

ParticleChromo3D+: A Web Server for ParticleChromo3D Algorithm for 3D Chromosome Structure Reconstruction

Abstract
:1. Introduction
2. Materials and Methods
2.1. Particle Swarm Optimization and ParticleChromo3D
- and are the position at a time stamp n and the position at the next time stamp , respectively.
- and are the velocity at a time stamp n and the velocity at the next time stamp , respectively.
- w is used to create inertia for the velocity. This helps reduce or increase the contribution of the individual particle’s past velocity.
- and are the local and global weights, respectively. These weights are used to tell each particle if it should prioritize its information or the swarm information.
- and are randomized values that increase the breadth of the geometries that the particles explore.
- contains the position of the chromatin bin for the optimum structure that the individual particle has found. Each particle has its own structure made up of each chromatin bin’s position.
- contains the position of the chromatin bin for the optimum structure that the swarm has found.
2.2. Scoring Intermittent 3D Structures
- is the distance between two chromatin bins found by our particle’s structure.
- is the expected distance between two chromatin bins based on the IF data.
- is the total number of chromatin bins.
- is a positive real number for alternating between the top and bottom loss functions. We set to .
- is the expected distance between two chromatin bins based on the IF data.
- is the information frequency between two chromatin bins i and j.
- is a conversion factor.
2.3. Scoring Final 3D Structures
- and are individual distances.
- and are sample means of the distances.
- and are the individual distances and converted into ranked variables.
- and are sample means of the ranked distances.
2.4. Containerization
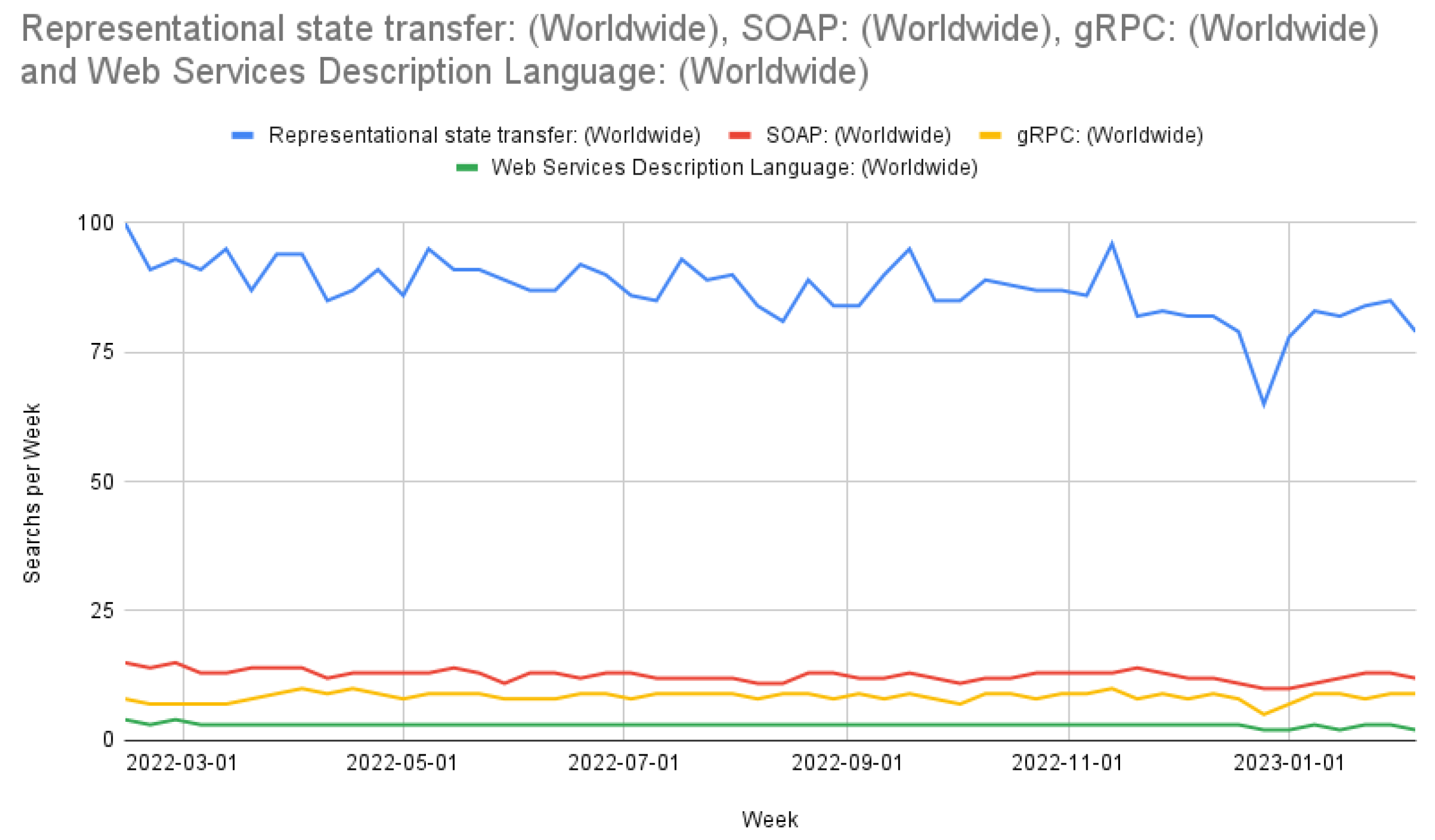
2.5. Representational State Transfer
3. Results
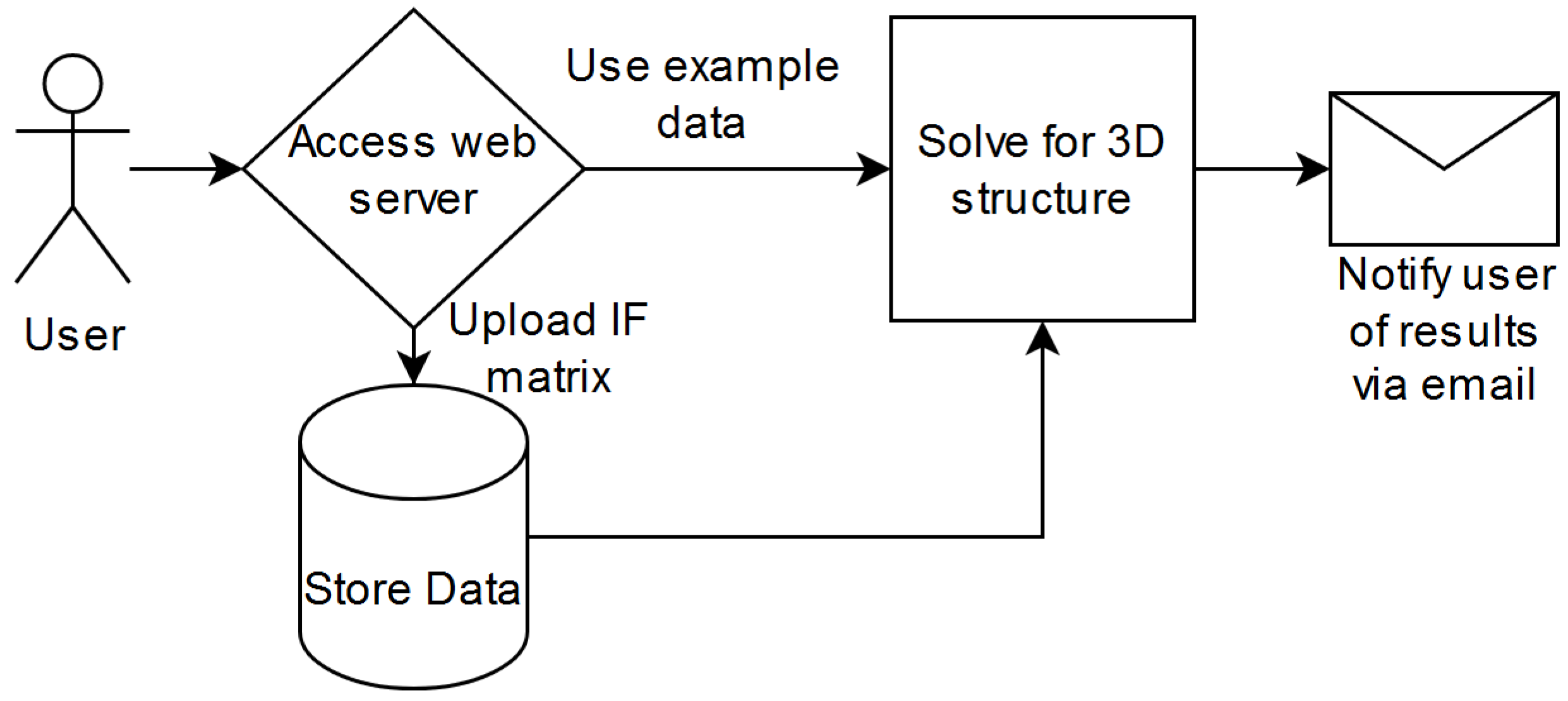
3.1. Usage
3.1.1. Front-End Access
3.1.2. Back-End API
http://biomlearn.uccs.edu:5001/uploadedThe download endpoint allows the user to download the contents of a known name PDB file.
biomlearn.uccs.edu:5001/download?ofname=${filename}
Lastly, the convert endpoint allows the user to convert 3xN matrices to square matrices so long as they are uploaded in a tab-separated values format. This endpoint can be used to format the IF data for use with ParticleChromo3D+ or any other square-matrix-based solution.
biomlearn.uccs.edu:5001/convert?filename=${filename}
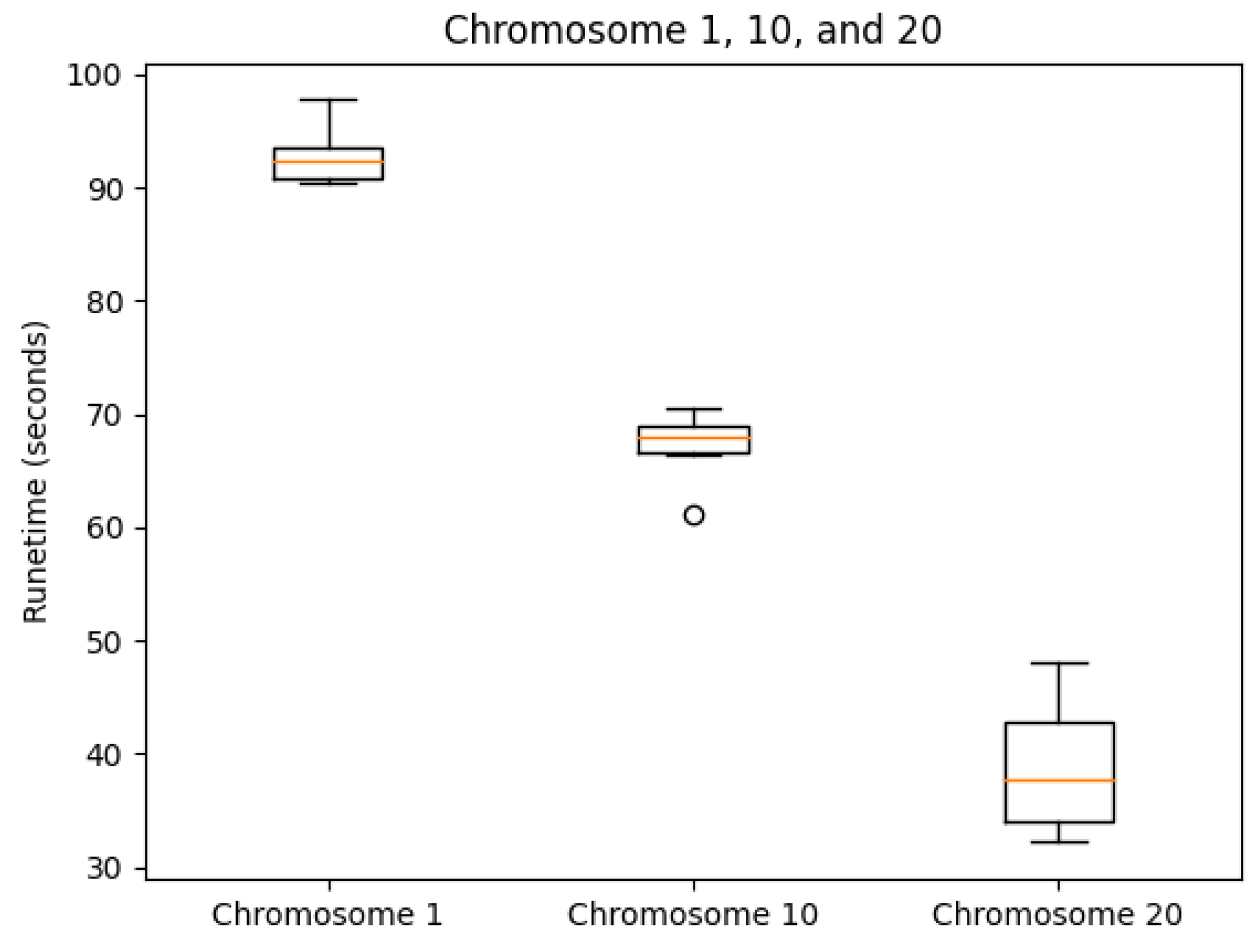
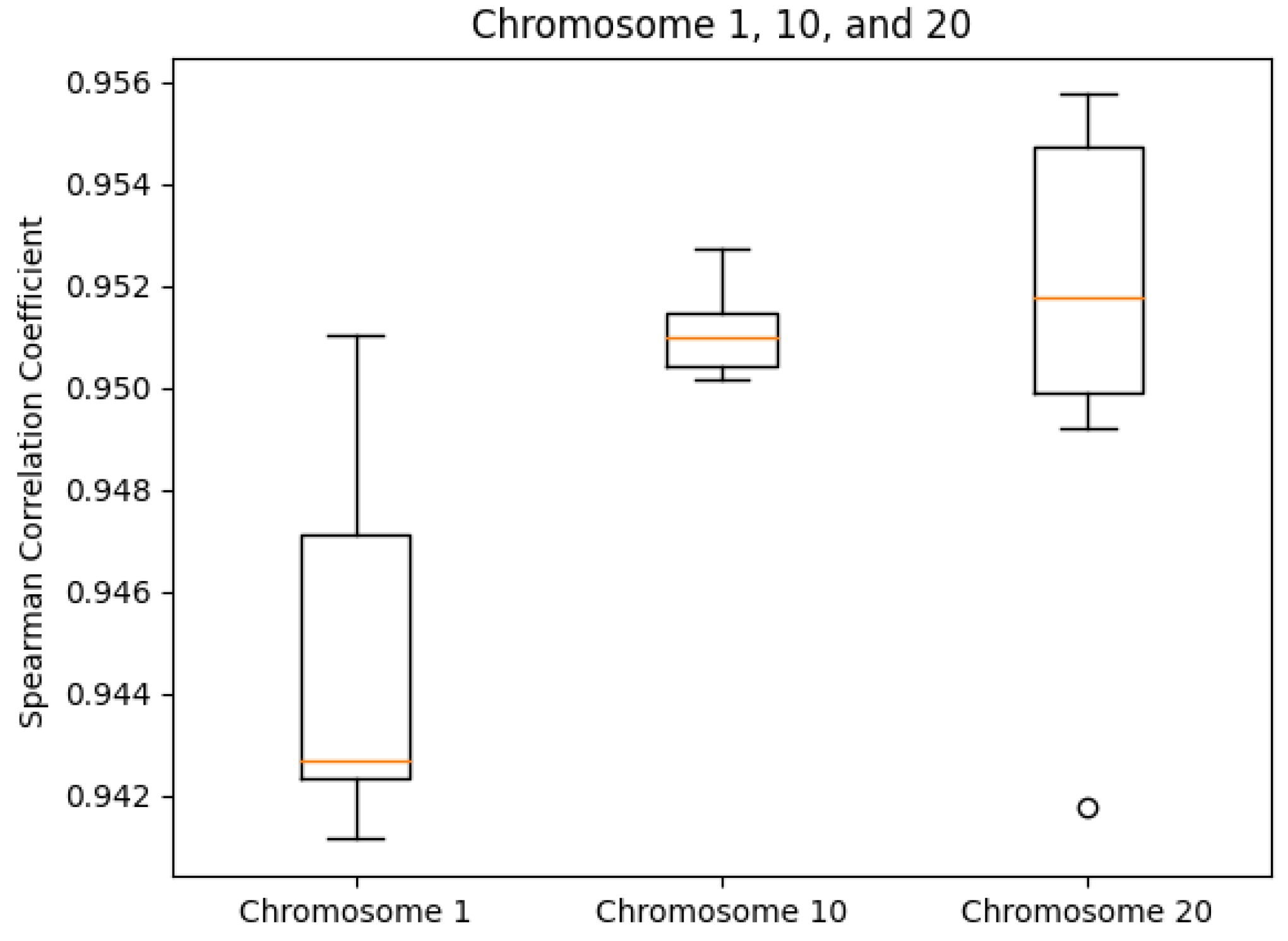
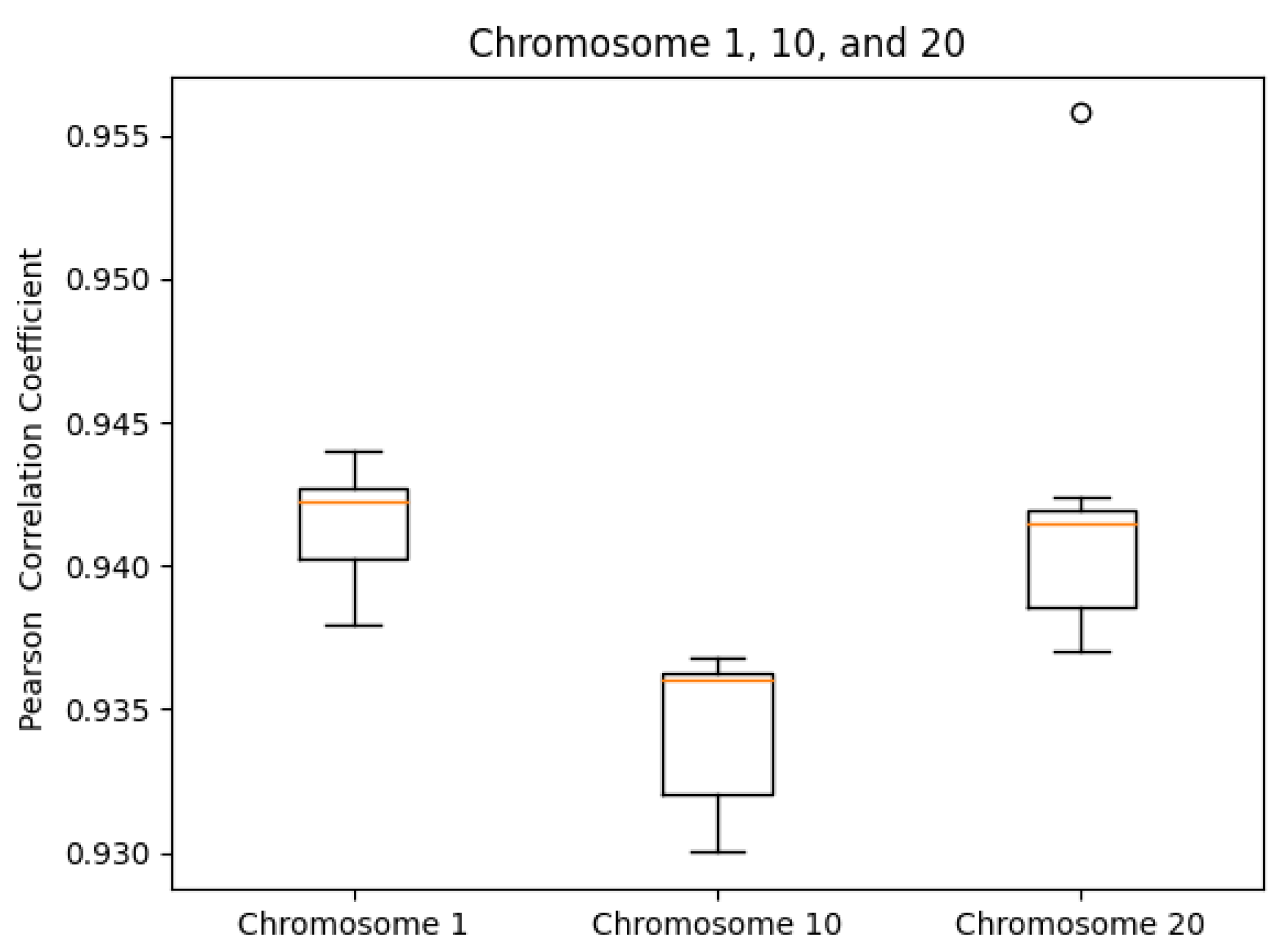
Examples of accessing all endpoints via curl can be found at in our GitHub repository under help/exampleCurlScript.bash.3.2. Consistency
3.3. Starting/Extending a Local ParticleChromo3D+ Server
3.3.1. Installation
docker image load -i particlechromo3d_image.tar.gz
docker tag \${IMAGE_ID} particlechromo3d:latest
Once the image has been loaded, it can be run ephemerally or persistently with the instructions on our GitHub homepage. The basic command to run our image is:
docker run -d \
-p 5001:5001 -p 8080:8080 \
-e SERVICE_EMAIL=${YOUR_SVC_EMAIL} \
-e HOSTNAME_BE=${YOUR_URL} \
-e SERVICE_EMAIL_KEY=${KEY} \
particlechromo3d:latestwhere:
- -d runs the container detached.
- -p [external:internal] sets the ports.
- -e SERVICE_EMAIL is the email address that will be used by the server.
- -e HOSTNAME_BE is the DNS name of the server.
- -e SERVICE_EMAIL_KEY is the password to the service email.
3.3.2. Extending the Image
docker build -t particlechromo3D:latest.If the user wants to make code changes, they should complete this before building the image.
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CLI | Command Line Interface |
| etc. | et cetera |
| FISH | fluorescence in situ hybridization |
| gRPC | gRPC Remote Procedure Calls |
| HTML | Hypertext Markup Language |
| i.e. | id est |
| IF | Interaction Frequency |
| JSON | JavaScript Object Notation |
| MSE | Mean Squared Error |
| PCC | Pearson Correlation Coefficient |
| REST | REpresentational State Transfer |
| RMSE | Root Mean Squared Error |
| SCC | Spearman Correlation Coefficient |
| SOAP | Simple Object Access Protocol |
| SSE | Sum of Squared Errors |
| VMs | virtual machines |
| WSDL | Web Service Description Language |
| 3C | Chromosome Conformation Capture |
References
- Sati, S.; Cavalli, G. Chromosome conformation capture technologies and their impact in understanding genome function. Chromosoma 2017, 126, 33–44. [Google Scholar] [CrossRef] [Green Version]
- Bolzer, A.; Kreth, G.; Solovei, I.; Koehler, D.; Saracoglu, K.; Fauth, C.; Müller, S.; Eils, R.; Cremer, C.; Speicher, M.R.; et al. Three-Dimensional Maps of All Chromosomes in Human Male Fibroblast Nuclei and Prometaphase Rosettes. PLoS Biol. 2005, 3, E157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masui, O.; Bonnet, I.; Le Baccon, P.; Brito, I.; Pollex, T.; Murphy, N.; Hupé, P.; Barillot, E.; Belmont, A.S.; Heard, E. Live-cell chromosome dynamics and outcome of X chromosome pairing events during ES cell differentiation. Cell 2011, 145, 447–458. [Google Scholar] [CrossRef] [Green Version]
- Dekker, J.; Rippe, K.; Dekker, M.; Kleckner, N. Capturing chromosome conformation. Science 2002, 295, 1306–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Wit, E.; De Laat, W. A decade of 3C technologies: Insights into nuclear organization. Genes Dev. 2012, 26, 11–24. [Google Scholar] [CrossRef] [Green Version]
- Denker, A.; de Laat, W. The second decade of 3C technologies: Detailed insights into nuclear organization. Genes Dev. 2016, 30, 1357–1382. [Google Scholar] [CrossRef] [Green Version]
- Lieberman-Aiden, E.; Van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Simonis, M.; Klous, P.; Splinter, E.; Moshkin, Y.; Willemsen, R.; De Wit, E.; Van Steensel, B.; De Laat, W. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture–on-chip (4C). Nat. Genet. 2006, 38, 1348–1354. [Google Scholar] [CrossRef]
- Dostie, J.; Richmond, T.A.; Arnaout, R.A.; Selzer, R.R.; Lee, W.L.; Honan, T.A.; Rubio, E.D.; Krumm, A.; Lamb, J.; Nusbaum, C.; et al. Chromosome Conformation Capture Carbon Copy (5C): A massively parallel solution for mapping interactions between genomic elements. Genome Res. 2006, 16, 1299–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalhor, R.; Tjong, H.; Jayathilaka, N.; Alber, F.; Chen, L. Genome architectures revealed by tethered chromosome confor-mation capture and population-based modeling. Nat. Biotechnol. 2012, 30, 90–98. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Fullwood, M.J.; Xu, H.; Mulawadi, F.H.; Velkov, S.; Vega, V.; Ariyaratne, P.N.; Mohamed, Y.B.; Ooi, H.S.; Tenna-koon, C.; et al. ChIA-PET tool for comprehensive chromatin interaction analysis with paired-end tag sequencing. Genome Biol. 2010, 11, R22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, K.; Forcato, M.; Ferrari, F. Hi-C analysis: From data generation to integration. Biophys. Rev. 2019, 11, 67–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacKay, K.; Kusalik, A. Computational methods for predicting 3D genomic organization from high-resolution chromo-some conformation capture data. Briefings Funct. Genom. 2020, 19, 292–308. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome 2009, 10, R25. [Google Scholar] [CrossRef] [Green Version]
- Durand, N.C.; Shamim, M.S.; Machol, I.; Rao, S.S.; Huntley, M.H.; Lander, E.S.; Aiden, E.L. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 2016, 3, 95–98. [Google Scholar] [CrossRef] [Green Version]
- Abdennur, N.; Mirny, L.A. Cooler: Scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics 2020, 36, 311–316. [Google Scholar] [CrossRef]
- Vadnais, D.; Middleton, M.; Oliwadare, O. ParticleChromo3D: A Particle Swarm Optimization algorithm for chromo-some 3D structure prediction from Hi-C data. BioData Mining. 2022, 15, 19. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Fu, L.Y.; Dong, P.F.; Deng, Z.L.; Li, J.X.; Wang, X.T.; Zhang, H.Y. The sequencing bias relaxed characteristics of Hi-C derived data and implications for chromatin 3D modeling. Nucleic Acids Res. 2013, 41, e183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, B.; Oluwadare, O.; Brown, P. ChromeBat: A Bio-Inspired Approach to 3D Genome Reconstruction. Genes 2021, 12, 1757. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, G.; Toh, K.C.; Sung, W.K. Inference of spatial organizations of chromosomes using semi-definite embed-ding approach and Hi-C data. In Proceedings of the Annual International Conference on Research in Computational Molecular Biology, Beijing, China, 7–10 April 2013; Springer: Berlin/Heidelberg, Germany, 2013; pp. 317–332. [Google Scholar]
- Adhikari, B.; Trieu, T.; Cheng, J. Chromosome3D: Reconstructing three-dimensional chromosomal structures from Hi-C interaction frequency data using distance geometry simulated annealing. BMC Genom. 2016, 17, 886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oluwadare, O.; Zhang, Y.; Cheng, J. A maximum likelihood algorithm for reconstructing 3D structures of human chro-mosomes from chromosomal contact data. BMC Genom. 2018, 19, 161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesne, A.; Riposo, J.; Roger, P.; Cournac, A.; Mozziconacci, J. 3D genome reconstruction from chromosomal contacts. Nat. Methods 2014, 11, 1141–1143. [Google Scholar] [CrossRef]
- Trieu, T.; Cheng, J. 3D genome structure modeling by Lorentzian objective function. Nucleic Acids Res. 2017, 45, 1049–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, C.; Zhang, Y.; Ouyang, Z. HSA: Integrating multi-track Hi-C data for genome-scale reconstruction of 3D chromatin structure. Genome Biol. 2016, 17, 40. [Google Scholar] [CrossRef] [Green Version]
- Trieu, T.; Cheng, J. MOGEN: A tool for reconstructing 3D models of genomes from chromosomal conformation capturing data. Bioinformatics 2016, 32, 1286–1292. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.; Deng, W.; Hu, H.; Ma, R.; Zhang, S.; Yang, J.; Peng, J.; Kaplan, T.; Zeng, J. Reconstructing spatial organizations of chromosomes through manifold learning. Nucleic Acids Res. 2018, 46, e50. [Google Scholar] [CrossRef] [Green Version]
- Varoquaux, N.; Ay, F.; Noble, W.S.; Vert, J.P. A statistical approach for inferring the 3D structure of the genome. Bioinformatics 2014, 30, i26–i33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Pierro, M.; Zhang, B.; Aiden, E.L.; Wolynes, P.G.; Onuchic, J.N. Transferable model for chromosome architecture. Proc. Natl. Acad. Sci. USA 2016, 113, 12168–12173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oluwadare, O.; Highsmith, M.; Cheng, J. An Overview of Methods for Reconstructing 3-D Chromosome and Genome Structures from Hi-C Data. Biol. Proced. Online 2019, 21, 7. [Google Scholar] [CrossRef]
- Paulsen, J.; Sekelja, M.; Oldenburg, A.R.; Barateau, A.; Briand, N.; Delbarre, E.; Shah, A.; Sørensen, A.L.; Vigouroux, C.; Buendia, B.; et al. Chrom3D: Three-dimensional genome modeling from Hi-C and nuclear lamin-genome contacts. Genome Biol. 2017, 18, 21. [Google Scholar] [CrossRef]
- Yaffe, E.; Tanay, A. Probabilistic modeling of Hi-C contact maps eliminates systematic biases to characterize global chromosomal architecture. Nat. Genet. 2011, 43, 1059–1065. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, J.; Eberhart, R.C. Particle swarm optimization. In Proceedings of the 1995 IEEE International Conference on Neural Networks, Perth, WA, Australia, 27 November–1 December 1995; Volume 4, pp. 1942–1948. [Google Scholar]
- Mohapatra, R.; Saha, S.; Dhavala, S.S. Adaswarm: A novel pso optimization method for the mathematical equivalence of error gradients. arXiv 2020, arXiv:2006.09875. [Google Scholar]
- Bonyadi, M.R.; Michalewicz, Z. Particle swarm optimization for single objective continuous space problems: A review. Evol. Comput. 2017, 25, 1–54. [Google Scholar] [CrossRef]
- Gupta, V.; Kling, H.; Yilmaz, K.; Martinez, G. Decomposition of the mean squared error and NSE performance criteria: Implications for improving hydrological modelling. J. Hydrol. 2009, 344, 80–91. [Google Scholar] [CrossRef] [Green Version]
- Nainggolan, R.; Perangin-angin, R.; Simarmata, E.; Tarigan, F. Improved the Performance of the K-Means Cluster Using the Sum of Squared Error (SSE) optimized by using the Elbow Method. J. Phys. Conf. Ser. 2018, 1361, 012015. [Google Scholar] [CrossRef]
- Huber, P.J. A Robust Version of the Probability Ratio Test. Ann. Math. Stat. 1965, 36, 1753–1758. [Google Scholar] [CrossRef]
- Li, Z.; Kihl, M.; Lu, Q.; Andersson, J.A. Performance Overhead Comparison between Hypervisor and Container Based Virtualization. In Proceedings of the IEEE 31st International Conference on Advanced Information Networking and Applications (AINA), Taipei, Taiwan, 27–29 March 2017; pp. 955–962. [Google Scholar]
- Parashar, M.; AbdelBaky, M.; Rodero, I.; Devarakonda, A. Cloud Paradigms and Practices for Computational and Da-ta-Enabled Science and Engineering. Comput. Sci. Eng. 2013, 15, 10–18. [Google Scholar] [CrossRef]
- Nogueira, A.F.; Ribeiro, C.; Rela, M.; Craske, A. Improving La Redoute’s CI/CD Pipeline and DevOps Processes by Applying Machine Learning Techniques. In Proceedings of the 11th International Conference on the Quality of Information and Communications Technology (QUATIC), Coimbra, Portugal, 4–7 September 2018; pp. 282–286. [Google Scholar]
- Rodríguez, C.; Báez, M.; Daniel, F.; Casati, F.; Trabucco, J.C.; Canali, L.; Percannella, G. REST APIs: A Large-Scale Analysis of Compliance with Principles and Best Practices. In Proceedings of the International Conference on Web Engineering, Lugano, Switzerland, 6–9 June 2016. [Google Scholar]
- Giretti, A. Understanding the gRPC Specification. In Beginning gRPC with ASP.NET Core 6; Apress: Berkeley, CA, USA, 2022. [Google Scholar]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [Green Version]
- Oluwadare, O.; Highsmith, M.; Turner, D.; Lieberman-Aiden, E.; Cheng, J. GSDB: A database of 3D chromosome and genome structures reconstructed from Hi-C data. BMC Mol. Cell Biol. 2020, 21, 30. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Type | Description |
|---|---|---|
| file | Text File | This is your IF matrix |
| Parameter | Type | Description |
|---|---|---|
| ifname | String | Name of the target IF matrix text file. |
| ss | Integer | Number of swarms desired. |
| itt | Integer | Iteration Count. This is an exit condition for the optimization script that defines the maximum iterations before stopping. |
| threshold | Double | This is an exit condition for the optimization script that defines the threshold of the error minimum before stopping. |
| randRange | Double | Range of values between which the (x, y, z) initial position can be randomly assigned for each chromatin bin. |
| lf | Integer [0, 3] | Bit mask for choosing the desired loss function. |
| outfile | String | Allows the user to tag the out put PDB file’s filename. |
| String | Defines the email address to be messaged upon job completion. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vadnais, D.; Oluwadare, O. ParticleChromo3D+: A Web Server for ParticleChromo3D Algorithm for 3D Chromosome Structure Reconstruction. Curr. Issues Mol. Biol. 2023, 45, 2549-2560. https://doi.org/10.3390/cimb45030167
Vadnais D, Oluwadare O. ParticleChromo3D+: A Web Server for ParticleChromo3D Algorithm for 3D Chromosome Structure Reconstruction. Current Issues in Molecular Biology. 2023; 45(3):2549-2560. https://doi.org/10.3390/cimb45030167
Chicago/Turabian StyleVadnais, David, and Oluwatosin Oluwadare. 2023. "ParticleChromo3D+: A Web Server for ParticleChromo3D Algorithm for 3D Chromosome Structure Reconstruction" Current Issues in Molecular Biology 45, no. 3: 2549-2560. https://doi.org/10.3390/cimb45030167
APA StyleVadnais, D., & Oluwadare, O. (2023). ParticleChromo3D+: A Web Server for ParticleChromo3D Algorithm for 3D Chromosome Structure Reconstruction. Current Issues in Molecular Biology, 45(3), 2549-2560. https://doi.org/10.3390/cimb45030167