| 40k | 2 m 31.42 s/12.88 MiB | 24 m 23.33 s/21.12 GiB | 14 m 57.86 s/24.39 GiB |
| 80k | 10 m 7.70 s/21.58 MiB | Out of maximum memory | 59 m 53.82 s/96.11 GiB |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, W.; Li, J.; Huang, S.; Li, X.; Zhang, X.; Hu, X.; Xiang, S.; Liu, C. GCEN: An Easy-to-Use Toolkit for Gene Co-Expression Network Analysis and lncRNAs Annotation. Curr. Issues Mol. Biol. 2022, 44, 1479-1487. https://doi.org/10.3390/cimb44040100
Chen W, Li J, Huang S, Li X, Zhang X, Hu X, Xiang S, Liu C. GCEN: An Easy-to-Use Toolkit for Gene Co-Expression Network Analysis and lncRNAs Annotation. Current Issues in Molecular Biology. 2022; 44(4):1479-1487. https://doi.org/10.3390/cimb44040100
Chicago/Turabian StyleChen, Wen, Jing Li, Shulan Huang, Xiaodeng Li, Xuan Zhang, Xiang Hu, Shuanglin Xiang, and Changning Liu. 2022. "GCEN: An Easy-to-Use Toolkit for Gene Co-Expression Network Analysis and lncRNAs Annotation" Current Issues in Molecular Biology 44, no. 4: 1479-1487. https://doi.org/10.3390/cimb44040100
APA StyleChen, W., Li, J., Huang, S., Li, X., Zhang, X., Hu, X., Xiang, S., & Liu, C. (2022). GCEN: An Easy-to-Use Toolkit for Gene Co-Expression Network Analysis and lncRNAs Annotation. Current Issues in Molecular Biology, 44(4), 1479-1487. https://doi.org/10.3390/cimb44040100