
Liraglutide Exerts Protective Effects by Downregulation of PPARγ, ACSL1 and SREBP-1c in Huh7 Cell Culture Models of Non-Alcoholic Steatosis and Drug-Induced Steatosis

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Culture
2.3. NAS and DIS Cell Culture Models
2.4. Measurement of the Hepatoprotective Effect of LIRA in NAS and DIS Cell Culture Models
2.5. Visualization of Fat Accumulation
2.6. Measurement of Triglyceride Levels
2.7. Total RNA Isolation and Reverse Transcription Polymerase Chain Reaction (RT-PCR) Analysis
2.8. Statistical Analyses
3. Results
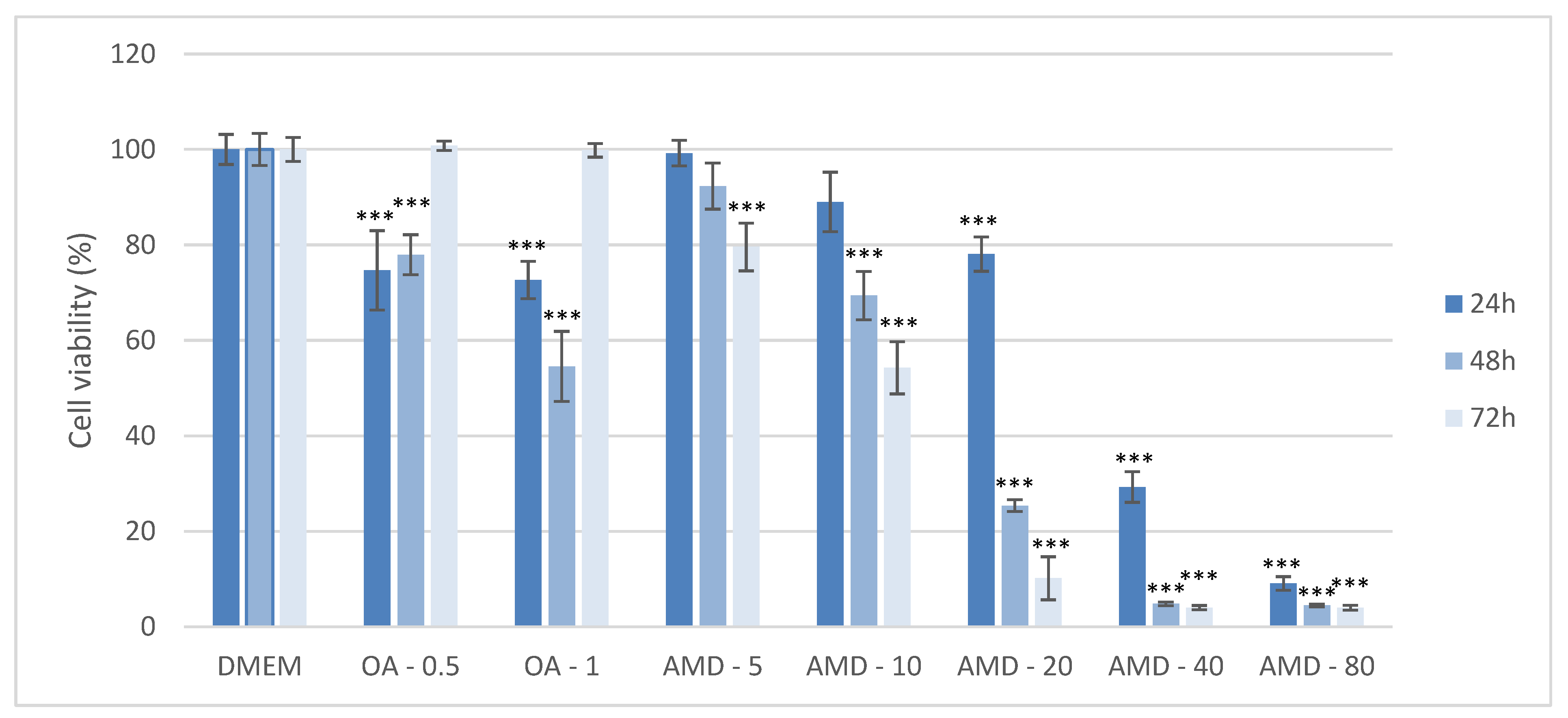
3.1. Establishment of the Cell Culture Model of DIS and NAS, and Assessment of the Effect of Amiodarone and Oleic Acid on Cell Viability
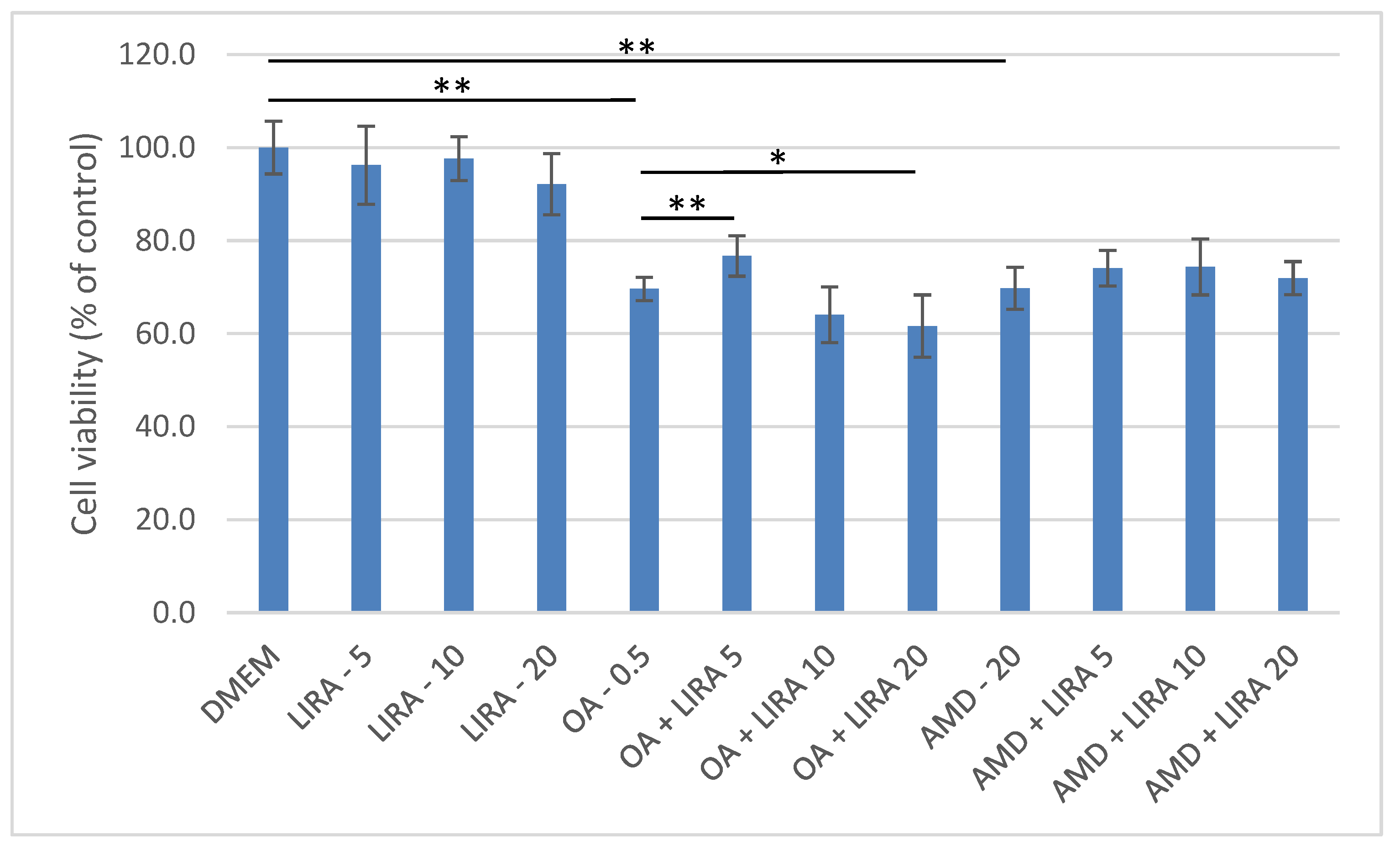
3.2. Assessment of the Protective Effect of LIRA (LIRA) on Cell Viability in DIS and NAS Cell Culture Models
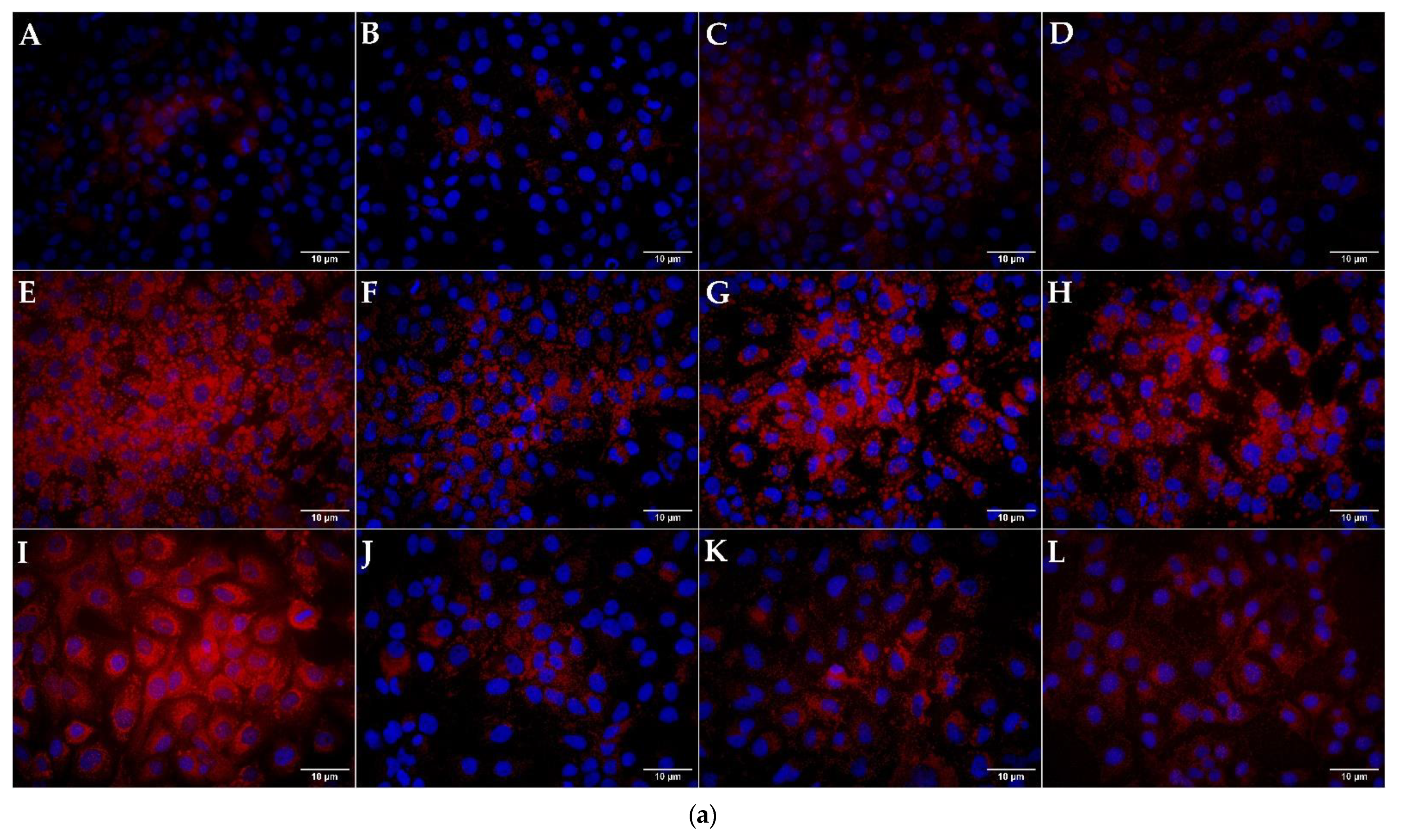
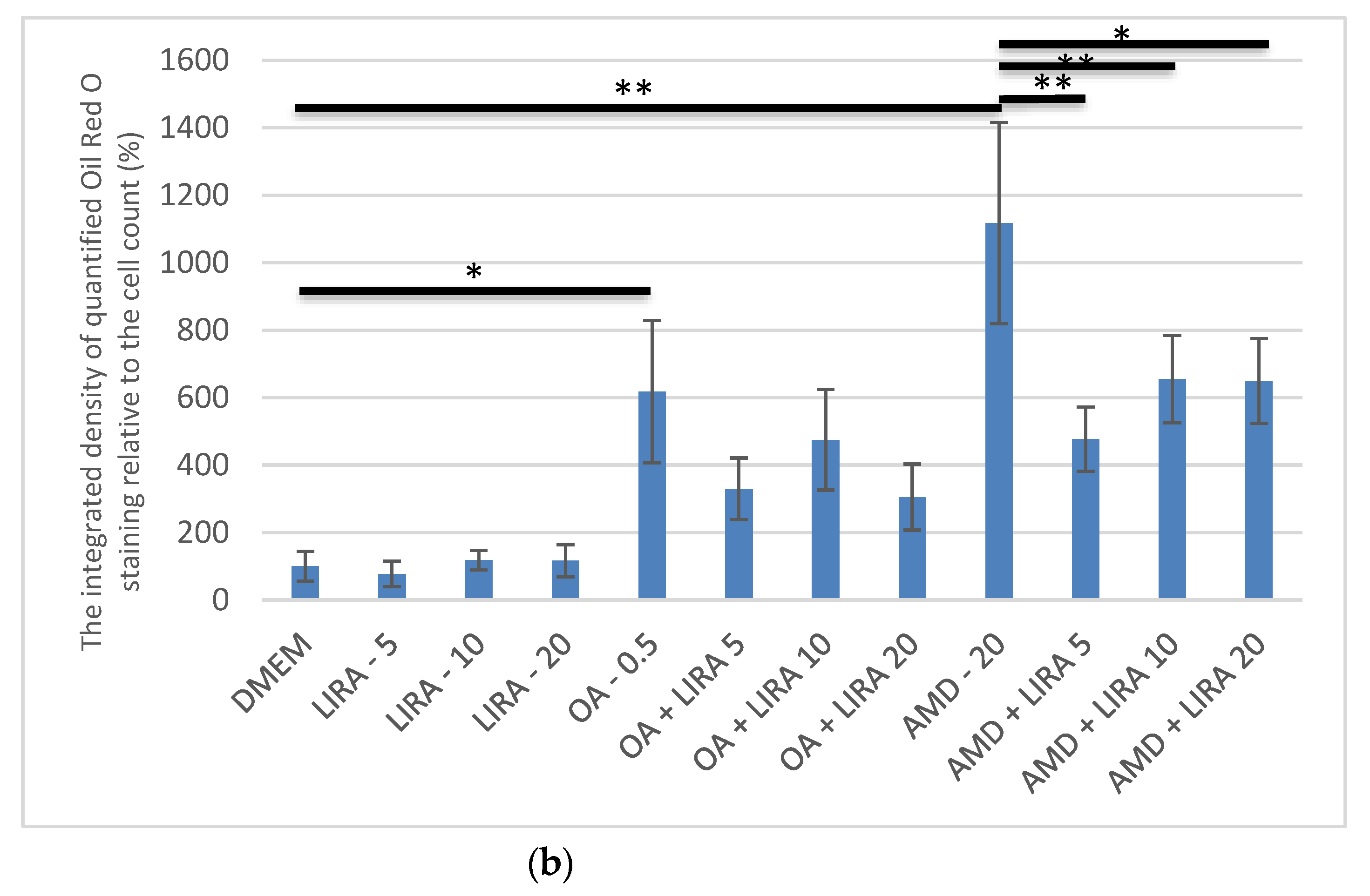
3.3. Quantification and Visualization of Lipid Accumulation in DIS and NAS Cell Culture Models, Incubated with Varying Concentrations of LIRA
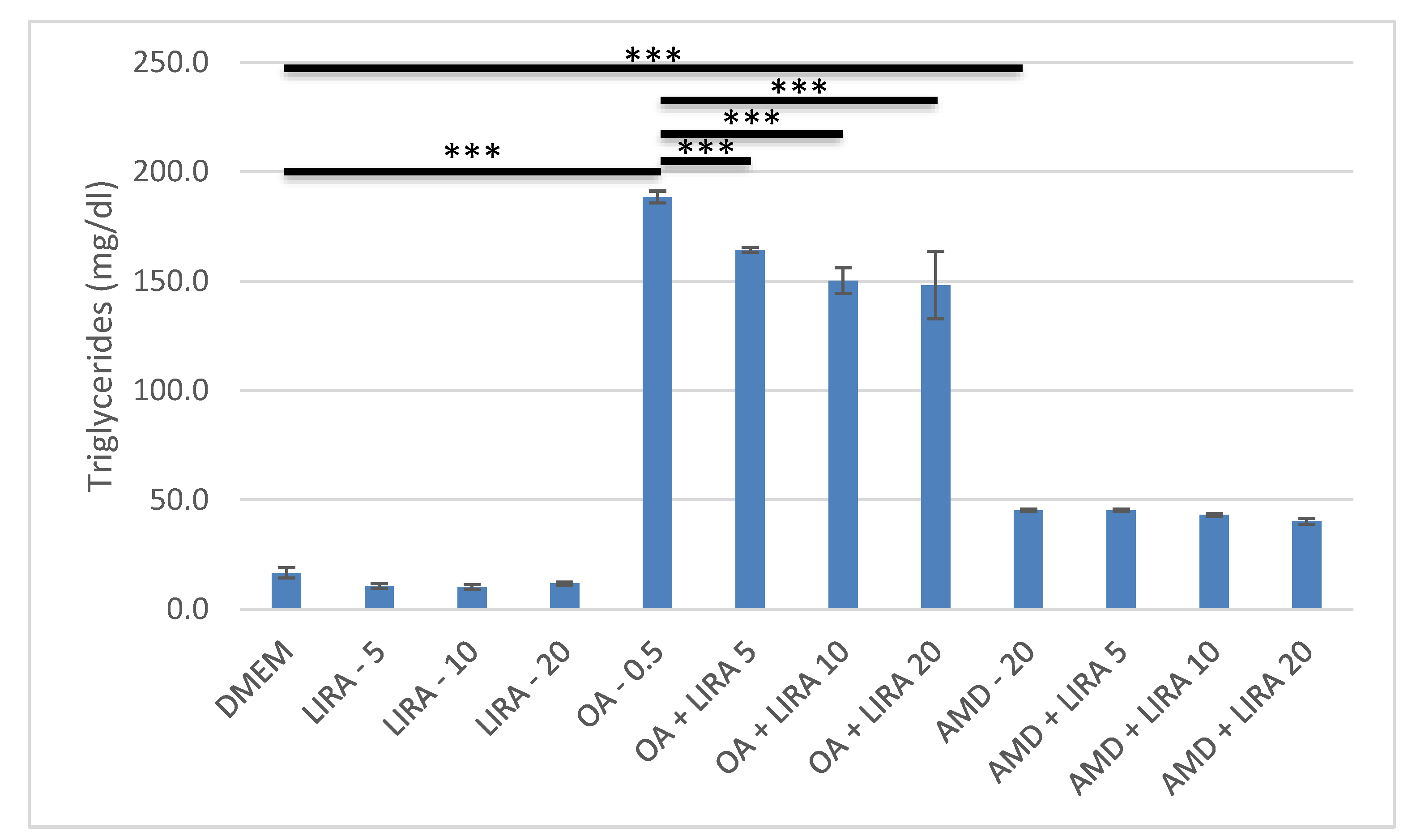
3.4. Assessment of the Effect of LIRA on Triglyceride Accumulation in NAS and DIS Cell Culture Models
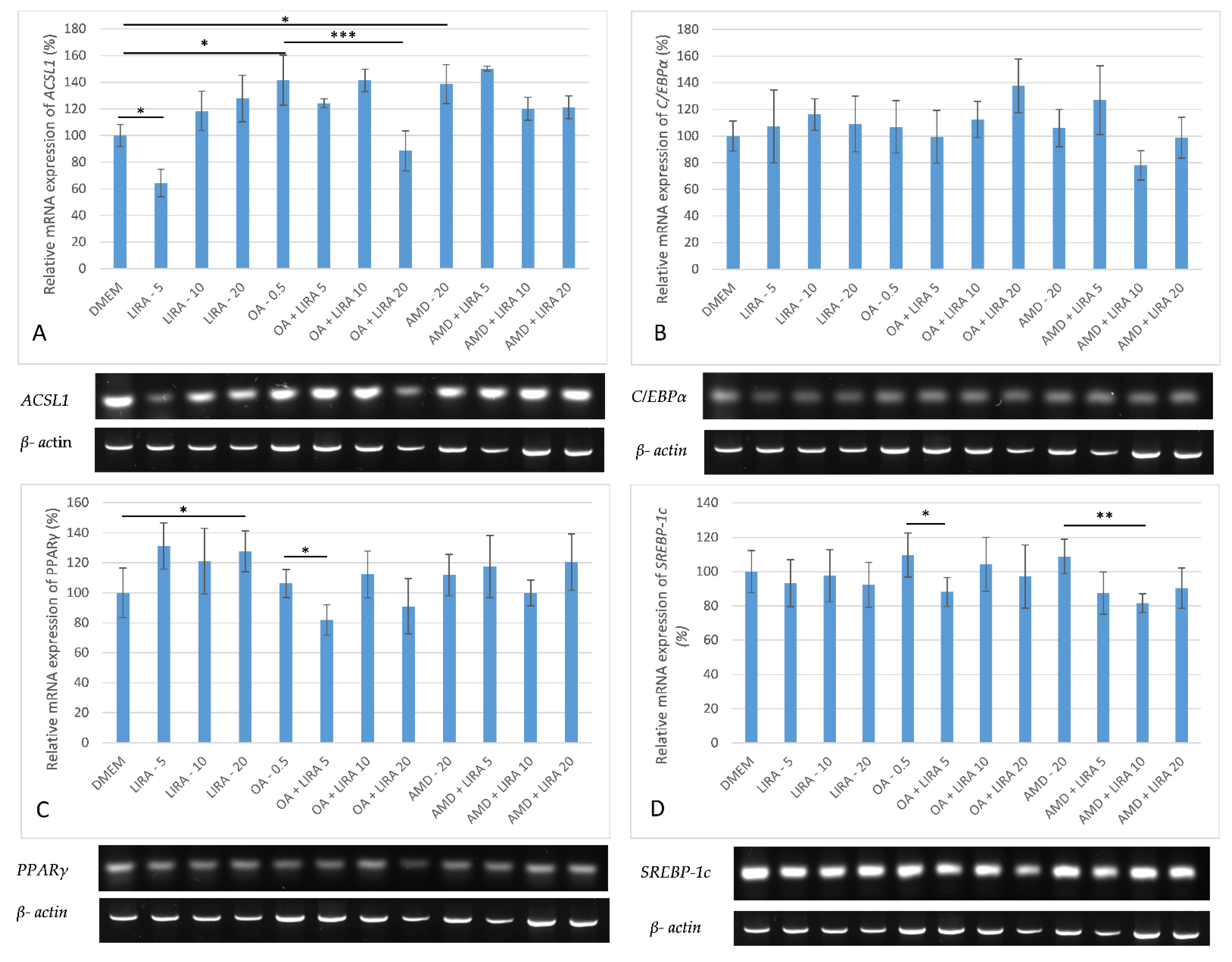
3.5. Assessment of the Effect of LIRA on Gene Expression in NAS and DIS Cell Culture Models
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, M.; Luo, Q.; Tao, Y.; Sun, X.; Liu, C. Pharmacotherapies for Drug-Induced Liver Injury: A Current Literature Review. Front. Pharmacol. 2021, 12, 806249. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.P.; Maddur, H.; Russo, M.W.; Wong, R.J.; Reddy, K.R.; Gastroenterology PPCotACo. ACG Clinical Guideline: Diagnosis and Management of Idiosyncratic Drug-Induced Liver Injury. Am. J. Gastroenterol. 2021, 116, 878–898. [Google Scholar] [CrossRef] [PubMed]
- Benić, M.S.; Nežić, L.; Vujić-Aleksić, V.; Mititelu-Tartau, L. Novel Therapies for the Treatment of Drug-Induced Liver Injury: A Systematic Review. Front. Pharmacol. 2021, 12, 785790. [Google Scholar] [CrossRef] [PubMed]
- Hoofnagle, J.H.; Björnsson, E.S. Drug-Induced Liver Injury—Types and Phenotypes. N. Engl. J. Med. 2019, 381, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, D.E.; Chalasani, N.P.; Lee, W.M.; Fontana, R.J.; Bonkovsky, H.L.; Watkins, P.B.; Hayashi, P.H.; Davern, T.J.; Navarro, V.; Reddy, R.; et al. Hepatic histological findings in suspected drug-induced liver injury: Systematic evaluation and clinical associations. Hepatology 2014, 59, 661–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miele, L.; Liguori, A.; Marrone, G.; Biolato, M.; Araneo, C.; Vaccaro, F.G.; Gasbarrini, A.; Grieco, A. Fatty liver and drugs: The two sides of the same coin. Eur. Rev. Med. Pharmacol. Sci. 2017, 21 (Suppl. S1), 86–94. [Google Scholar]
- Akbay, E.; Erdem, B.; Ünlü, A.; Durukan, A.B.; Onur, M.A. Effects of N-acetyl cysteine, vitamin E and vitamin C on liver glutathione levels following amiodarone treatment in rats. Kardiochir. Torakochirurgia. Pol. 2019, 16, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Vitins, A.P.; Kienhuis, A.S.; Speksnijder, E.N.; Roodbergen, M.; Luijten, M.; van der Ven, L.T. Mechanisms of amiodarone and valproic acid induced liver steatosis in mouse In Vivo act as a template for other hepatotoxicity models. Arch. Toxicol. 2014, 88, 1573–1588. [Google Scholar] [CrossRef]
- Anthérieu, S.; Rogue, A.; Fromenty, B.; Guillouzo, A.; Robin, M.A. Induction of vesicular steatosis by amiodarone and tetracycline is associated with up-regulation of lipogenic genes in HepaRG cells. Hepatology 2011, 53, 1895–1905. [Google Scholar] [CrossRef]
- Rabinowich, L.; Shibolet, O. Drug Induced Steatohepatitis: An Uncommon Culprit of a Common Disease. Biomed Res. Int. 2015, 2015, 168905. [Google Scholar] [CrossRef] [Green Version]
- Massart, J.; Begriche, K.; Buron, N.; Porceddu, M.; Borgne-Sanchez, A.; Fromenty, B. Drug-Induced Inhibition of Mitochondrial Fatty Acid Oxidation and Steatosis. Curr. Pathobiol. Rep. 2013, 1, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.; Choi, H.K.; Lee, H.; Moon, K.S.; Oh, J.H.; Lee, J.; Shin, J.G.; Kim, D.H. Increased hepatic acylcarnitines after oral administration of amiodarone in rats. J. Appl. Toxicol. 2020, 40, 1004–1013. [Google Scholar] [CrossRef] [PubMed]
- Decara, J.; Arrabal, S.; Beiroa, D.; Rivera, P.; Vargas, A.; Serrano, A.; Pavón, F.J.; Ballesteros, J.; Dieguez, C.; Nogueiras, R.; et al. Antiobesity efficacy of GLP-1 receptor agonist liraglutide is associated with peripheral tissue-specific modulation of lipid metabolic regulators. Biofactors 2016, 42, 600–611. [Google Scholar] [CrossRef] [PubMed]
- Attia, S.L.; Softic, S.; Mouzaki, M. Evolving Role for Pharmacotherapy in NAFLD/NASH. Clin. Transl. Sci. 2021, 14, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Mahjoubin-Tehran, M.; De Vincentis, A.; Mikhailidis, D.P.; Atkin, S.L.; Mantzoros, C.S.; Jamialahmadi, T.; Sahebkar, A. Non-alcoholic fatty liver disease and steatohepatitis: State of the art on effective therapeutics based on the gold standard method for diagnosis. Mol. Metab. 2021, 50, 101049. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Ao, N.; Yang, J.; Wang, X.; Jin, S.; Du, J. The preventive effect of liraglutide on the lipotoxic liver injury via increasing autophagy. Ann. Hepatol. 2020, 19, 44–52. [Google Scholar] [CrossRef]
- Yu, P.; Xu, X.; Zhang, J.; Xia, X.; Xu, F.; Weng, J.; Lai, X.; Shen, Y. Liraglutide Attenuates Nonalcoholic Fatty Liver Disease through Adjusting Lipid Metabolism via SHP1/AMPK Signaling Pathway. Int. J. Endocrinol. 2019, 2019, 1567095. [Google Scholar] [CrossRef]
- Ji, J.; Feng, M.; Huang, Y.; Niu, X. Liraglutide inhibits receptor for advanced glycation end products (RAGE)/reduced form of nicotinamide-adenine dinucleotide phosphate (NAPDH) signaling to ameliorate non-alcoholic fatty liver disease (NAFLD) In Vivo and Vitro. Bioengineered 2022, 13, 5091–5102. [Google Scholar] [CrossRef]
- Ghazanfar, H.; Kandhi, S.D.; Nawaz, I.; Javed, N.; Abraham, M.C.; Farag, M.; Mahasamudram, J.; Patel, V.B.; Altaf, F.; Patel, H. Role of Glucagon-Like Peptide-1 Receptor Agonists in the Management of Non-Alcoholic Steatohepatitis: A Clinical Review Article. Cureus 2021, 13, e15141. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Hull, D.; Guo, K.; Barton, D.; Hazlehurst, J.M.; Gathercole, L.L.; Nasiri, M.; Yu, J.; Gough, S.C.; Newsome, P.N. Glucagon-like peptide 1 decreases lipotoxicity in non-alcoholic steatohepatitis. J. Hepatol. 2016, 64, 399–408. [Google Scholar] [CrossRef] [Green Version]
- Gastaldelli, A.; Cusi, K. From NASH to diabetes and from diabetes to NASH: Mechanisms and treatment options. JHEP Rep. 2019, 1, 312–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniels, S.J.; Leeming, D.J.; Detlefsen, S.; Bruun, M.F.; Hjuler, S.T.; Henriksen, K.; Hein, P.; Karsdal, M.A.; Brockbank, S.; Cruwys, S. Biochemical and histological characterisation of an experimental rodent model of non-alcoholic steatohepatitis—Effects of a peroxisome proliferator-activated receptor gamma (PPAR-γ) agonist and a glucagon-like peptide-1 analogue. Biomed. Pharmacother. 2019, 111, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazelhurst, J.M.; Guo, K.; Abouda, G.; Aldersley, M.A.; et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): A multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016, 387, 679–690. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.G.; Yang, T.L. Liraglutide reduces fatty degeneration in hepatic cells via the AMPK/SREBP1 pathway. Exp. Ther. Med. 2015, 10, 1777–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.G.; Luo, Y.; Wang, H.; Li, J.Y.; Yang, J.; Liu, Y.X.; Qu, H.Q.; Wang, B.L.; Zhu, M. Liraglutide Ameliorates Lipotoxicity-Induced Oxidative Stress by Activating the NRF2 Pathway in HepG2 Cells. Horm. Metab. Res. 2020, 52, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S.; Lee, B.W. Beneficial effect of anti-diabetic drugs for nonalcoholic fatty liver disease. Clin. Mol. Hepatol. 2020, 26, 430–443. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Cohen, D.E. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J. Gastroenterol. 2013, 48, 434–441. [Google Scholar] [CrossRef] [Green Version]
- Yasmin, T.; Rahman, M.M.; Khan, F.; Kabir, F.; Nahar, K.; Lasker, S.; Islam, M.D.; Hossain, M.M.; Hasan, R.; Rana, S.; et al. Metformin treatment reverses high fat diet- induced non-alcoholic fatty liver diseases and dyslipidemia by stimulating multiple antioxidant and anti-inflammatory pathways. Biochem. Biophys. Rep. 2021, 28, 101168. [Google Scholar] [CrossRef]
- Kuna, L.; Bozic, I.; Kizivat, T.; Bojanic, K.; Mrso, M.; Kralj, E.; Smolic, R.; Wu, G.Y.; Smolic, M. Models of Drug Induced Liver Injury (DILI)—Current Issues and Future Perspectives. Curr. Drug Metab. 2018, 19, 830–838. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.A.; Mells, J.; Dunham, R.M.; Grakoui, A.; Handy, J.; Saxena, N.K.; Anania, F.A. Glucagon-like peptide-1 receptor is present on human hepatocytes and has a direct role in decreasing hepatic steatosis in vitro by modulating elements of the insulin signaling pathway. Hepatology 2010, 51, 1584–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavez, C.P.; Cusi, K.; Kadiyala, S. The Emerging Role of Glucagon-like Peptide-1 Receptor Agonists for the Management of NAFLD. J. Clin. Endocrinol. Metab. 2022, 107, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Ji, L.; Zhu, C.; Xiao, Y.; Zhang, J.; Lu, J.; Yin, J.; Wei, L. Liraglutide Alleviates Hepatic Steatosis by Activating the TFEB-Regulated Autophagy-Lysosomal Pathway. Front. Cell Dev. Biol. 2020, 8, 602574. [Google Scholar] [CrossRef]
- Li, X.; Wu, X.; Jia, Y.; Fu, J.; Zhang, L.; Jiang, T.; Liu, J.; Wang, G. Liraglutide Decreases Liver Fat Content and Serum Fibroblast Growth Factor 21 Levels in Newly Diagnosed Overweight Patients with Type 2 Diabetes and Nonalcoholic Fatty Liver Disease. J. Diabetes Res. 2021, 2021, 3715026. [Google Scholar] [CrossRef]
- Song, T.; Jia, Y.; Li, Z.; Wang, F.; Ren, L.; Chen, S. Effects of Liraglutide on Nonalcoholic Fatty Liver Disease in Patients with Type 2 Diabetes Mellitus: A Systematic Review and Meta-Analysis. Diabetes Ther. 2021, 12, 1735–1749. [Google Scholar] [CrossRef] [PubMed]
- Ricchi, M.; Odoardi, M.R.; Carulli, L.; Anzivino, C.; Ballestri, S.; Pinetti, A.; Fantoni, L.I.; Marra, F.; Bertolotti, M.; Banni, S.; et al. Differential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured hepatocytes. J. Gastroenterol. Hepatol. 2009, 24, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Van Summeren, A.; Renes, J.; Bouwman, F.G.; Noben, J.P.; van Delft, J.H.; Kleinjans, J.C.; Mariman, E.C. Proteomics investigations of drug-induced hepatotoxicity in HepG2 cells. Toxicol. Sci. 2011, 120, 109–122. [Google Scholar] [CrossRef] [Green Version]
- Volarevic, M.; Wu, C.H.; Smolic, R.; Andorfer, J.H.; Wu, G.Y. A novel G418 conjugate results in targeted selection of genetically protected hepatocytes without bystander toxicity. Bioconjug. Chem. 2007, 18, 1965–1971. [Google Scholar] [CrossRef] [PubMed]
- Zjalić, M.; Mustapić, M.; Glumac, Z.; Prološčić, I.; Blažetić, S.; Vuković, A.; Masud, M.; Billah, M.; Khan, A.; Šegota, S.; et al. Construction of AC/DC magnetic syringe device for stimulated drug release, injection and ejection of nanocarriers and testing cytotoxicity. MethodsX 2021, 8, 101312. [Google Scholar] [CrossRef] [PubMed]
- Rueden, C.T.; Schindelin, J.; Hiner, M.C.; DeZonia, B.E.; Walter, A.E.; Arena, E.T.; Eliceiri, K.W. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinform. 2017, 18, 529. [Google Scholar] [CrossRef]
- Zhao, F.; Xie, P.; Jiang, J.; Zhang, L.; An, W.; Zhan, Y. The effect and mechanism of tamoxifen-induced hepatocyte steatosis In Vitro. Int. J. Mol. Sci. 2014, 15, 4019–4030. [Google Scholar] [CrossRef] [Green Version]
- Felser, A.; Lindinger, P.W.; Schnell, D.; Kratschmar, D.V.; Odermatt, A.; Mies, S.; Jenö, P.; Krähenbühl, S. Hepatocellular toxicity of benzbromarone: Effects on mitochondrial function and structure. Toxicology 2014, 324, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Allard, J.; Bucher, S.; Massart, J.; Ferron, P.J.; Le Guillou, D.; Loyant, R.; Daniel, Y.; Launay, Y.; Buron, N.; Begriche, K.; et al. Drug-induced hepatic steatosis in absence of severe mitochondrial dysfunction in HepaRG cells: Proof of multiple mechanism-based toxicity. Cell Biol. Toxicol. 2021, 37, 151–175. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Hua, J.; Barritt, G.; Liu, Y.; Tang, B.Z.; Tang, Y. Live Imaging and Quantitation of Lipid Droplets and Mitochondrial Membrane Potential Changes with Aggregation-Induced Emission Luminogens in an In Vitro Model of Liver Steatosis. Chembiochem 2019, 20, 1256–1259. [Google Scholar] [CrossRef]
- Sanoh, S.; Yamachika, Y.; Tamura, Y.; Kotake, Y.; Yoshizane, Y.; Ishida, Y.; Tateno, C.; Ohta, S. Assessment of amiodarone-induced phospholipidosis in chimeric mice with a humanized liver. J. Toxicol. Sci. 2017, 42, 589–596. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, Y.; Fukusato, T. Histopathology of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J. Gastroenterol. 2014, 20, 15539–15548. [Google Scholar] [CrossRef]
- Tandra, S.; Yeh, M.M.; Brunt, E.M.; Vuppalanchi, R.; Cummings, O.W.; Ünalp-Arida, A.; Wilson, L.A.; Chalasani, N. Presence and significance of microvesicular steatosis in nonalcoholic fatty liver disease. J. Hepatol. 2011, 55, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Li, X.; Meng, H.; Chen, L.; Meng, F. ACSL1 affects Triglyceride Levels through the PPARγ Pathway. Int. J. Med. Sci. 2020, 17, 720–727. [Google Scholar] [CrossRef] [Green Version]
- Li, L.O.; Ellis, J.M.; Paich, H.A.; Wang, S.; Gong, N.; Altshuller, G.; Thresher, R.J.; Koves, T.R.; Watkins, S.M.; Muoio, D.M.; et al. Liver-specific loss of long chain acyl-CoA synthetase-1 decreases triacylglycerol synthesis and beta-oxidation and alters phospholipid fatty acid composition. J. Biol. Chem. 2009, 284, 27816–27826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, S.; Wu, F.; Chen, M.; Li, Y.; You, M.; Zhang, Y.; Yang, P.; Wei, L.; Ruan, X.Z.; Zhao, L.; et al. Inhibition of Fatty Acid Translocase (FAT/CD36) Palmitoylation Enhances Hepatic Fatty Acid β-Oxidation by Increasing Its Localization to Mitochondria and Interaction with Long-Chain Acyl-CoA Synthetase 1. Antioxid. Redox Signal. 2022, 36, 1081–1100. [Google Scholar] [CrossRef]
- Yang, Z.; Yu, G.L.; Zhu, X.; Peng, T.H.; Lv, Y.C. Critical roles of FTO-mediated mRNA m6A demethylation in regulating adipogenesis and lipid metabolism: Implications in lipid metabolic disorders. Genes Dis. 2022, 9, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Nakajima, T.; Gonzalez, F.J.; Tanaka, N. PPARs as Metabolic Regulators in the Liver: Lessons from Liver-Specific PPAR-Null Mice. Int. J. Mol. Sci. 2020, 21, 2061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szalowska, E.; van der Burg, B.; Man, H.Y.; Hendriksen, P.J.; Peijnenburg, A.A. Model steatogenic compounds (amiodarone, valproic acid, and tetracycline) alter lipid metabolism by different mechanisms in mouse liver slices. PLoS ONE 2014, 9, e86795. [Google Scholar] [CrossRef] [Green Version]
- Guzmán, C.; Benet, M.; Pisonero-Vaquero, S.; Moya, M.; García-Mediavilla, M.V.; Martínez-Chantar, M.L.; Gonzállez-Gallego, J.; Castell, J.V.; Sánchez-Campos, S.; Jover, R. The human liver fatty acid binding protein (FABP1) gene is activated by FOXA1 and PPARα; and repressed by C/EBPα: Implications in FABP1 down-regulation in nonalcoholic fatty liver disease. Biochim. Biophys. Acta 2013, 1831, 803–818. [Google Scholar] [CrossRef]
- Jackson, S.H.; Martin, T.S.; Jones, J.D.; Seal, D.; Emanuel, F. Liraglutide (victoza): The first once-daily incretin mimetic injection for type-2 diabetes. P T 2010, 35, 498–529. [Google Scholar]
- Ao, N.; Ma, Z.; Yang, J.; Jin, S.; Zhang, K.; Luo, E.; Du, J. Liraglutide ameliorates lipotoxicity-induced inflammation through the mTORC1 signalling pathway. Peptides 2020, 133, 170375. [Google Scholar] [CrossRef]
- Petit, J.M.; Cercueil, J.P.; Loffroy, R.; Denimal, D.; Bouillet, B.; Fourmont, C.; Chevallier, O.; Duvillard, L.; Vergés, B. Effect of Liraglutide Therapy on Liver Fat Content in Patients with Inadequately Controlled Type 2 Diabetes: The Lira-NAFLD Study. J. Clin. Endocrinol. Metab. 2017, 102, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Ma, X.; Xuan, X.; Deng, H.; Chen, Q.; Yuan, L. Liraglutide Attenuates Non-Alcoholic Fatty Liver Disease in Mice by Regulating the Local Renin-Angiotensin System. Front. Pharmacol. 2020, 11, 432. [Google Scholar] [CrossRef] [Green Version]
- Flock, G.; Baggio, L.L.; Longuet, C.; Drucker, D.J. Incretin receptors for glucagon-like peptide 1 and glucose-dependent insulinotropic polypeptide are essential for the sustained metabolic actions of vildagliptin in mice. Diabetes 2007, 56, 3006–3013. [Google Scholar] [CrossRef]
- Ni, X.X.; Li, X.Y.; Wang, Q.; Hua, J. Regulation of peroxisome proliferator-activated receptor-gamma activity affects the hepatic stellate cell activation and the progression of NASH via TGF-β1/Smad signaling pathway. J. Physiol. Biochem. 2021, 77, 35–45. [Google Scholar] [CrossRef]
- Liu, X.; Xu, J.; Rosenthal, S.; Zhang, L.J.; McCubbin, R.; Meshgin, N.; Shang, L.; Koyama, Y.; Ma, H.Y.; Sharma, S.; et al. Identification of Lineage-Specific Transcription Factors That Prevent Activation of Hepatic Stellate Cells and Promote Fibrosis Resolution. Gastroenterology 2020, 158, 1728–1744.e14. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Guo, C.; Wu, J. The Agonists of Peroxisome Proliferator-Activated Receptor-γ for Liver Fibrosis. Drug Des. Devel Ther. 2021, 15, 2619–2628. [Google Scholar] [CrossRef]
- Quintero, P.; Arrese, M. Nuclear control of inflammation and fibrosis in nonalcoholic steatohepatitis: Therapeutic potential of dual peroxisome proliferator-activated receptor alpha/delta agonism. Hepatology 2013, 58, 1881–1884. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.L.; Ding, D.; Yin, W.H.; Peng, J.Y.; Hou, C.J.; Liu, X.P.; Chen, Y.I. TSA increases C/EBP-α expression by increasing its lysine acetylation in hepatic stellate cells. Mol. Med. Rep. 2017, 16, 6088–6093. [Google Scholar] [CrossRef] [Green Version]
- Mei, S.; Wang, X.; Zhang, J.; Qian, J.; Ji, J. In Vivo transfection of C/EBP-alpha gene could ameliorate CCL(4)-induced hepatic fibrosis in mice. Hepatol. Res. 2007, 37, 531–539. [Google Scholar] [CrossRef]
- Tao, L.L.; Cheng, Y.Y.; Ding, D.; Mei, S.; Xu, J.W.; Yu, J.; Ou-Yang, Q.; Deng, L.; Chen, Q.; Li, Q.Q.; et al. C/EBP-α ameliorates CCl(4)-induced liver fibrosis in mice through promoting apoptosis of hepatic stellate cells with little apoptotic effect on hepatocytes in vitro and In Vivo. Apoptosis 2012, 17, 492–502. [Google Scholar] [CrossRef]
- Wang, X.; Huang, G.; Mei, S.; Qian, J.; Ji, J.; Zhang, J. Over-expression of C/EBP-alpha induces apoptosis in cultured rat hepatic stellate cells depending on p53 and peroxisome proliferator-activated receptor-gamma. Biochem. Biophys. Res. Commun. 2009, 380, 286–291. [Google Scholar] [CrossRef]
- Liu, H.; Zhan, Y.L.; Luo, G.J.; Zou, L.L.; Li, Y.; Lu, H.Y. Liraglutide and Insulin Have Contrary Effects on Adipogenesis of Human Adipose-Derived Stem Cells via Wnt Pathway. Diabetes Metab. Syndr. Obes. 2020, 13, 3075–3087. [Google Scholar] [CrossRef]
- Sanz, C.; Vázquez, P.; Blázquez, C.; Barrio, P.A.; Alvarez, M.e.M.; Blázquez, E. Signaling and biological effects of glucagon-like peptide 1 on the differentiation of mesenchymal stem cells from human bone marrow. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E634–E643. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Denaturation | Annealing | Elongation |
|---|---|---|---|
| β-actin | 94 °C for 3 min | 56.7 °C for 45 s | 72 °C for 1 min in 30 cycles |
| ACSL1 | 94 °C for 3 min | 61 °C for 45 s | 72 °C for 1 min in 30 cycles |
| C/EBPα | 94 °C for 3 min | 61 °C for 45 s | 72 °C for 1 min in 30 cycles |
| PPARγ | 94 °C for 3 min | 61 °C for 45 s | 72 °C for 1 min in 30 cycles |
| SREBP-1c | 94 °C for 3 min | 61 °C for 45 s | 72 °C for 1 min in 30 cycles |
| Gene | Primer Sequences (5′-3′) |
|---|---|
| β-actin | Forward GCACCACACCTTCTACAATG Reverse TGCTTGCTGATCCACATCTG |
| ACSL1 | Forward GGAGTGGGCTGCAGTGAC Reverse GGGCTTGCATTGTCCTGT |
| C/EBPα | Forward CGCCTTCAACGACGAGTTCCTG Reverse CGCCTTGGCCTTCTCCTGCT |
| PPARγ | Forward ACCAAAGTGCAATCAAAGTGGA Reverse ATGAGGGAGTTGGAAGGCTCT |
| SREBP-1c | Forward CGGAACCATCTTGGCAACAGT Reverse CGCTTCTCAATGGCGTTGT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Omanovic Kolaric, T.; Kizivat, T.; Mihaljevic, V.; Zjalic, M.; Bilic-Curcic, I.; Kuna, L.; Smolic, R.; Vcev, A.; Wu, G.Y.; Smolic, M. Liraglutide Exerts Protective Effects by Downregulation of PPARγ, ACSL1 and SREBP-1c in Huh7 Cell Culture Models of Non-Alcoholic Steatosis and Drug-Induced Steatosis. Curr. Issues Mol. Biol. 2022, 44, 3465-3480. https://doi.org/10.3390/cimb44080239
Omanovic Kolaric T, Kizivat T, Mihaljevic V, Zjalic M, Bilic-Curcic I, Kuna L, Smolic R, Vcev A, Wu GY, Smolic M. Liraglutide Exerts Protective Effects by Downregulation of PPARγ, ACSL1 and SREBP-1c in Huh7 Cell Culture Models of Non-Alcoholic Steatosis and Drug-Induced Steatosis. Current Issues in Molecular Biology. 2022; 44(8):3465-3480. https://doi.org/10.3390/cimb44080239
Chicago/Turabian StyleOmanovic Kolaric, Tea, Tomislav Kizivat, Vjera Mihaljevic, Milorad Zjalic, Ines Bilic-Curcic, Lucija Kuna, Robert Smolic, Aleksandar Vcev, George Y. Wu, and Martina Smolic. 2022. "Liraglutide Exerts Protective Effects by Downregulation of PPARγ, ACSL1 and SREBP-1c in Huh7 Cell Culture Models of Non-Alcoholic Steatosis and Drug-Induced Steatosis" Current Issues in Molecular Biology 44, no. 8: 3465-3480. https://doi.org/10.3390/cimb44080239
APA StyleOmanovic Kolaric, T., Kizivat, T., Mihaljevic, V., Zjalic, M., Bilic-Curcic, I., Kuna, L., Smolic, R., Vcev, A., Wu, G. Y., & Smolic, M. (2022). Liraglutide Exerts Protective Effects by Downregulation of PPARγ, ACSL1 and SREBP-1c in Huh7 Cell Culture Models of Non-Alcoholic Steatosis and Drug-Induced Steatosis. Current Issues in Molecular Biology, 44(8), 3465-3480. https://doi.org/10.3390/cimb44080239