
Mitochondrial DNA and Inflammation in Alzheimer’s Disease

Abstract
:
1. Introduction
2. The Role of Microglia in Neurodegeneration
3. Mitochondria, between Neurodegeneration and Neuroinflammation
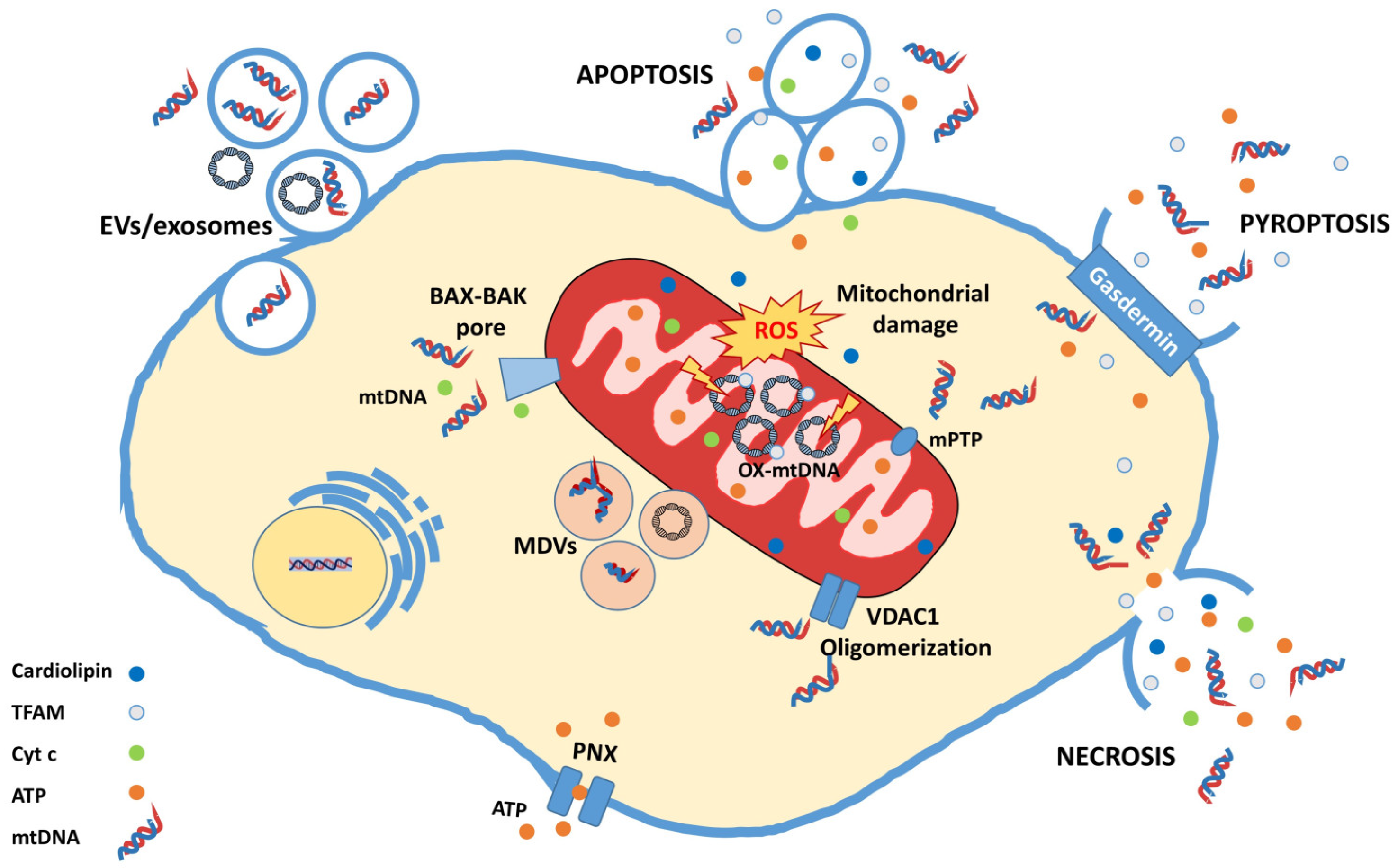
4. mtDNA, a Mitochondrial DAMP with Great Potential
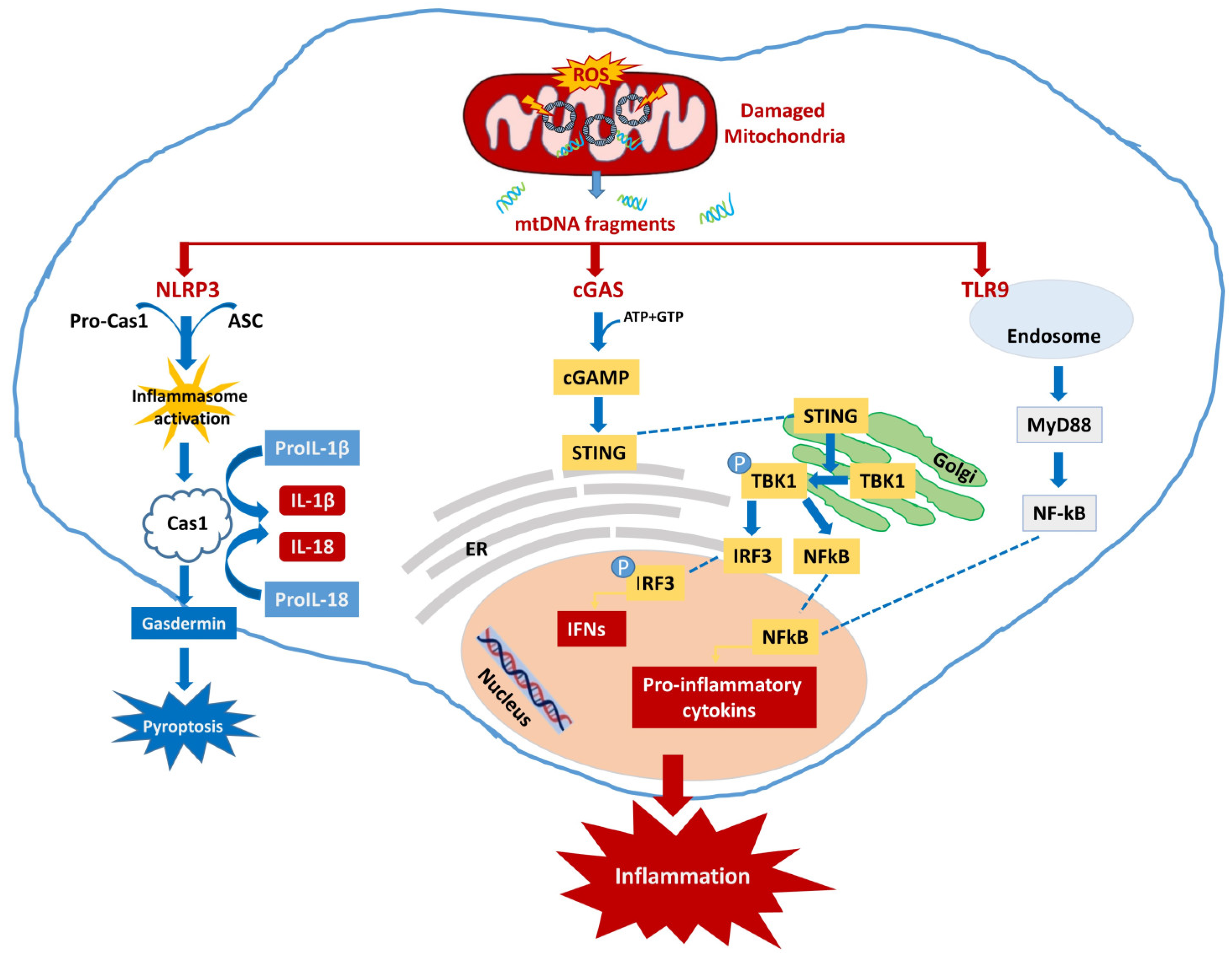
4.1. Neuroinflammation Activated by mtDNA
4.1.1. cGAS-STING
4.1.2. NLRP3
4.1.3. TLR
5. Therapeutic Strategies
Drugs with Mitochondrial Action
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, S.; Govindarajulu, M.; Jones, E.; Suppiramaniam, V.; Moore, T.; Dhanasekaran, M. Mitochondrial dysfunction and the role of Mitophagy in Alzheimer’s Disease. In Alzheimer’s Disease & Treatment; MedDocs Publishers LLC: Reno, NV, USA, 2018. [Google Scholar]
- Chakravorty, A.; Jetto, C.T.; Manjithaya, R. Dysfunctional Mitochondria and Mitophagy as Drivers of Alzheimer’s Disease Pathogenesis. Front. Aging Neurosci. 2019, 11, 311. [Google Scholar] [CrossRef] [PubMed]
- Wirths, O.; Breyhan, H.; Marcello, A.; Cotel, M.C.; Brück, W.; Bayer, T.A. Inflammatory changes are tightly associated with neurodegeneration in the brain and spinal cord of the APP/PS1KI mouse model of Alzheimer’s disease. Neurobiol. Aging 2010, 31, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Wiste, H.J.; Lesnick, T.G.; Weigand, S.D.; Knopman, D.S.; Vemuri, P.; Pankratz, V.S.; Senjem, M.L.; Gunter, J.L.; Mielke, M.M.; et al. Brain β-amyloid load approaches a plateau. Neurology 2013, 80, 890–896. [Google Scholar] [CrossRef] [PubMed]
- Govindarajulu, M.; Ramesh, S.; Beasley, M.; Lynn, G.; Wallace, C.; Labeau, S.; Pathak, S.; Nadar, R.; Moore, T.; Dhanasekaran, M. Role of cGAS–Sting Signaling in Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 8151. [Google Scholar] [CrossRef]
- Moya, G.E.; Rivera, P.D.; Dittenhafer-Reed, K.E. Evidence for the Role of Mitochondrial DNA Release in the Inflammatory Response in Neurological Disorders. Int. J. Mol. Sci. 2021, 22, 7030. [Google Scholar] [CrossRef]
- Lin, M.M.; Liu, N.; Qin, Z.H.; Wang, Y. Mitochondrial-derived damage-associated molecular patterns amplify neuroinflammation in neurodegenerative diseases. Acta Pharmacol. Sin. 2022, 43, 2439–2447. [Google Scholar] [CrossRef]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimers Dement 2016, 12, 719–732. [Google Scholar] [CrossRef]
- Deus, C.M.; Tavares, H.; Beatriz, M.; Mota, S.; Lopes, C. Mitochondrial Damage-Associated Molecular Patterns Content in Extracellular Vesicles Promotes Early Inflammation in Neurodegenerative Disorders. Cells 2022, 11, 2364. [Google Scholar] [CrossRef]
- Vezzani, B.; Carinci, M.; Patergnani, S.; Pasquin, M.P.; Guarino, A.; Aziz, N.; Pinton, P.; Simonato, M.; Giorgi, C. The Dichotomous Role of Inflammation in the CNS: A Mitochondrial Point of View. Biomolecules 2020, 10, 1437. [Google Scholar] [CrossRef]
- Di Filippo, M.; Chiasserini, D.; Tozzi, A.; Picconi, B.; Calabresi, P. Mitochondria and the link between neuroinflammation and neurodegeneration. J. Alzheimers Dis. 2010, 20 (Suppl. S2), S369–S379. [Google Scholar] [CrossRef] [PubMed]
- Bernaus, A.; Blanco, S.; Sevilla, A. Glia Crosstalk in Neuroinflammatory Diseases. Front. Cell. Neurosci. 2020, 14, 209. [Google Scholar] [CrossRef] [PubMed]
- Andronie-Cioara, F.L.; Ardelean, A.I.; Nistor-Cseppento, C.D.; Jurcau, A.; Jurcau, M.C.; Pascalau, N.; Marcu, F. Molecular Mechanisms of Neuroinflammation in Aging and Alzheimer’s Disease Progression. Int. J. Mol. Sci. 2023, 24, 1869. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.J.; Lyons, D.A. Glia as architects of central nervous system formation and functions. Science 2018, 362, 181–185. [Google Scholar] [CrossRef]
- Javanmehr, N.; Saleki, K.; Alijanizadeh, P.; Rezaei, N. Microglia dynamics in aging-related neurobehavioral and neuroinflammatory diseases. J. Neuroinflamm. 2022, 19, 273. [Google Scholar] [CrossRef]
- Vela, J.M.; Dalmau, I.; Gonzalez, B.; Castellano, B. Morphology and distribution of microglial cells in the young and adult mouse cerebellum. J. Comp. Neurol. 1995, 361, 602–616. [Google Scholar] [CrossRef]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Jin, J.; Guo, J.; Cai, H.; Zhao, C.; Wang, H.; Liu, Z.; Ge, Z.M. M2-Like Microglia Polarization Attenuates Neuropathic Pain Associated with Alzheimer’s Disease. J. Alzheimers Dis. 2020, 76, 1255–1265. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef]
- Rodríguez-Gómez, J.A.; Kavanagh, E.; Engskog-Vlachos, P.; Engskog, M.K.R.; Herrera, A.J.; Espinosa-Oliva, A.M.; Joseph, B.; Hajji, N.; Venero, J.L.; Burguillos, M.A. Microglia: Agents of the CNS Pro-Inflammatory Response. Cells 2020, 9, 1717. [Google Scholar] [CrossRef]
- Jurga, A.M.; Paleczna, M.; Kuter, K.Z. Overview of General and Discriminating Markers of Differential Microglia Phenotypes. Front. Cell. Neurosci. 2020, 14, 198. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Xiao, L.; Zhong, Z.; Wang, L.; Li, Z.; Pan, X.; Liu, Z. Astaxanthin acts via LRP-1 to inhibit inflammation and reverse lipopolysaccharide-induced M1/M2 polarization of microglial cells. Oncotarget 2017, 8, 69370–69385. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Liu, W.; Yin, J.; Chen, Y.; Guo, S.; Fan, H.; Li, X.; Zhang, X.; He, X.; Duan, C. TSG-6 attenuates inflammation-induced brain injury via modulation of microglial polarization in SAH rats through the SOCS3/STAT3 pathway. J. Neuroinflamm. 2018, 15, 231. [Google Scholar] [CrossRef] [PubMed]
- Bersano, A.; Engele, J.; Schäfer, M. Neuroinflammation and Brain Disease. BMC Neurol. 2023, 23, 227. [Google Scholar] [CrossRef]
- Subhramanyam, C.S.; Wang, C.; Hu, Q.; Dheen, S.T. Microglia-mediated neuroinflammation in neurodegenerative diseases. Semin. Cell Dev. Biol. 2019, 94, 112–120. [Google Scholar] [CrossRef]
- Carson, M.J.; Thrash, J.C.; Walter, B. The cellular response in neuroinflammation: The role of leukocytes, microglia and astrocytes in neuronal death and survival. Clin. Neurosci. Res. 2006, 6, 237–245. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef]
- Prat, A.; Biernacki, K.; Wosik, K.; Antel, J.P. Glial cell influence on the human blood-brain barrier. Glia 2001, 36, 145–155. [Google Scholar] [CrossRef]
- Koehler, R.C.; Gebremedhin, D.; Harder, D.R. Role of astrocytes in cerebrovascular regulation. J. Appl. Physiol. 2006, 100, 307–317. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Barres, B.A. Reactive astrocytes: Production, function, and therapeutic potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.S.; Sohn, D.H. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw. 2018, 18, e27. [Google Scholar] [CrossRef] [PubMed]
- Cianciulli, A.; Porro, C.; Calvello, R.; Trotta, T.; Lofrumento, D.D.; Panaro, M.A. Microglia mediated neuroinflammation: Focus on PI3K modulation. Biomolecules 2020, 10, 137. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, S.P.; Kamath, S.P.; Choolani, M.; Lu, J.; Dheen, S.T. microRNA-200b modulates microglia-mediated neuroinflammation via the cJun/MAPK pathway. J. Neurochem. 2014, 130, 388–401. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, B.; Kumar, D.; Cruz-Martins, N.; Sellamuthu, S. Role of TREM2 in Alzheimer’s Disease: A Long Road Ahead. Mol. Neurobiol. 2021, 58, 5239–5252. [Google Scholar] [CrossRef]
- Lee, C.Y.D.; Daggett, A.; Gu, X.; Jiang, L.L.; Langfelder, P.; Li, X.; Wang, N.; Zhao, Y.; Park, C.S.; Cooper, Y.; et al. Elevated TREM2 Gene Dosage Reprograms Microglia Responsivity and Ameliorates Pathological Phenotypes in Alzheimer’s Disease Models. Neuron 2018, 97, 1032–1048.e1035. [Google Scholar] [CrossRef]
- Galizzi, G.; Deidda, I.; Amato, A.; Calvi, P.; Terzo, S.; Caruana, L.; Scoglio, S.; Mulè, F.; Di Carlo, M. Aphanizomenon flos-aquae (AFA) Extract Prevents Neurodegeneration in the HFD Mouse Model by Modulating Astrocytes and Microglia Activation. Int. J. Mol. Sci. 2023, 24, 4731. [Google Scholar] [CrossRef]
- Yoo, S.M.; Park, J.; Kim, S.H.; Jung, Y.K. Emerging perspectives on mitochondrial dysfunction and inflammation in Alzheimer’s disease. BMB Rep. 2020, 53, 35–46. [Google Scholar] [CrossRef]
- Chen, C.H.; Zhou, W.; Liu, S.; Deng, Y.; Cai, F.; Tone, M.; Tone, Y.; Tong, Y.; Song, W. Increased NF-kB signaling up-regulates BACE1 expression and its therapeutic potential in Alzheimer’s disease. Int. J. Neuropsychopharmacol. 2012, 15, 77–90. [Google Scholar] [CrossRef]
- Cai, Z.; Hussain, M.D.; Yan, L.J. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int. J. Neurosci. 2014, 124, 307–321. [Google Scholar] [CrossRef]
- Antico Arciuch, V.G.; Elguero, M.E.; Poderoso, J.J.; Carreras, M.C. Mitochondrial regulation of cell cycle and proliferation. Antioxid. Redox Signal. 2012, 16, 1150–1180. [Google Scholar] [CrossRef] [PubMed]
- Verdin, E.; Hirschey, M.D.; Finley, L.W.S.; Haigis, M.C. Sirtuin regulation of mitochondria: Energy production, apoptosis, and signaling. Trends Biochem. Sci. 2010, 35, 669–675. [Google Scholar] [CrossRef] [PubMed]
- Billups, B.; Forsythe, I.D. Presynaptic Mitochondrial Calcium Sequestration Influences Transmission at Mammalian Central Synapses. J. Neurosci. 2002, 22, 5840–5847. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670–14675. [Google Scholar] [CrossRef] [PubMed]
- Golpich, M.; Amini, E.; Mohamed, Z.; Azman Ali, R.; Mohamed Ibrahim, N.; Ahmadiani, A. Mitochondrial Dysfunction and Biogenesis in Neurodegenerative diseases: Pathogenesis and Treatment. CNS Neurosci. Ther. 2017, 23, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Jetto, C.T.; Nambiar, A.; Manjithaya, R. Mitophagy and Neurodegeneration: Between the Knowns and the Unknowns. Front. Cell Dev. Biol. 2022, 10, 837337. [Google Scholar] [CrossRef]
- Mutisya, E.M.; Bowling, A.C.; Beal, M.F. Cortical Cytochrome Oxidase Activity Is Reduced in Alzheimer’s Disease. J. Neurochem. 1994, 63, 2179–2184. [Google Scholar] [CrossRef]
- Misrani, A.; Tabassum, S.; Yang, L. Mitochondrial Dysfunction and Oxidative Stress in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 17588. [Google Scholar] [CrossRef]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef]
- Raha, S.; Robinson, B.H. Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem. Sci. 2000, 25, 502–508. [Google Scholar] [CrossRef]
- Picca, A.; Calvani, R.; Coelho-Junior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial Dysfunction, Oxidative Stress, and Neuroinflammation: Intertwined Roads to Neurodegeneration. Antioxidants 2020, 9, 647. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Gupta, R.; Blanco, L.P.; Yang, S.; Shteinfer-Kuzmine, A.; Wang, K.; Kang, H. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science 2019, 366, 1531–1536. [Google Scholar] [CrossRef] [PubMed]
- Galizzi, G.; Di Carlo, M. Insulin and Its Key Role for Mitochondrial Function/Dysfunction and Quality Control: A Shared Link between Dysmetabolism and Neurodegeneration. Biology 2022, 11, 943. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498. [Google Scholar] [CrossRef]
- Arnoult, D.; Soares, F.; Tattoli, I.; Girardin, S.E. Mitochondria in innate immunity. EMBO Rep. 2011, 12, 901–910. [Google Scholar] [CrossRef]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef]
- Bajwa, E.; Pointer, C.B.; Klegeris, A. The Role of Mitochondrial Damage-Associated Molecular Patterns in Chronic Neuroinflammation. Mediat. Inflamm. 2019, 2019, 4050796. [Google Scholar] [CrossRef]
- Patil, V.; Cuenin, C.; Chung, F.; Rodríguez-Aguilera, J.R.; Fernandez-Jimenez, N.; Romero-Garmendia, I.; Bilbao, J.R.; Cahais, V.; Rothwell, J.; Herceg, Z. Human mitochondrial DNA is extensively methylated in a non-CpG context. Nucleic Acids Res. 2019, 47, 10072–10085. [Google Scholar] [CrossRef]
- Van Der Wijst, M.G.P.; Van Tilburg, A.Y.; Ruiters, M.H.J.; Rots, M.G. Experimental mitochondria-targeted DNA methylation identifies GpC methylation, not CpG methylation, as potential regulator of mitochondrial gene expression. Sci. Rep. 2017, 7, 177. [Google Scholar] [CrossRef]
- Luna-Sánchez, M.; Bianchi, P.; Quintana, A. Mitochondria-Induced Immune Response as a Trigger for Neurodegeneration: A Pathogen from Within. Int. J. Mol. Sci. 2021, 22, 8523. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, B.; Xu, L.; Yu, S.; Fu, J.; Wang, J.; Yan, X.; Su, J. ROS-Induced mtDNA Release: The Emerging Messenger for Communication between Neurons and Innate Immune Cells during Neurodegenerative Disorder Progression. Antioxidants 2021, 10, 1917. [Google Scholar] [CrossRef]
- Garcia, N.; Garcia, J.J.; Correa, F.; Chavez, E. The permeability transition pore as a pathway for the release of mitochondrial DNA. Life Sci. 2005, 76, 2873–2880. [Google Scholar] [CrossRef]
- McArthur, K.; Whitehead, L.W.; Heddleston, J.M.; Li, L.; Padman, B.S.; Oorschot, V.; Geoghegan, N.D.; Chappaz, S.; Davidson, S.; Chin, H.S.; et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 2018, 359, eaao6047. [Google Scholar] [CrossRef]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075. [Google Scholar] [CrossRef]
- Torralba, D.; Baixauli, F.; Villarroya-Beltri, C.; Fernández-Delgado, I.; Latorre-Pellicer, A.; Acín-Pérez, R.; Martín-Cófreces, N.B.; Jaso-Tamame, Á.L.; Iborra, S.; Jorge, I.; et al. Priming of dendritic cells by DNA-containing extracellular vesicles from activated T cells through anti-gen-driven contacts. Nat. Commun. 2018, 9, 2658. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.; Dixit, V.M. Signaling in innate immunity and inflammation. Cold Spring Harb. Perspect. Biol. 2012, 4, a006049. [Google Scholar] [CrossRef] [PubMed]
- Bader, V.; Winklhofer, K. Mitochondria at the interface between neurodegeneration and neuroinflammation. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2020; pp. 163–171. [Google Scholar]
- Voet, S.; Srinivasan, S.; Lamkanfi, M.; van Loo, G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. 2019, 11, e10248. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef]
- Chen, Q.; Sun, L.; Chen, Z. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.-T.; Grishin, N.V.; et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef] [PubMed]
- Gulen, M.F.; Samson, N.; Keller, A.; Schwabenland, M.; Liu, C.; Glück, S.; Thacker, V.V.; Favre, L.; Mangeat, B.; Kroese, L.J.; et al. cGAS–STING drives ageing-related inflammation and neurodegeneration. Nature 2023, 620, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Shang, G.; Gui, X.; Zhang, X.; Bai, X.-c.; Chen, Z.J. Structural basis of STING binding with and phosphorylation by TBK1. Nature 2019, 567, 394–398. [Google Scholar] [CrossRef]
- Barber, G.N. STING-dependent cytosolic DNA sensing pathways. Trends Immunol. 2014, 35, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.M.; Moore, Z.; Minter, M.R.; Crack, P.J. Type-I interferon pathway in neuroinflammation and neurodegeneration: Focus on Alzheimer’s disease. J. Neural Transm. 2018, 125, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Ma, G.; Li, X.; Zhao, J.; Zhao, Z.; Zeng, J. Activation of innate immune cGAS-STING pathway contributes to Alzheimer’s pathogenesis in 5 × FAD mice. Nat. Aging 2023, 3, 202–212. [Google Scholar] [CrossRef]
- Hou, Y.; Wei, Y.; Lautrup, S.; Yang, B.; Wang, Y.; Cordonnier, S.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A. NAD(+) supplementation reduces neuroinflammation and cell senescence in a transgenic mouse model of Alzheimer’s disease via cGAS-STING. Proc. Natl. Acad. Sci. USA 2021, 118, e2011226118. [Google Scholar] [CrossRef] [PubMed]
- Sijia, H.; Xin, L.; Namrata, M.; Anindita, B.; Hu, W.; Shangang, Z.; Feng, L.; Xianlin, H. Microglial cGAS deletion protects against amyloid-β induced Alzheimer’s disease pathogenesis. bioRxiv 2023, 2023, 552300. [Google Scholar] [CrossRef]
- Taylor, J.M.; Minter, M.R.; Newman, A.G.; Zhang, M.; Adlard, P.A.; Crack, P.J. Type-1 interferon signaling mediates neuro-inflammatory events in models of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 1012–1023. [Google Scholar] [CrossRef]
- Roy, E.R.; Wang, B.; Wan, Y.; Chiu, G.; Cole, A.; Yin, Z.; Propson, N.E.; Xu, Y.; Jankowsky, J.L.; Liu, Z.; et al. Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J. Clin. Investig. 2020, 130, 1912–1930. [Google Scholar] [CrossRef]
- Mesquita, S.D.; Ferreira, A.C.; Gao, F.; Coppola, G.; Geschwind, D.H.; Sousa, J.C.; Correia-Neves, M.; Sousa, N.; Palha, J.A.; Marques, F. The choroid plexus transcriptome reveals changes in type I and II interferon responses in a mouse model of Alzheimer’s disease. Brain Behav. Immun. 2015, 49, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Udeochu, J.C.; Amin, S.; Huang, Y.; Fan, L.; Torres, E.R.S.; Carling, G.K.; Liu, B.; McGurran, H.; Coronas-Samano, G.; Kauwe, G.; et al. Tau activation of microglial cGAS–IFN reduces MEF2C-mediated cognitive resilience. Nat. Neurosci. 2023, 26, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Baruch, K.; Deczkowska, A.; David, E.; Castellano, J.M.; Miller, O.; Kertser, A.; Berkutzki, T.; Barnett-Itzhaki, Z.; Bezalel, D.; Wyss-Coray, T.; et al. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science 2014, 346, 89–93. [Google Scholar] [CrossRef]
- Minter, M.R.; Moore, Z.; Zhang, M.; Brody, K.M.; Jones, N.C.; Shultz, S.R.; Taylor, J.M.; Crack, P.J. Deletion of the type-1 interferon receptor in APPSWE/PS1ΔE9 mice preserves cognitive function and alters glial phenotype. Acta Neuropathol. Commun. 2016, 4, 72. [Google Scholar] [CrossRef] [PubMed]
- Minter, M.R.; Main, B.S.; Brody, K.M.; Zhang, M.; Taylor, J.M.; Crack, P.J. Soluble amyloid triggers a myeloid differentiation factor 88 and interferon regulatory factor 7 dependent neuronal type-1 interferon response in vitro. J. Neuroinflammation 2015, 12, 71. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Seo, G.J.; Choi, Y.J.; Kwak, M.-J.; Ge, J.; Rodgers, M.A.; Shi, M.; Leslie, B.J.; Hopfner, K.-P.; Ha, T.; et al. Crosstalk between the cGAS DNA sensor and Beclin-1 autophagy protein shapes innate antimicrobial immune responses. Cell Host Microbe 2014, 15, 228–238. [Google Scholar] [CrossRef]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef]
- Man, S.M.; Kanneganti, T.-D. Regulation of inflammasome activation. Immunol. Rev. 2015, 265, 6–21. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Agostini, L.; Martinon, F.; Burns, K.; McDermott, M.F.; Hawkins, P.N.; Tschopp, J. NALP3 forms an IL-1β-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 2004, 20, 319–325. [Google Scholar] [CrossRef]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome activation and regulation: Toward a better understanding of complex mechanisms. Cell Discov. 2020, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Gustin, A.; Kirchmeyer, M.; Koncina, E.; Felten, P.; Losciuto, S.; Heurtaux, T.; Tardivel, A.; Heuschling, P.; Dostert, C. NLRP3 Inflammasome Is Expressed and Functional in Mouse Brain Microglia but Not in Astrocytes. PLoS ONE 2015, 10, e0130624. [Google Scholar] [CrossRef] [PubMed]
- Missiroli, S.; Patergnani, S.; Caroccia, N.; Pedriali, G.; Perrone, M.; Previati, M.; Wieckowski, M.R.; Giorgi, C. Mitochondria-associated membranes (MAMs) and inflammation. Cell Death Dis. 2018, 9, 329. [Google Scholar] [CrossRef] [PubMed]
- Kesavardhana, S.; Kanneganti, T.-D. Mechanisms governing inflammasome activation, assembly and pyroptosis induction. Int. Immunol. 2017, 29, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.; Satija, G.; Madan, A.; Garg, M.; Alam, M.M.; Shaquiquzzaman, M.; Khanna, S.; Tiwari, P.; Parvez, S.; Iqubal, A.; et al. Role of NLRP3 Inflammasome and Its Inhibitors as Emerging Therapeutic Drug Candidate for Alzheimer’s Disease: A Review of Mechanism of Activation, Regulation, and Inhibition. Inflammation 2023, 46, 56–87. [Google Scholar] [CrossRef]
- Canzone, L.; Pei, L.; Yao, S.; Wu, Y.; Shang, Y. NLRP3 Inflammasome in Neurological Diseases, from Functions to Therapies. Front. Cell. Neurosci. 2017, 11, 63. [Google Scholar]
- Aglietti, R.A.; Estevez, A.; Gupta, A.; Ramirez, M.G.; Liu, P.S.; Kayagaki, N.; Ciferri, C.; Dixit, V.M.; Dueber, E.C. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc. Natl. Acad. Sci. USA 2016, 113, 7858–7863. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.K.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2010, 12, 222–230. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nat. Cell Biol. 2010, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Barczuk, J.; Siwecka, N.; Lusa, W.; Rozpędek-Kamińska, W.; Kucharska, E.; Majsterek, I. Targeting NLRP3-Mediated Neuroinflammation in Alzheimer’s Disease Treatment. Int. J. Mol. Sci. 2022, 23, 8979. [Google Scholar] [CrossRef] [PubMed]
- Lučiūnaitė, A.; McManus, R.M.; Jankunec, M.; Rácz, I.; Dansokho, C.; Dalgėdienė, I.; Schwartz, S.; Brosseron, F.; Heneka, M.T. Soluble Aβ Oligomers and Protofibrils Induce NLRP3 Inflammasome Activation in Microglia. J. Neurochem. 2020, 155, 650–661. [Google Scholar] [CrossRef] [PubMed]
- Halle, A.; Hornung, V.; Petzold, G.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Flores, J.; Fillion, M.L.; LeBlanc, A.C. Caspase-1 inhibition improves cognition without significantly altering amyloid and inflammation in aged Alzheimer disease mice. Cell Death Dis. 2022, 13, 864. [Google Scholar] [CrossRef]
- Furman, D.; Chang, J.; Lartigue, L. Expression of specific inflammasome gene modules stratifies older individuals into two extreme clinical and immunological states. Nature Med. 2017, 23, 174–184. [Google Scholar] [CrossRef]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Griffin, W.S.; Stanley, L.C.; Ling, C.; White, L.; MacLeod, V.; Perrot, L.J.; White, C.L.; Araoz, C. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc. Natl. Acad. Sci. USA 1989, 86, 7611–7615. [Google Scholar] [CrossRef]
- Santos, A.N.; Ewers, M.; Minthon, L.; Simm, A.; Silber, R.E.; Blennow, K.; Prvulovic, D.; Hansson, O.; Hampel, H. Amyloid-β Oligomers in Cerebrospinal Fluid are Associated with Cognitive Decline in Patients with Alzheimer’s Disease. J. Alzheimers Dis. 2012, 29, 171–176. [Google Scholar] [CrossRef]
- Swardfager, W.; Lanctôt, K.; Rothenburg, L.; Wong, A.; Cappell, J.; Herrmann, N. A meta-analysis of cytokines in Alzheimer’s disease. Biol. Psychiatry 2010, 68, 930–941. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.E.; Iyer, S.; Thangavel, R.; Kempuraj, D.; Selvakumar, G.P.; Raikwar, S.P.; Zaheer, S.; Zaheer, A. Co-Localization of Glia Maturation Factor with NLRP3 Inflammasome and Autophagosome Markers in Human Alzheimer’s Disease Brain. J. Alzheimers Dis. 2017, 60, 1143–1160. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.G.; Ito, K.; Skinner, R.D.; Mrak, R.E.; Rovnaghi, C.R.; Van Eldik, L.J.; Griffin, W.S. In vivo and in vitro evidence supporting a role for the inflammatory cytokine interleukin-1 as a driving force in Alzheimer pathogenesis. Neurobiol. Aging 1996, 17, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Kitazawa, M.; Cheng, D.; Tsukamoto, M.R.; Koike, M.A.; Wes, P.D.; Vasilevko, V.; Cribbs, D.H.; LaFerla, F.M. Blocking IL-1 Signaling Rescues Cognition, Attenuates Tau Pathology, and Restores Neuronal β-Catenin Pathway Function in an Alzheimer’s Disease Model. J. Immunol. 2011, 187, 6539–6549. [Google Scholar] [CrossRef] [PubMed]
- Bossù, P.; Ciaramella, A.; Salani, F.; Bizzoni, F.; Varsi, E.; Di Iulio, F.; Giubilei, F.; Gianni, W.; Trequattrini, A.; Moro, M.L.; et al. Interleukin-18 produced by peripheral blood cells is increased in Alzheimer’s disease and correlates with cognitive impairment. Brain Behav. Immun. 2008, 22, 487–492. [Google Scholar] [CrossRef]
- Ojala, J.O.; Sutinen, E.M.; Salminen, A.; Pirttilä, T. Interleukin-18 increases expression of kinases involved in tau phosphorylation in SH-SY5Y neuroblastoma cells. J. Neuroimmunol. 2008, 205, 86–93. [Google Scholar] [CrossRef]
- Brosseron, F.; Krauthausen, M.; Kummer, M.; Heneka, M.T. Body fluid cytokine levels in mild cognitive impairment and Alzheimer’s disease: A comparative overview. Mol. Neurobiol. 2014, 50, 534–544. [Google Scholar] [CrossRef]
- Sudduth, T.L.; Schmitt, F.A.; Nelson, P.T.; Wilcock, D.M. Neuroinflammatory phenotype in early Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1051–1059. [Google Scholar] [CrossRef]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef]
- Hanslik, K.L.; Ulland, T.K. The Role of Microglia and the Nlrp3 Inflammasome in Alzheimer’s Disease. Front. Neurol. 2020, 11, 570711. [Google Scholar] [CrossRef]
- He, X.F.; Xu, J.H.; Li, G.; Li, M.Y.; Li, L.L.; Pei, Z.; Zhang, L.Y.; Hu, X.Q. NLRP3-dependent microglial training impaired the clearance of amyloid-beta and aggravated the cognitive decline in Alzheimer’s disease. Cell Death Dis. 2020, 11, 849. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, C.; Rubio Araiz, A.; Bryson, K.J.; Finucane, O.; Larkin, C.; Mills, E.L.; Robertson, A.A.B.; Cooper, M.A.; O’Neill, L.A.J.; Lynch, M.A. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-beta and cognitive function in APP/PS1 mice. Brain Behav. Immun. 2017, 61, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhuang, L.; Luo, X.; Liang, J.; Sun, E.; He, Y. Protection of MCC950 against Alzheimer’s disease via inhibiting neuronal pyroptosis in SAMP8 mice. Exp. Brain Res. 2020, 238, 2603–2614. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Guan, Y.; Liang, B.; Ding, P.; Hou, X.; Wei, W.; Ma, Y. Therapeutic potential of MCC950, a specific inhibitor of NLRP3 inflammasome. Eur. J. Pharmacol. 2022, 928, 175091. [Google Scholar] [CrossRef]
- Kuwar, R.; Rolfe, A.; Di, L.; Blevins, H.; Xu, Y.; Sun, X.; Bloom, G.S.; Zhang, S.; Sun, D. A Novel Inhibitor Targeting NLRP3 Inflammasome Reduces Neuropathology and Improves Cognitive Function in Alzheimer’s Disease Transgenic Mice. J. Alzheimers Dis. 2021, 82, 1769–1783. [Google Scholar] [CrossRef]
- Dong, D.; Ren, A.; Yang, Y.; Su, J.; Liu, L.; Zhuo, W.; Liang, Y. VX-765 Alleviates β-Amyloid Deposition and Secondary Degeneration in the Ipsilateral Hippocampus and Ameliorates Cognitive Decline after Focal Cortical Infarction in Rats. J. Mol. Neurosci. 2022, 72, 2389–2397. [Google Scholar] [CrossRef]
- Flores, J.; Noël, A.; Foveau, B.; Lynham, J.; Lecrux, C.; LeBlanc, A.C. Caspase-1 inhibition alleviates cognitive impairment and neuropathology in an Alzheimer’s disease mouse model. Nat. Commun. 2018, 9, 3916. [Google Scholar] [CrossRef]
- El-Zayat, S.R.; Sibaii, H.; Mannaa, F.A. Toll-like receptors activation, signaling, and targeting: An overview. Bull. Natl. Res. Cent. 2019, 43, 187. [Google Scholar] [CrossRef]
- Frederiksen, H.R.; Haukedal, H.; Freude, K. Cell Type Specific Expression of Toll-Like Receptors in Human Brains and Implications in Alzheimer’s Disease. BioMed Res. Int. 2019, 2019, 7420189. [Google Scholar] [CrossRef]
- Jiménez-Dalmaroni, M.J.; Gerswhin, M.E.; Adamopoulos, I.E. The critical role of toll-like receptors—From microbial recognition to autoimmunity: A comprehensive review. Autoimmun. Rev. 2016, 15, 1–8. [Google Scholar] [CrossRef]
- Al-Ghraiybah, N.F.; Wang, J.; Alkhalifa, A.E.; Roberts, A.B.; Raj, R.; Yang, E.; Kaddoumi, A. Glial Cell-Mediated Neuroinflammation in Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 10572. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed]
- Latz, E.; Schoenemeyer, A.; Visintin, A.; Fitzgerald, K.A.; Monks, B.G.; Knetter, C.F.; Lien, E.; Nilsen, N.J.; Espevik, T.; Golenbock, D.T. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat. Immunol. 2004, 5, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-Z.; Liu, Z.; Liu, J.; Ren, J.-X.; Sun, T.-S. Mitochondrial DNA induces inflammation and increases TLR9/NF-κB expression in lung tissue. Int. J. Mol. Med. 2014, 33, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Nuevo, A.; Díaz-Ramos, A.; Noguera, E.; Díaz-Sáez, F.; Duran, X.; Muñoz, J.P.; Romero, M.; Plana, N.; Sebastián, D.; Tezze, C.; et al. Mitochondrial DNA and TLR9 drive muscle inflammation upon Opa1 deficiency. EMBO J. 2018, 37, e96553. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Signaling to NF-κB by toll-like receptors. Trends Mol. Med. 2007, 13, 460–469. [Google Scholar] [CrossRef]
- Heilig, R.; Lee, J.; Tait, S.W.G. Mitochondrial DNA in cell death and inflammation. Biochem. Soc. Trans. 2023, 51, 457–472. [Google Scholar] [CrossRef]
- Amcheslavsky, A.; Zou, W.; Bar-Shavit, Z. Toll-like receptor 9 regulates tumor necrosis factor-alpha expression by different mechanisms. Implications for osteoclastogenesis. J. Biol. Chem. 2004, 279, 54039–54045. [Google Scholar] [CrossRef]
- Priviero, F.; Calmasini, F.; Dela Justina, V.; Wenceslau, C.F.; McCarthy, C.G.; Webb, R.C. Macrophage-Specific Toll Like Receptor 9 (TLR9) Causes Corpus Cavernosum Dysfunction in Mice Fed a High Fat Diet. J. Sex. Med. 2021, 18, 723–731. [Google Scholar] [CrossRef]
- Adhikarla, S.V.; Jha, N.K.; Goswami, V.K.; Sharma, A.; Bhardwaj, A.; Dey, A.; Villa, C.; Kumar, Y.; Jha, S.K. TLR-Mediated Signal Transduction and Neurodegenerative Disorders. Brain Sci. 2021, 11, 1373. [Google Scholar] [CrossRef]
- Di, J.M.; Pang, J.; Pu, X.Y.; Zhang, Y.; Liu, X.P.; Fang, Y.Q.; Ruan, X.X.; Gao, X. Toll-like receptor 9 agonists promote IL-8 and TGF-beta1 production via activation of nuclear factor kappaB in PC-3 cells. Cancer Genet. Cytogenet 2009, 192, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Doi, Y.; Mizuno, T.; Maki, Y.; Jin, S.; Mizoguchi, H.; Ikeyama, M.; Doi, M.; Michikawa, M.; Takeuchi, H.; Suzumura, A. Microglia activated with the toll-like receptor 9 ligand CpG attenuate oligomeric amyloid {beta} neurotoxicity in in vitro and in vivo models of Alzheimer’s disease. Am. J. Pathol. 2009, 175, 2121–2132. [Google Scholar] [CrossRef] [PubMed]
- Scholtzova, H.; Kascsak, R.J.; Bates, K.A.; Boutajangout, A.; Kerr, D.J.; Meeker, H.C.; Mehta, P.D.; Spinner, D.S.; Wisniewski, T. Induction of Toll-Like Receptor 9 Signaling as a Method for Ameliorating Alzheimer’s Disease-Related Pathology. J. Neurosci. 2009, 29, 61846–61854. [Google Scholar] [CrossRef] [PubMed]
- Scholtzova, H.; Chianchiano, P.; Pan, J.; Sun, Y.; Goñi, F.; Mehta, P.D.; Wisniewski, T. Amyloid β and Tau Alzheimer’s disease related pathology is reduced by Toll-like receptor 9 stimulation. Acta Neuropathol. Commun. 2014, 2, 101. [Google Scholar] [CrossRef]
- Scholtzova, H.; Do, E.; Dhakal, S.; Sun, Y.; Liu, S.; Mehta, P.D.; Wisniewski, T. Innate Immunity Stimulation via Toll-Like Receptor 9 Ameliorates Vascular Amyloid Pathology in Tg-SwDI Mice with Associated Cognitive Benefits. J. Neurosci. 2017, 37, 936–959. [Google Scholar] [CrossRef]
- Patel, A.G.; Nehete, P.N.; Krivoshik, S.R.; Pei, X.; Cho, E.L.; Nehete, B.P.; Ramani, M.D.; Shao, Y.; Williams, L.E.; Wisniewski, T.; et al. Innate immunity stimulation via CpG oligodeoxynucleotides ameliorates Alzheimer’s disease pathology in aged squirrel monkeys. Brain 2021, 144, 2146–2165. [Google Scholar] [CrossRef]
- Heikenwalder, M.; Polymenidou, M.; Junt, T.; Sigurdson, C.; Wagner, H.; Akira, S.; Zinkernagel, R.; Aguzzi, A. Lymphoid follicle destruction and immunosuppression after repeated CpG oligodeoxynucleotide administration. Nat. Med. 2004, 10, 187–192. [Google Scholar] [CrossRef]
- Campbell, J.D.; Cho, Y.; Foster, M.L.; Kanzler, H.; Kachura, M.A.; Lum, J.A.; Ratcliffe, M.J.; Sathe, A.; Leishman, A.J.; Bahl, A.; et al. CpG-containing immunostimulatory DNA sequences elicit TNF-alpha-dependent toxicity in rodents but not in humans. J. Clin. Investig. 2009, 119, 2564–2576. [Google Scholar] [CrossRef]
- Swanson, C.J.; Zhang, Y.; Dhadda, S.; Wang, J.; Kaplow, J.; Lai, R.Y.K.; Lannfelt, L.; Bradley, H.; Rabe, M.; Koyama, A.; et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Abeta protofibril antibody. Alzheimers Res. Ther. 2021, 13, 80. [Google Scholar] [CrossRef]
- Van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in early Alzheimer’s disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef]
- Shadfar, S.; Hwang, C.J.; Lim, M.-S.; Choi, D.-Y.; Hong, J.T. Involvement of inflammation in Alzheimer’s disease pathogenesis and therapeutic potential of anti-inflammatory agents. Arch. Pharm. Res. 2015, 38, 2106–2119. [Google Scholar] [CrossRef] [PubMed]
- Breitner, J.C.; Baker, L.D.; Montine, T.J.; Meinert, C.L.; Lyketsos, C.G.; Ashe, K.H.; Brandt, J.; Craft, S.; Evans, D.E.; ADAPT Research Group; et al. Extended results of the Alzheimer’s disease anti-inflammatory prevention trial. Alzheimers Dement. 2011, 7, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Porrini, V.; Lanzillotta, A.; Branca, C.; Benarese, M.; Parrella, E.; Lorenzini, L.; Calza, L.; Flaibani, R.; Spano, P.; Imbimbo, B.; et al. CHF5074 (CSP-1103) induces microglia alternative activation in plaque-free Tg2576 mice and primary glial cultures exposed to beta-amyloid. Neuroscience 2015, 302, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.; Sharma, S.; Winston, J.; Nunez, M.; Bottini, G.; Franceschi, M.; Scarpini, E.; Frigerio, E.; Fiorentini, F.; Fernandez, M.; et al. CHF5074 reduces biomarkers of neuroinflammation in patients with mild cognitive impairment: A 12-week, double-blind, placebo-controlled study. Curr. Alzheimer Res. 2013, 10, 742–753. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, Z.; Zheng, Y.; Yu, Q.; Zeng, M.; Bai, L.; Yang, L.; Guo, M.; Jiang, X.; Gan, J. Inhibitors of the NLRP3 inflammasome pathway as promising therapeutic candidates for inflammatory diseases. Int. J. Mol. Med. 2023, 51, 35. [Google Scholar] [CrossRef]
- Farré-Alins, V.; Narros-Fernández, P.; Palomino-Antolín, A.; Decouty-Pérez, C.; Lopez-Rodriguez, A.B.; Parada, E.; Muñoz-Montero, A.; Gómez-Rangel, V.; López-Muñoz, F.; Ramos, E.; et al. Melatonin Reduces NLRP3 Inflammasome Activation by Increasing α7 nAChR-Mediated Autophagic Flux. Antioxidants 2020, 9, 1299. [Google Scholar] [CrossRef]
- Wade, A.G.; Farmer, M.; Harari, G.; Fund, N.; Laudon, M.; Nir, T.; Frydman-Marom, A.; Zisapel, N. Add-on prolonged-release melatonin for cognitive function and sleep in mild to moderate Alzheimer’s disease: A 6-month, randomized, placebo-controlled, multicenter trial. Clin. Interv. Aging 2014, 9, 947–961. [Google Scholar] [CrossRef]
- Schneider, L.S.; Laudon, M.; Nir, T.; Caceres, J.; Ianniciello, G.; Capulli, M.; Zisapel, N. A polymorphism cluster at the 2q12 locus may predict response to Piromelatine in patients with mild Alzheimer’s disease. J. Prev. Alzheimers Dis. 2022, 9, 247–254. [Google Scholar] [CrossRef]
- Shabalina, I.G.; Vyssokikh, M.Y.; Gibanova, N.; Csikasz, R.I.; Edgar, D.; Hallden-Waldemarson, A.; Rozhdestvenskaya, Z.; Bakeeva, L.E.; Vays, V.B.; Pustovidko, A.V.; et al. Improved Health-Span and Lifespan in mtDNA Mutator Mice Treated with the Mitochondrially Targeted Antioxidant SkQ1. Aging 2017, 9, 315–339. [Google Scholar] [CrossRef]
- Johri, A. Disentangling Mitochondria in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 11520. [Google Scholar] [CrossRef]
- McManus, M.J.; Murphy, M.P.; Franklin, J.L. The mitochondria-targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2011, 31, 15703–15715. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Wang, X.; Xiang, Q.; Meng, X.; Peng, Y.; Du, N.; Liu, Z.; Sun, Q.; Wang, C.; Liu, X. Astaxanthin alleviates brain aging in rats by attenuating oxidative stress and increasing BDNF levels. Food Funct. 2013, 5, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zou, J.; Wang, Y.; Wang, J.; Ji, X.; Zhang, T.; Chu, Y.; Cui, R.; Zhang, G.; Shi, G.; et al. Hydralazine inhibits neuroinflammation and oxidative stress in APP/PS1 mice via TLR4/NF-κB and Nrf2 pathways. Neuropharmacology 2023, 3, 240, 109706. [Google Scholar] [CrossRef] [PubMed]
- Mari, M.; Morales, A.; Colell, A.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Mitochondrial glutathione, a key survival antioxidant. Antioxid. Redox Signal. 2009, 11, 2685–2700. [Google Scholar] [CrossRef] [PubMed]
- Studer, R.; Baysang, G.; Brack, C. N-Acetyl-L-Cystein downregulates beta-amyloid precursor protein gene transcription in human neuroblastoma cells. Biogerontology 2001, 2, 55–60. [Google Scholar] [CrossRef]
- Olivieri, G.; Baysang, G.; Meier, F.; Müller-Spahn, F.; Stähelin, H.B.; Brockhaus, M.; Brack, C.H. N-acetyl-L-cysteine protects SHSY5Y neuroblastoma cells from oxidative stress and cell cytotoxicity: Effects on beta-amyloid secretion and tau phosphorylation. J. Neurochem. 2001, 76, 224–233. [Google Scholar] [CrossRef]
- Fu, A.L.; Dong, Z.H.; Sun, M.J. Protective effect of N-acetyl-L-cysteine on amyloid beta-peptide-induced learning and memory deficits in mice. Brain Res. 2006, 1109, 201–206. [Google Scholar] [CrossRef]
- Remington, R.; Bechtel, C.; Larsen, D.; Samar, A.; Doshanjh, L.; Fishman, P.; Luo, Y.; Smyers, K.; Page, R.; Morrell, C.; et al. A phase II randomized clinical trial of a nutritional formulation for cognition and mood in Alzheimer’s Disease. J. Alzheimers Dis. 2015, 45, 395–405. [Google Scholar] [CrossRef]
- Jang, S.Y.; Kang, H.T.; Hwang, E.S. Nicotinamide-induced mitophagy: Event mediated by high NAD+/NADH ratio and SIRT1 protein activation. J. Biol. Chem. 2012, 287, 19304–19314. [Google Scholar] [CrossRef]
- Long, A.N.; Owens, K.; Schlappal, A.E.; Kristian, T.; Fishman, P.S.; Schuh, R.A. Effect of nicotinamide mononucleotide on brain mitochondrial respiratory deficits in an Alzheimer’s disease-relevant murine model. BMC Neurol. 2015, 15, 19. [Google Scholar] [CrossRef]
- Kiss, T.; Nyúl-Tóth, Á.; Balasubramanian, P.; Tarantini, S.; Ahire, C.; Yabluchanskiy, A.; Csipo, T.; Farkas, E.; Wren, J.D.; Garman, L.; et al. Nicotinamide mononucleotide (NMN) supplementation promotes neurovascular rejuvenation in aged mice: Transcriptional footprint of SIRT1 activation, mitochondrial protection, anti-inflammatory, and anti-apoptotic effects. Geroscience 2020, 42, 527–546. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hu, X.; Yang, Y.; Takata, T.; Sakurai, T. Nicotinamide mononucleotide protects against β-amyloid oligomer-induced cognitive impairment and neuronal death. Brain Res. 2016, 1643, 1–9. [Google Scholar] [CrossRef]
- Li, A.; Zhang, S.; Li, J.; Liu, K.; Huang, F.; Liu, B. Metformin and resveratrol inhibit Drp1-mediated mitochondrial fission and prevent ER stress-associated NLRP3 inflammasome activation in the adipose tissue of diabetic mice. Mol. Cell. Endocrinol. 2016, 434, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Torabi, N.; Noursadeghi, E.; Shayanfar, F.; Nazari, M.; Fahanik-Babaei, J.; Saghiri, R.; Khodagholi, F.; Eliassi, A. Intranasal insulin improves the structure-function of the brain mitochondrial ATP-sensitive Ca2+ activated potassium channel and respiratory chain activities under diabetic conditions. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166075. [Google Scholar] [CrossRef] [PubMed]
- Wardelmann, K.; Blümel, S.; Rath, M.; Alfine, E.; Chudoba, C.; Schell, M.; Cai, W.; Hauffe, R.; Warnke, K.; Flore, T.; et al. Insulin action in the brain regulates mitochondrial stress responses and reduces diet-induced weight gain. Mol. Metab. 2019, 21, 68–81. [Google Scholar] [CrossRef]
- Saunders, A.M.; Burns, D.K.; Gottschalk, W.K. Reassessment of Pioglitazone for Alzheimer’s Disease. Front. Neurosci. 2021, 15, 666958. [Google Scholar] [CrossRef]
- Vieira, R.; Souto, S.B.; Sánchez-López, E.; Machado, A.L.; Severino, P.; Jose, S.; Santini, A.; Fortuna, A.; García, M.L.; Silva, A.M. Sugar-Lowering Drugs for Type 2 Diabetes Mellitus and Metabolic Syndrome—Review of Classical and New Compounds: Part-I. Pharmaceuticals 2019, 12, 152. [Google Scholar] [CrossRef]
- Bharath, L.P.; Agrawal, M.; McCambridge, G.; Nicholas, D.A.; Hasturk, H.; Liu, J.; Jiang, K.; Liu, R.; Guo, Z.; Deeney, J. Metformin Enhances Autophagy and Normalizes Mitochondrial Function to Alleviate Aging-Associated Inflammation. Cell Metab. 2020, 32, 44–55. [Google Scholar] [CrossRef]
- Koenig, A.M.; Mechanic-Hamilton, D.; Xie, S.X.; Combs, M.F.; Cappola, A.R.; Xie, L.; Detre, J.A.; Wolk, D.A.; Arnold, S.E. Effects of the Insulin Sensitizer Metformin in Alzheimer Disease. Alzheimer Dis. Assoc. Disord. 2017, 31, 107–113. [Google Scholar] [CrossRef]
- Luchsinger, J.A.; Perez, T.; Chang, H.; Mehta, P.; Steffener, J.; Pradabhan, G.; Ichise, M.; Manly, J.; Devanand, D.P.; Bagiella, E. Metformin in Amnestic Mild Cognitive Impairment: Results of a Pilot Randomized Placebo Controlled Clinical Trial. J. Alzheimers Dis. 2016, 51, 501–514. [Google Scholar] [CrossRef]
- Aroda, V.R.; Edelstein, S.L.; Goldberg, R.B.; Knowler, W.C.; Marcovina, S.M.; Orchard, T.J.; Bray, G.A.; Schade, D.S.; Temprosa, M.G.; White, N.H.; et al. Long-Term Metformin Use and Vitamin B12 Deficiency in the Diabetes Prevention Program Outcomes Study. J. Clin. Endocrinol. Metab. 2016, 101, 1754–1761. [Google Scholar] [CrossRef] [PubMed]
- Mantik, K.E.K.; Kim, S.; Gu, B.; Moon, S.; Kwak, H.-B.; Park, D.-H.; Kang, J.-H. Repositioning of Anti-Diabetic Drugs against Dementia: Insight from Molecular Perspectives to Clinical Trials. Int. J. Mol. Sci. 2023, 24, 11450. [Google Scholar] [CrossRef] [PubMed]
- Aisen, P.S.; Schafer, K.A.; Grundman, M.; Pfeiffer, E.; Sano, M.; Davis, K.L.; Farlow, M.R.; Jin, S.; Thomas, R.G.; Thal, L.J.; et al. Effects of rofecoxib or naproxen vs. placebo on Alzheimer disease progression: A randomized controlled trial. JAMA 2003, 289, 2819–2826. [Google Scholar] [CrossRef] [PubMed]
- Bentham, P.; Gray, R.; Sellwood, E.; Hills, R.; Crome, P.; Raftery, J. Aspirin in Alzheimer’s disease (AD2000): A randomised open-label trial. Lancet Neurol. 2008, 7, 41–49. [Google Scholar] [PubMed]
- Scharf, S.; Mander, A.; Ugoni, A.; Vajda, F.; Christophidis, N. A double-blind, placebo-controlled trial of diclofenac/misoprostol in Alzheimer’s disease. Neurology 1999, 53, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Thal, L.J.; Ferris, S.H.; Kirby, L.; Block, G.A.; Lines, C.R.; Yuen, E.; Assaid, C.; Nessly, M.L.; Norman, B.A.; Baranak, C.C.; et al. A randomized, double-blind, study of rofecoxib in patients with mild cognitive impairment. Neuropsychopharmacology 2005, 30, 1204–1215. [Google Scholar] [CrossRef]
- Zheng, Y.; Chen, X.X.; Zhang, D.Y.; Wang, W.J.; Peng, K.; Li, Z.Y.; Mao, Z.W.; Tan, C.P. Activation of the cGAS-STING pathway by a mitochondrial DNA-targeted emissive rhodium(iii) metallointercalator. Chem Sci. 2023, 14, 6890–6903. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Drug | Activity | Patients | ClinicalTrials.gov ID |
|---|---|---|---|
| Melatonin | Antioxidant; mPTP inhibitor. | 60 partecipants Age: 18 years and older with AD dementia or MCI | NCT04522960 |
| Piromelatine | 225 participants; Age: 60–85 years with cognitive decline | NCT05267535 Phase II/III | |
| Coenzyme Q10 | Antioxidant | 100 participants; Age: 50–80 years with MCI or AD | NCT06040905 |
| Astaxanthin | Antioxidant; mPTP inhibitor. | 50 participants; Age: 60–90 years with dementia | NCT05015374 |
| Hydralazine | Activates autophagy; Regulates antioxidant production by NRF2 | 424 participants; Age: 49 Years and older with diagnosis of AD | NCT04842552 Phase III |
| Glutathione (GlyNAC) | Antioxidant | 52 participants; Age: 55–85 years with progressive memory loss | NCT04740580 Early Phase I |
| Nicotinamide mononucleotide (MIB-626) | Induces mitophagy; Preserves mitochondrial function | 50 participants; Age: 55–85 years with AD | NCT05040321 Phase I/II |
| Nicotinamide Riboside | Boosts NAD+ levels; Activates mitophagy; Regulates mitochondrial function | 50 participants; Age: 55–89 years with MCI or Mild AD | NCT04430517 Early Phase I |
| 80 participants; Age: 50–85 years with AD | NCT05617508 | ||
| Insulin | Regulates mitochondrial function | 40 participants; Age: 55–85 years with mild MCI or Mild AD | NCT05006599 Phase II |
| Metformin | Antioxidant; Preserves mitochondrial morphology; Induces mitophagy. | 242 participants; Age: 60–80 Years with MCI | NCT04511416 Phase III |
| 600 participants; Age: 60–79 Years with risk factors for dementia | NCT05109169 Phase II | ||
| 326 participants; Age: 55–90 years with MCI | NCT04098666 Phase II/III |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galizzi, G.; Di Carlo, M. Mitochondrial DNA and Inflammation in Alzheimer’s Disease. Curr. Issues Mol. Biol. 2023, 45, 8586-8606. https://doi.org/10.3390/cimb45110540
Galizzi G, Di Carlo M. Mitochondrial DNA and Inflammation in Alzheimer’s Disease. Current Issues in Molecular Biology. 2023; 45(11):8586-8606. https://doi.org/10.3390/cimb45110540
Chicago/Turabian StyleGalizzi, Giacoma, and Marta Di Carlo. 2023. "Mitochondrial DNA and Inflammation in Alzheimer’s Disease" Current Issues in Molecular Biology 45, no. 11: 8586-8606. https://doi.org/10.3390/cimb45110540
APA StyleGalizzi, G., & Di Carlo, M. (2023). Mitochondrial DNA and Inflammation in Alzheimer’s Disease. Current Issues in Molecular Biology, 45(11), 8586-8606. https://doi.org/10.3390/cimb45110540