

Parkin, as a Regulator, Participates in Arsenic Trioxide-Triggered Mitophagy in HeLa Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Plasmid Constructs
2.3. Establishment of the AcGFP1-Parkin Mutant (C431S) HeLa Stable Cell Lines
2.4. Reagents and Antibodies
2.5. Cytotoxicity Assay
2.6. Colony Formation Assays
2.7. Analysis of Cell Cycle and Apoptosis
2.8. Transmission Electron Microscopy
2.9. Mitochondrial Staining
2.10. Reactive Oxygen Species Detection
2.11. Western Blot Analysis
2.12. Statistical Analysis
3. Result
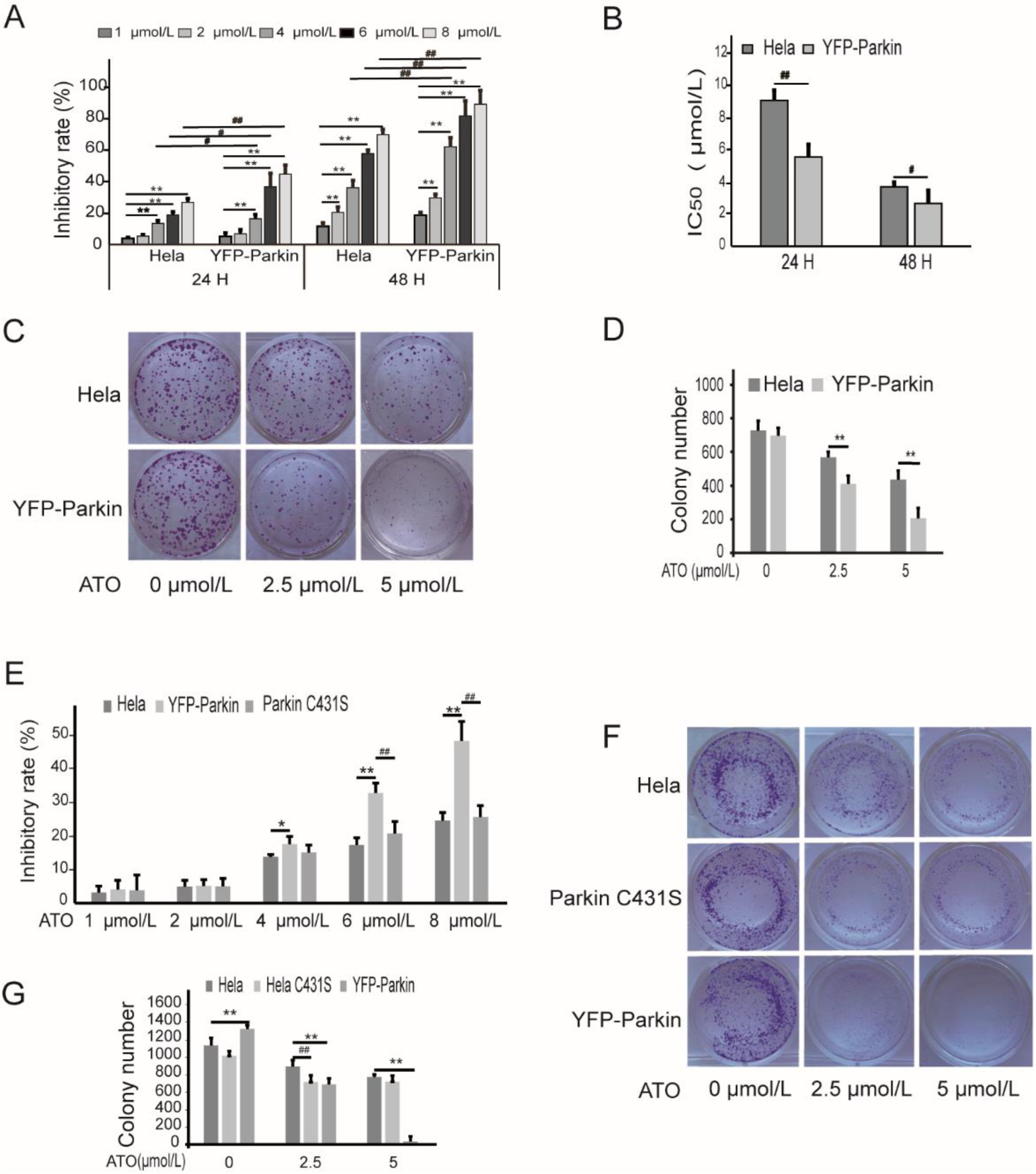
3.1. Parkin Aggravates the Proliferation Inhibition of HeLa Cells by ATO Treatment
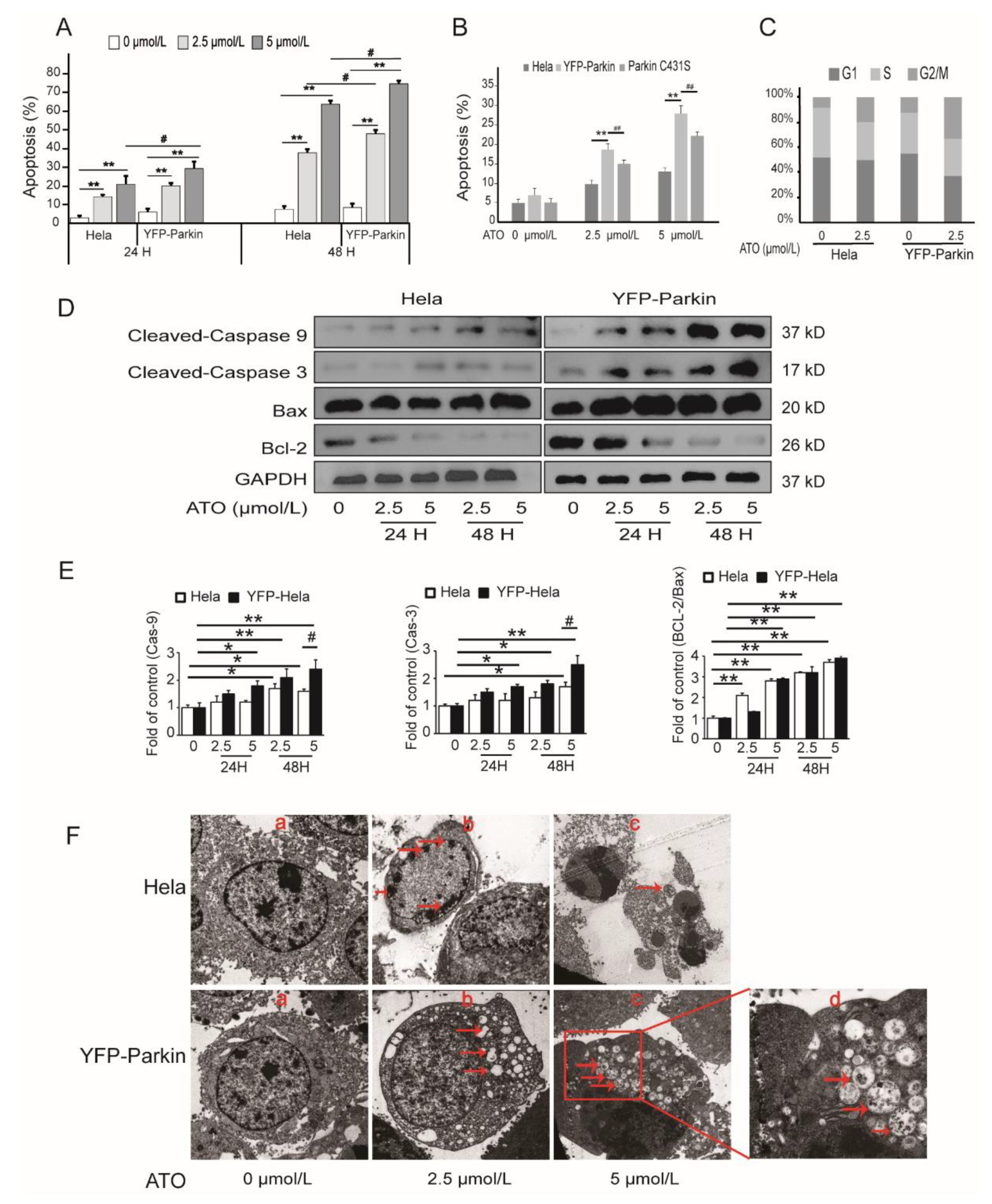
3.2. Parkin Promotes ATO-Induced Apoptosis of HeLa Cells
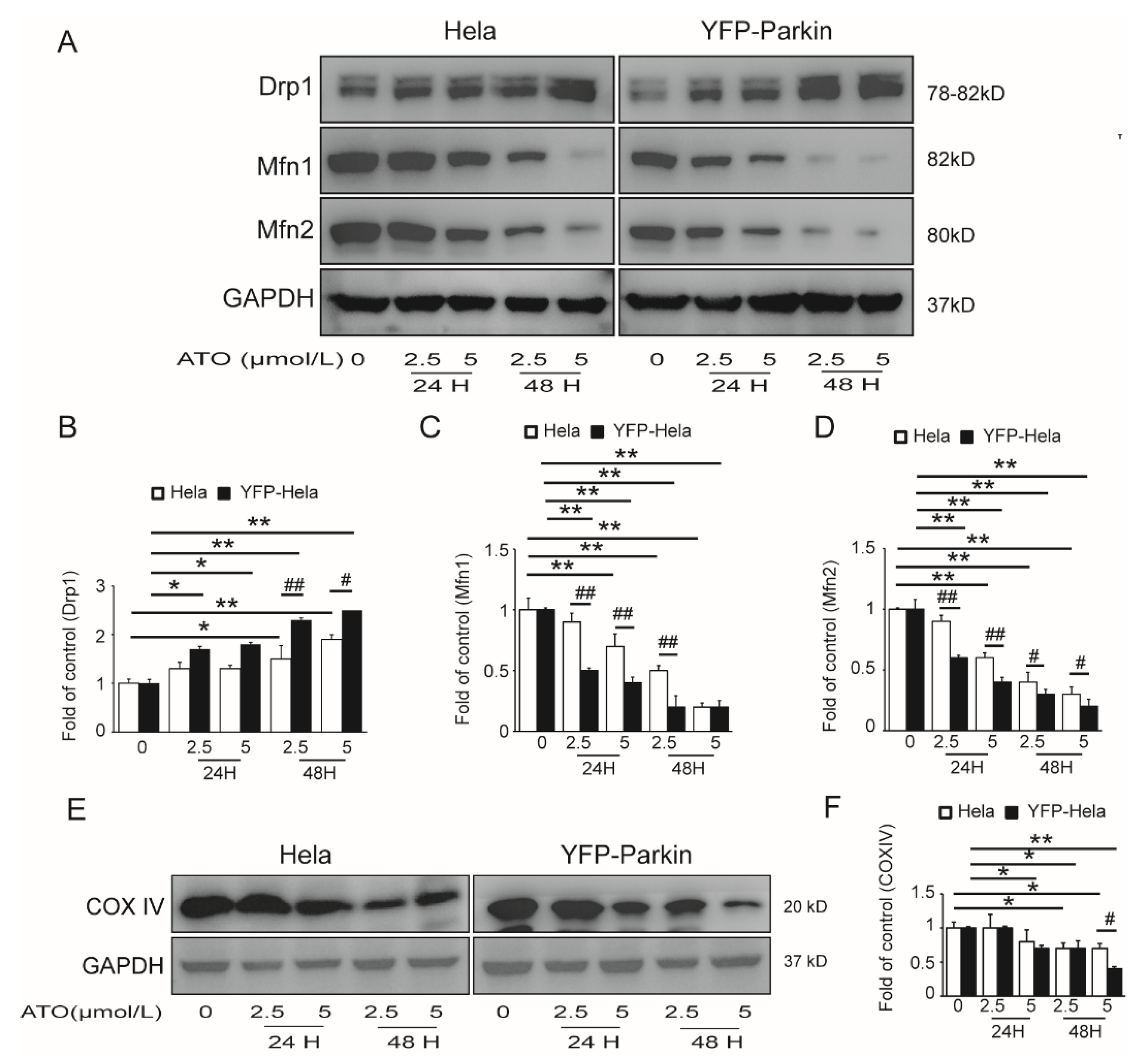
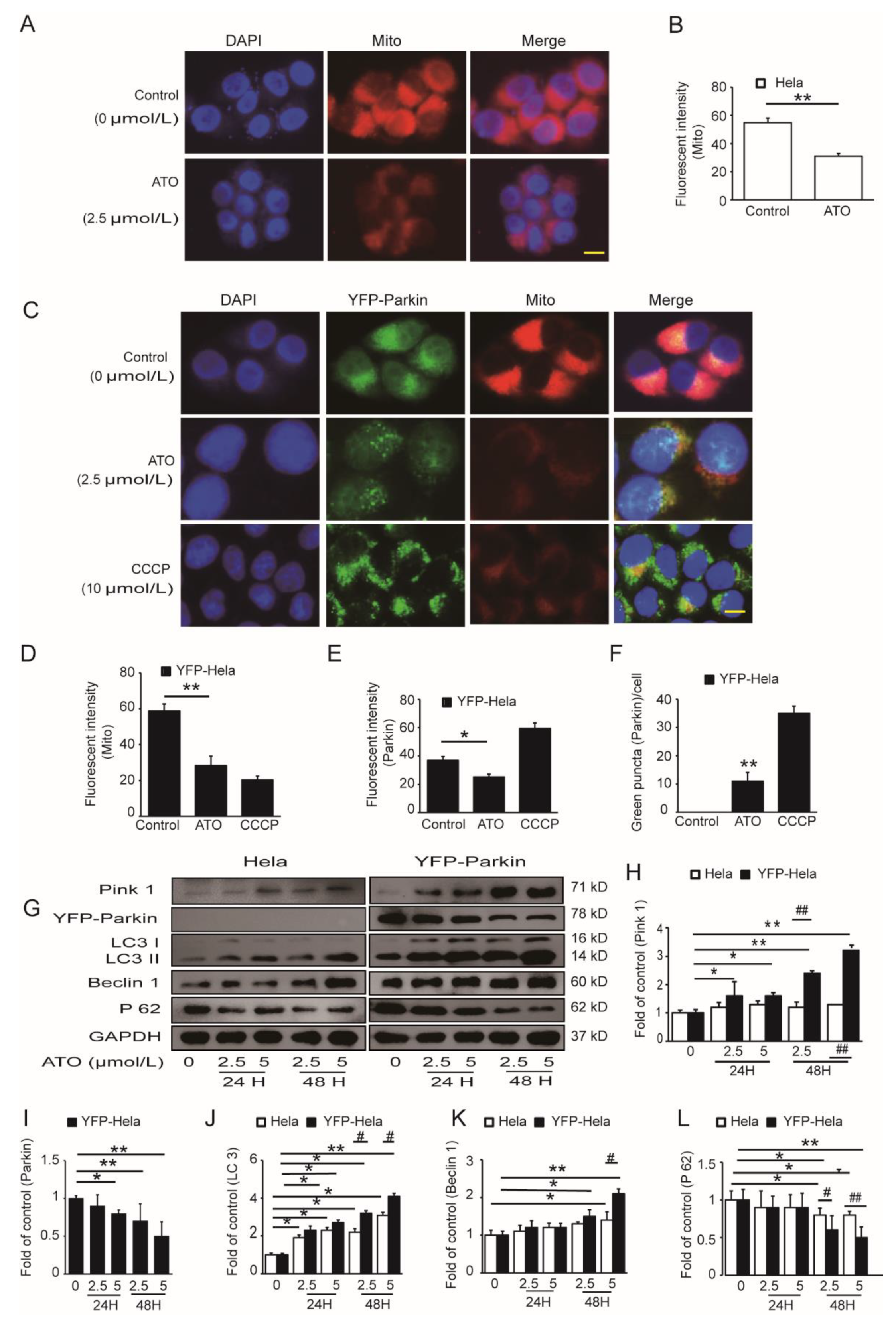
3.3. PINK1/Parkin Pathway Was Involved in the ATO-Induced Mitochondrial Damage and Mitophagy
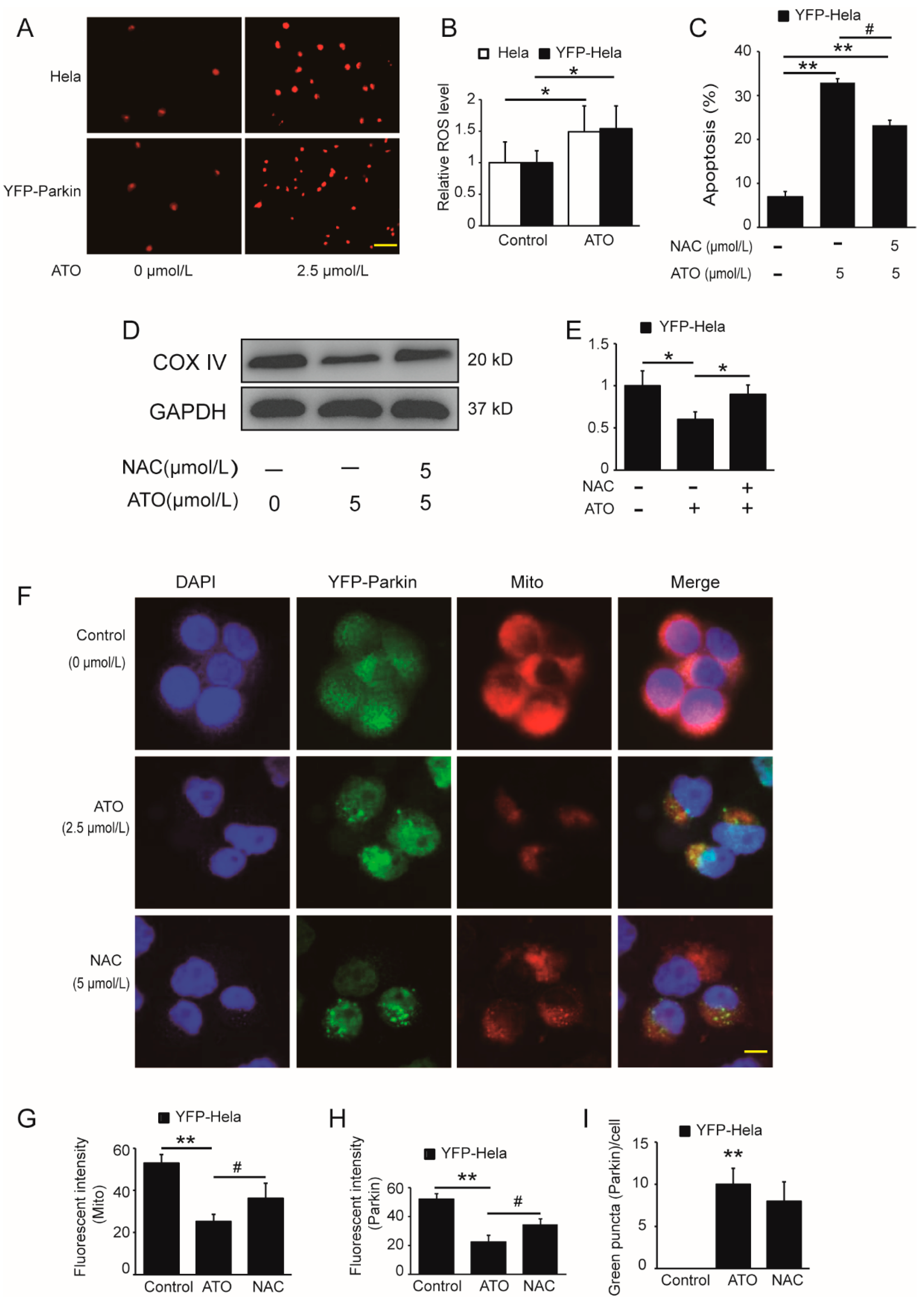
3.4. ROS Activate the PINK1/Parkin Pathway to Mediate Mitophagy and Mitophagic Death in ATO-Exposed HeLa Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Bernardini, J.P.; Lazarou, M.; Dewson, G. Parkin and mitophagy in cancer. Oncogene 2017, 36, 1313–1327. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.L.; Yu, W.J.; Lv, P.; Liang, X.Y.; Sun, S.Q.; Zhang, Y.M. Parkin overexpression alleviates cardiac aging through facilitating K63-polyubiquitination of TBK1 to facilitate mitophagy. Mol. Basis Dis. 2021, 1867, 165997. [Google Scholar] [CrossRef] [PubMed]
- Shiiba, I.; Takeda, K.; Nagashima, S.; Ito, N.; Tokuyama, T.; Yamashita, S.I.; Kanki, T.; Komatse, Y.; Urano, Y.; Fujikawa, Y.; et al. MITOL promotes cell survival by degrading Parkin during mitophagy. EMBO Rep. 2021, 22, e49097. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Tsai, C.W. PINK1/Parkin-mediated mitophagy pathway is related to neuroprotection by carnosic acid in SH-SY5Y cells. Food Chem. Toxicol. 2019, 125, 430–437. [Google Scholar] [CrossRef]
- Sun, D.Z.; Song, C.Q.; Xu, Y.M.; Wang, R.; Liu, W.; Liu, Z.; Dong, X.S. Involement of PINK1/Parkin-mediated mitophagy in paraquat-induced apoptosis in human lung epithelial-like A549 cells. Toxicol. Vitr. 2019, 53, 148–159. [Google Scholar] [CrossRef]
- Wang, B.; Yin, X.Q.; Gan, W.D.; Pan, F.; Li, A.Y.; Xiang, Z.; Han, X.D.; Li, D.M. PRCC-TFE3 fusion-mediated PRKN/parkin-dependent mitophagy promotes cell survival and proliferation in PRCC-TFE3 translocation renal cell carcinoma. Autophagy 2021, 17, 2475–2493. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 5, 795–803. [Google Scholar] [CrossRef]
- Wang, J.; Hu, T.; Wang, Q.; Chen, R.W.; Xie, Y.X.; Chang, H.Y.; Cheng, J. Repression of the AURKA-CXCL5 axis induces autophagic cell death and promotes radiosensitivity in non-amsll-cell lung cancer. Cancer Lett. 2021, 509, 89–104. [Google Scholar] [CrossRef]
- Berger, A.K.; Cortese, G.P.; Amodeo, K.D.; Weihofen, A.; Letai, A.; LaVoie, M.J. Parkin selectively alters the intrinsic threshold for mitochondrial cytochrome c release. Hum. Mol. Genet. 2009, 18, 4317–4328. [Google Scholar] [CrossRef]
- Carroll, R.G.; Hollville, E.; Martin, S.J. Parkin sensitizes toward apoptosis induced by mitochondrial depolarization through Promoting degradation of Mcl-1. Cell Rep. 2014, 9, 1538–1553. [Google Scholar] [CrossRef]
- Bin, J.G.; Bai, T.Z.; Zhao, Q.X.; Duan, X.H.; Deng, S.X.; Xu, Y.J. Parkin overexpression reduces inflammation-mediated cardiomyocyte apoptosis through activating Nrf2/ARE signaling pathway. J. Recept. Signal Transduct. 2020, 41, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.N.; Berger, A.K.; Cortese, G.P.; LaVoie, M.J. The ubiquitin E3 ligase parkin regulated the proapoptotic function of Bax. Proc. Natl. Acad. Sci. USA 2012, 16, 6283–6288. [Google Scholar] [CrossRef] [PubMed]
- Hovllville, E.; Carroll, R.G.; Cullen, S.P.; Martin, S.M. Bcl-2 family proteins participate in mitochondrial quality control by regulating Parkin/PINK1-Dependent mitophagy. Mol. Cell 2014, 55, 451–466. [Google Scholar] [CrossRef] [PubMed]
- Kapadia, M.; Snoo, M.L.D.; Kalia, L.V.; Kalia, S.K. Regulation of Parkin-dependent mitophagy by Bcl-2-associated athanogene (BAG) family members. Neural Regen. Res. 2021, 16, 684–685. [Google Scholar]
- Cheng, Y.H.; Chang, L.W.; Tsou, T.C. Mitogen-activated protein kinases mediate arsenic-induced down-regulation of surviving in human lung adenocarcinoma cells. Mol. Toxicol. 2006, 80, 310–318. [Google Scholar]
- Gu, S.Y.; Chen, C.Z.; Jiang, X.J.; Zhang, Z.Z. ROS-mediated endoplasmic reticulum stress and mitochondrial dysfunction underlie apoptosis induced by resveratrol and arsenic trioxide in A549 cells. Chemico-Biol. Interact. 2016, 245, 100–109. [Google Scholar] [CrossRef]
- Laka, K.; Makgoo, L.L.; Mbita, Z.K. Survivin splice variants in arsenic trioxide (As2O3)-induced deactivation of PI3K and MAPK cell signaling pathways in MCF-7 cells. Genes 2019, 10, 41. [Google Scholar] [CrossRef]
- Cheng, J.; Wei, H.L.; Chen, J.; Xie, B. Antitumor effect of arsenic trioxide in human K562 and K562/ADM cells by autophagy. Toxicol. Mech. Methods 2012, 22, 512–519. [Google Scholar] [CrossRef]
- Li, C.L.; Wei, H.L.; Chen, J.; Wang, B.; Xie, B.; Fan, L.L.; Li, L.L. Arsenic trioxide induces autophagy and antitumoreffects in Burkitt’s lymphoma Raji cells. Oncol. Rep. 2014, 32, 1557–1563. [Google Scholar] [CrossRef]
- Li, C.L.; Wei, H.L.; Chen, J.; Wang, B.; Xie, B.; Fan, L.L.; Li, L.L. Ebb-and-flow of macroautophagy in Raji cells induced by starvation and arsenic trioxide. Asian Pac. J. Cancer Prev. 2014, 15, 5715–5719. [Google Scholar] [CrossRef]
- Zhang, P.Y.; Zhao, Z.X.; Zhang, W.J.; He, A.L.; Lei, B.; Zhang, W.G.; Chen, Y.X. Leukemia-associated gene MLAA-34 reduces arsenic trioxide-induced apoptosis in Hela cells via activation of the Wnt/β-catenin signaling pathway. PLoS ONE 2017, 12, e0186868. [Google Scholar] [CrossRef] [PubMed]
- Niu, Z.D.; Zhang, W.Y.; Gu, X.Y.; Zhang, X.N.; Qi, Y.M.; Zhang, Y.M. Mitophagy inhibits proliferation by decreasing cyclooxygenase-2 (COX-2) in arsenic trioxide-treated HepG2 cells. Environ. Toxicol. Pharmacol. 2016, 45, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Funakoshi, T.; Unuma, K.; Aki, T.; Uemura, K. Activation of the ubiquitin-proteasome system against arsenic trioxide cardiotoxicity involves ubiquitin ligase Parkin for mitochondrial homeostasis. Toxicology 2014, 322, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhou, C.M.; Yi, J.; Sun, J.J.; Xie, B.; Zhang, Z.W.; Wang, Q.F.; Chen, G.; Jin, S.Y.; Hou, J.X.; et al. Metformin and arsenic trioxide synergize to trigger Parkin/pink1 -dependent mitophagic cell death in human cervical cancer Hela cells. J. Cancer 2021, 12, 6310–6319. [Google Scholar] [CrossRef]
- Zhang, F.; Peng, W.X.; Zhang, J.; Dong, W.T.; Wu, J.H.; Wang, T.; Xie, Z.H. P53 and Parkin co-regulate mitophagy in bone marrw mesenchymal stem cells to promote the repair of early steroid-induced osteonecrosis of the femoral head. Cell Death Dis. 2020, 11, 42. [Google Scholar] [CrossRef] [PubMed]
- Shires, S.E.; Quiles, J.M.; Najor, R.H.; Leon, L.J.; Cortez, M.Q.; Lampert, M.A.; Mark, A.; Gustafsson, A.B. Nuclear Parkin activates the ERRα transcriptional program and drices widespread changes in gene expression following hypoxia. Sci. Rep. 2020, 10, 8499. [Google Scholar] [CrossRef]
- Chen, Y.P.; Shih, P.C.; Feng, C.W.; Wu, C.C.; Tsui, K.H.; Lin, Y.H.; Kuo, H.M.; Wen, Z.H. Paraxin activates excessive mitophagy and mitochondria-mediated apoptosis in human ovarian cancer by inducing reactive oxygen species. Antioxidants 2021, 10, 1883. [Google Scholar] [CrossRef]
- Okatsu, K.; Koyano, F.; Kimura, M.; Kosako, H.; Saeki, Y.; Tanaka, K.; Matsuda, N. Phosphorylated ubiquitin chain is the genuine Parkin receptor. J. Cell Biol. 2015, 1, 111–128. [Google Scholar] [CrossRef]
- Xu, J.; Wang, L.M.; Zhang, L.H.; Zheng, F.; Wang, F.; Leng, J.H.; Wang, K.Y.; Héroux, P.; Shen, H.M.; Wu, Y.H.; et al. Mon-2-ethylhexyl phthalate drives progression of PINK1-parkin-mediated mitophagy via increasing mitochondrial ROS to exacerbate cytotoxicity. Redox Biol. 2021, 38, 101776. [Google Scholar] [CrossRef]
- Xiao, B.; Deng, X.; Grace, G.L.; Xie, S.P.; Zhou, Z.D.; Lim, K.L.; Tan, E.K. Superoxide drives progression of Parkin/PINK1-dependent mitophagy following translocation of Parkin to mitochondria. Cell Death Dis. 2017, 8, e3097. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Yi, J.; Xie, B.; Chen, J.; Zhang, X.; Wang, L.; Wang, J.; Hou, J.; Wei, H. Parkin, as a Regulator, Participates in Arsenic Trioxide-Triggered Mitophagy in HeLa Cells. Curr. Issues Mol. Biol. 2022, 44, 2759-2771. https://doi.org/10.3390/cimb44060189
Zhang Z, Yi J, Xie B, Chen J, Zhang X, Wang L, Wang J, Hou J, Wei H. Parkin, as a Regulator, Participates in Arsenic Trioxide-Triggered Mitophagy in HeLa Cells. Current Issues in Molecular Biology. 2022; 44(6):2759-2771. https://doi.org/10.3390/cimb44060189
Chicago/Turabian StyleZhang, Zhewen, Juan Yi, Bei Xie, Jing Chen, Xueyan Zhang, Li Wang, Jingyu Wang, Jinxia Hou, and Hulai Wei. 2022. "Parkin, as a Regulator, Participates in Arsenic Trioxide-Triggered Mitophagy in HeLa Cells" Current Issues in Molecular Biology 44, no. 6: 2759-2771. https://doi.org/10.3390/cimb44060189
APA StyleZhang, Z., Yi, J., Xie, B., Chen, J., Zhang, X., Wang, L., Wang, J., Hou, J., & Wei, H. (2022). Parkin, as a Regulator, Participates in Arsenic Trioxide-Triggered Mitophagy in HeLa Cells. Current Issues in Molecular Biology, 44(6), 2759-2771. https://doi.org/10.3390/cimb44060189