
Genome-Wide Mining of Selaginella moellendorffii for Hevein-like Lectins and Their Potential Molecular Mimicry with SARS-CoV-2 Spike Glycoprotein

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
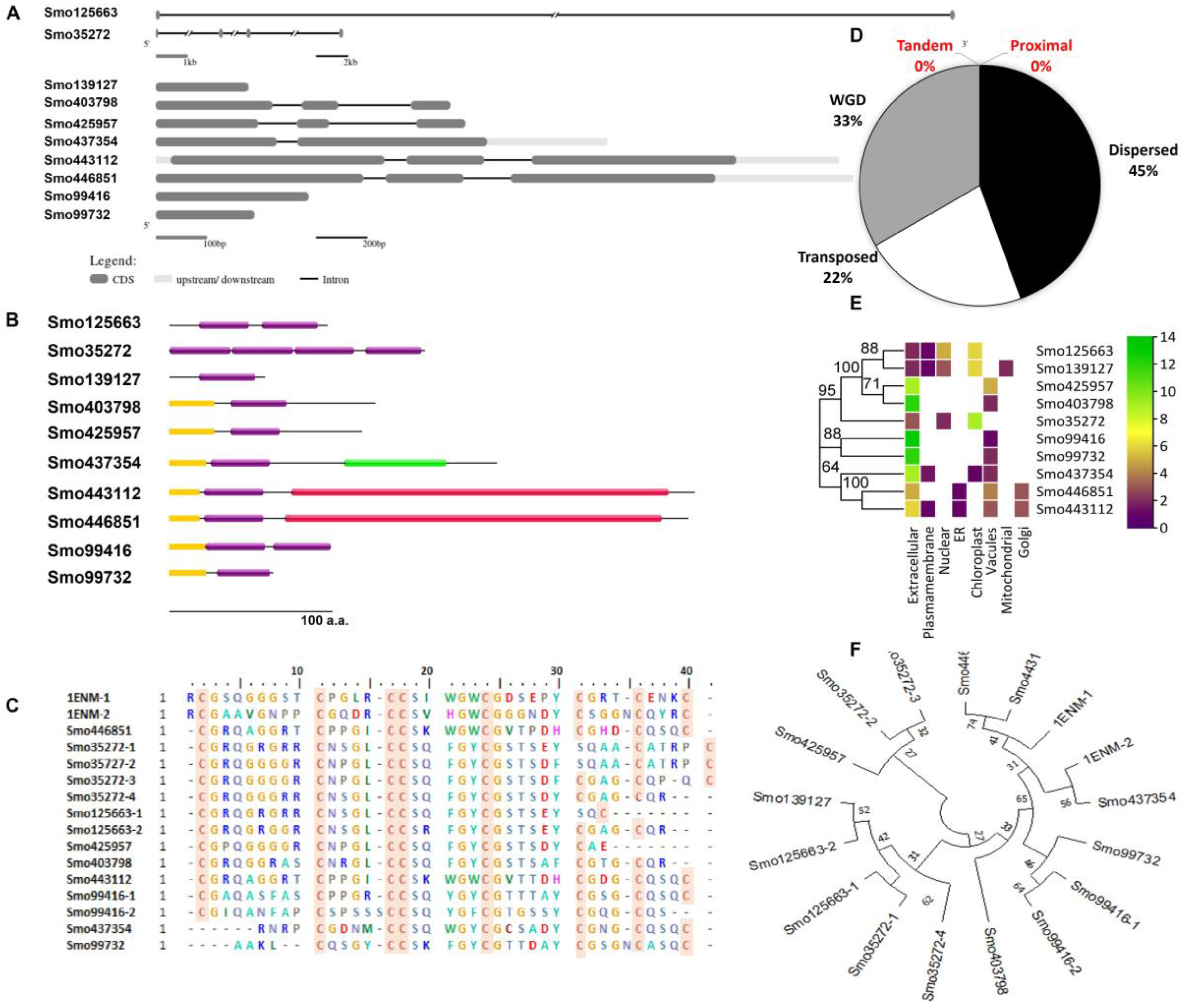
2.1. Screening the Spike Moss Genome for Putative Hevein-like Lectin Genes
2.2. Analysis of Hevein-like Gene Expansion and Evolutionary Relationship
2.3. Expressional Profile of Hevein-like Genes Based on Publicly Available Resources
2.4. Characterization of Hevein-like Homologs
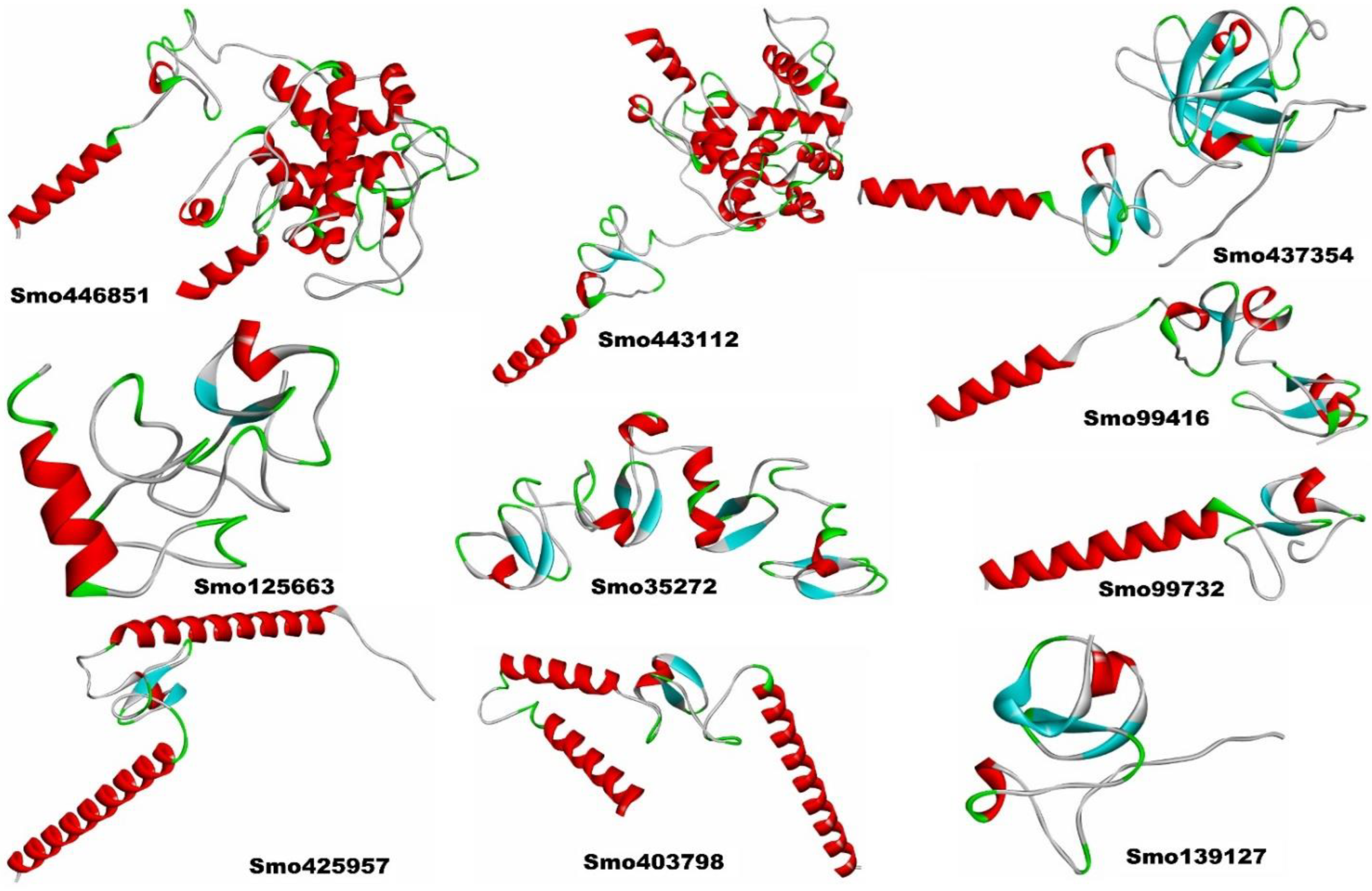
2.5. Secondary Structure Prediction, Structural Modeling, and Validation of Hevein-like Homologs
2.6. Retrieval and Pzre-Processing of SARS-CoV-2 Spike Glycoprotein and the Lectin Structures
2.7. Identifying the Binding Sites of the Spike Glycoprotein and Lectins for Macromolecular and Ligand Docking
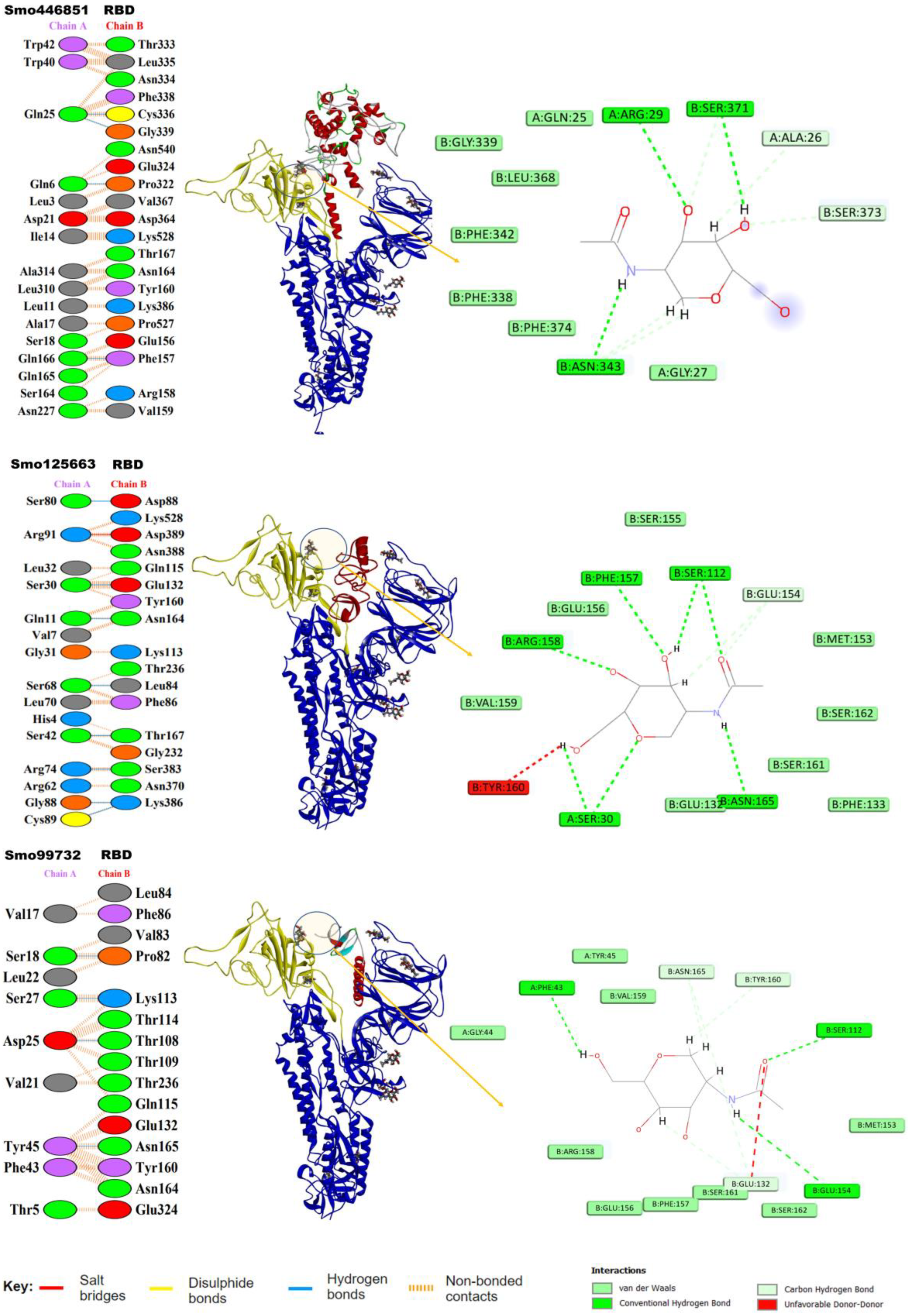
2.8. In Silico Molecular Docking
2.9. In Silico Mutant Spike Protein Interactions
2.10. Hotspot Analysis of the SARS-CoV-2 RBD–Lectin Complex
2.11. Normal-State Analyses via Torsional Coordinate-Association
2.12. Molecular Dynamics Simulation (MDS)
3. Results
3.1. General Overview of Hevein-like Lectins
3.2. Expression Profile of Hevein-like Genes in Different Organs
3.3. Structural Model Building of the Lectins and Their Secondary Structures
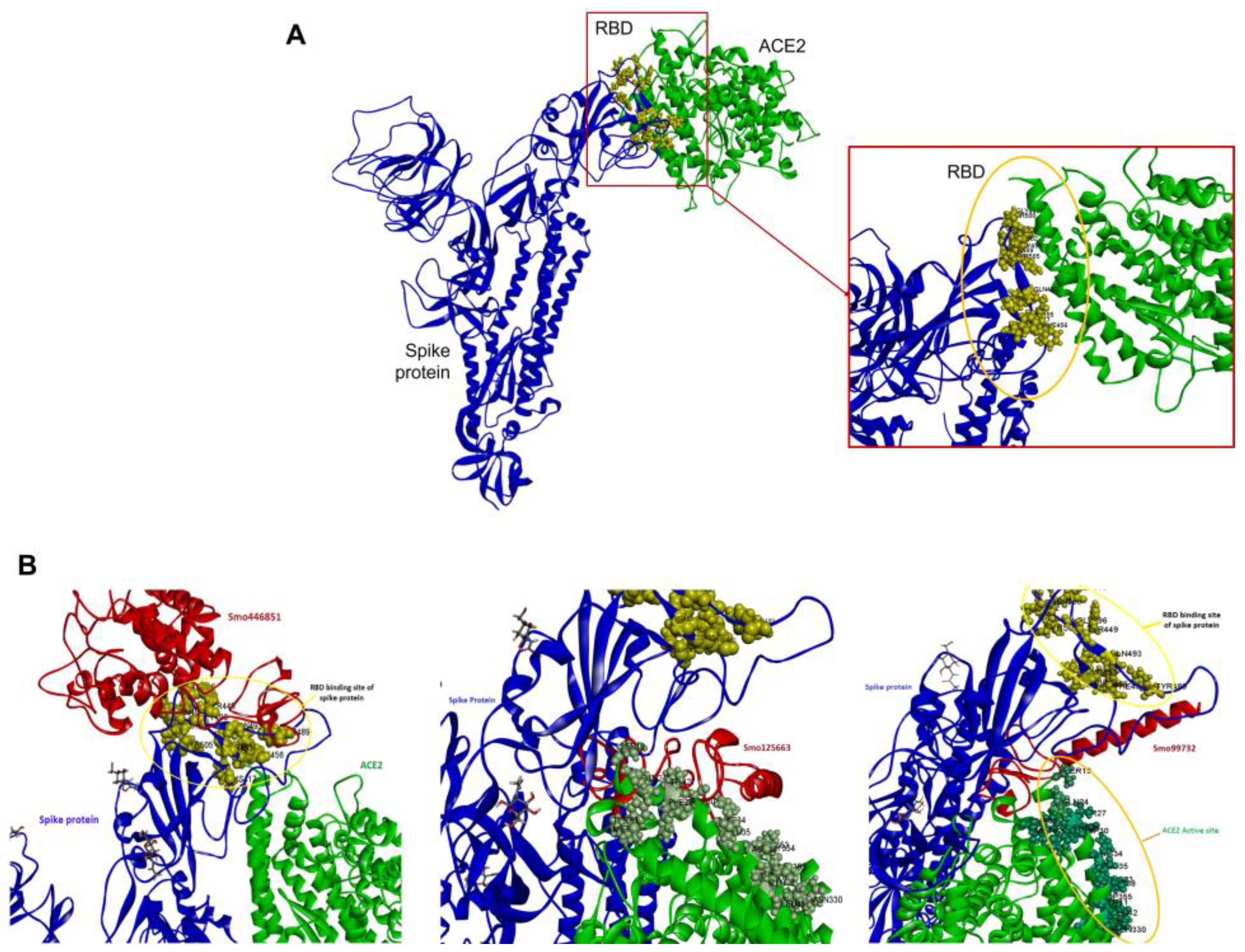
3.4. Identifying the Binding Sites of the S Glycoprotein and Lectins for Macromolecular and Ligand Docking
3.5. Molecular Docking of Lectins with SARS-CoV-2 Spike Protein
3.6. Hotspot Analysis of RBD–Lectin Complex
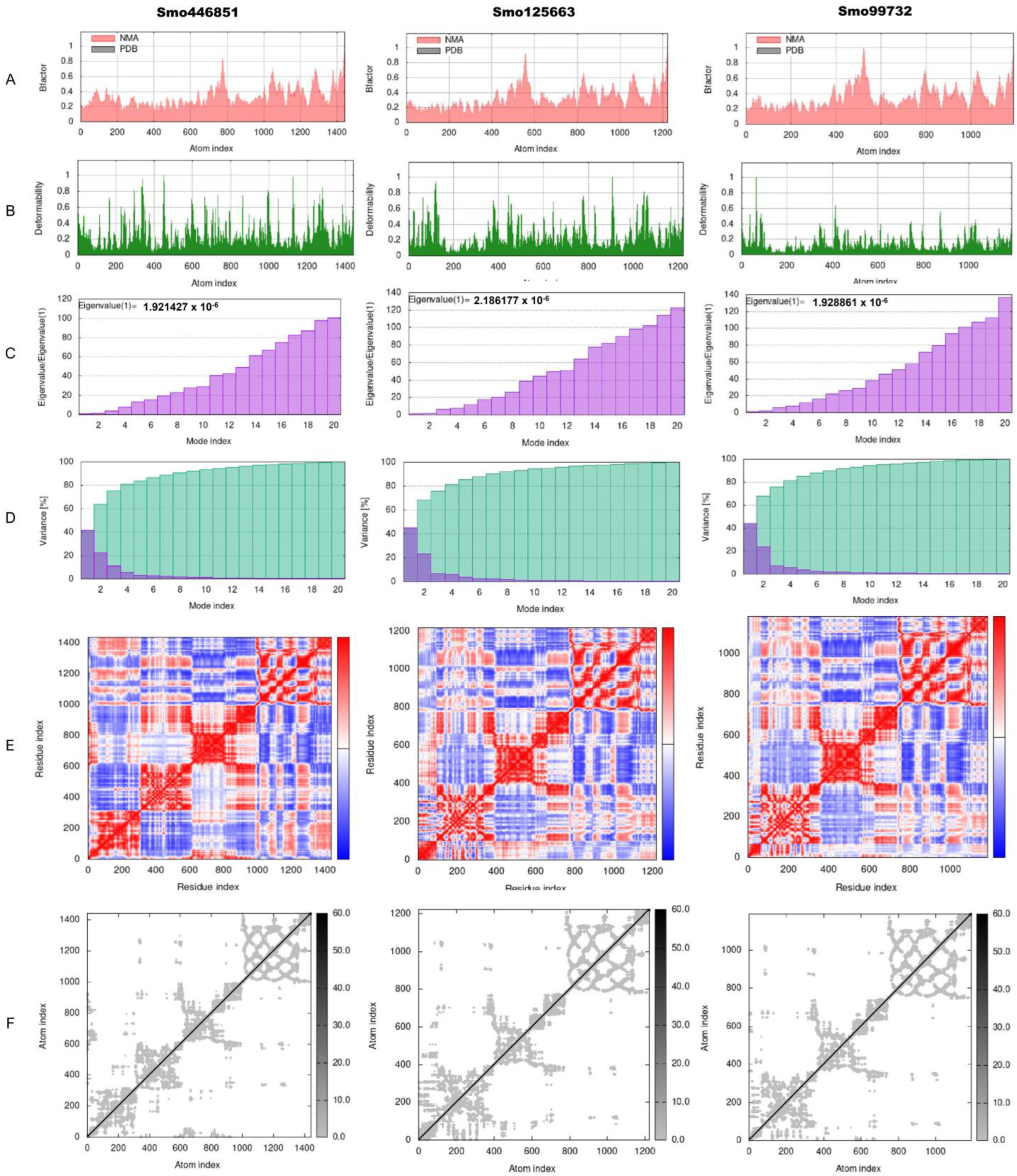
3.7. Normal-State Analyses via Torsional Coordinate-Association
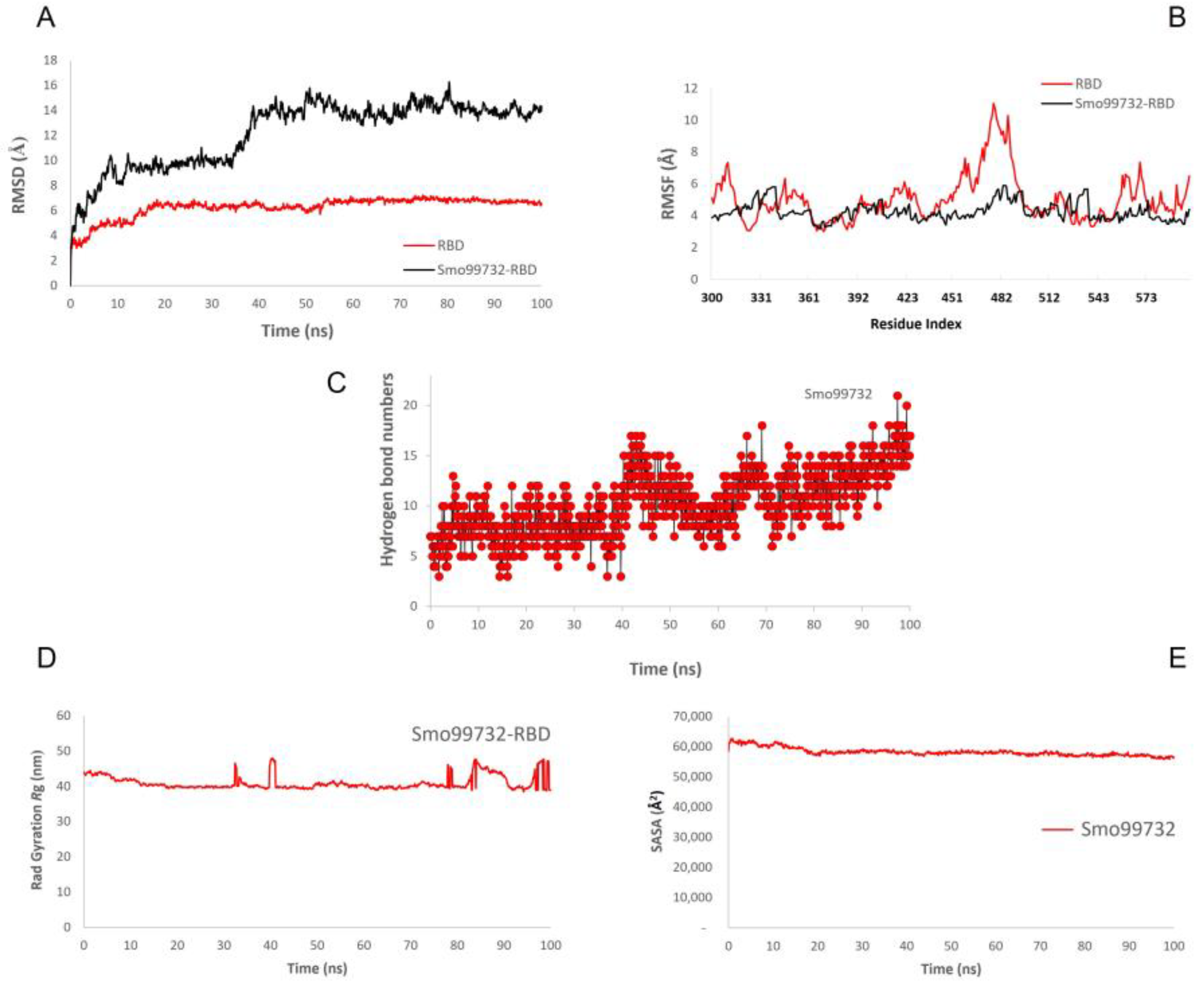
3.8. Molecular Dynamics Simulation
3.9. Mutant Spike Protein Interaction with Lectins
4. Discussion
4.1. Selaginella Moellendorffii Hevein Lectins
4.2. Hevein Lectin Interaction with SARS-CoV-2 Spike Protein’s RBD
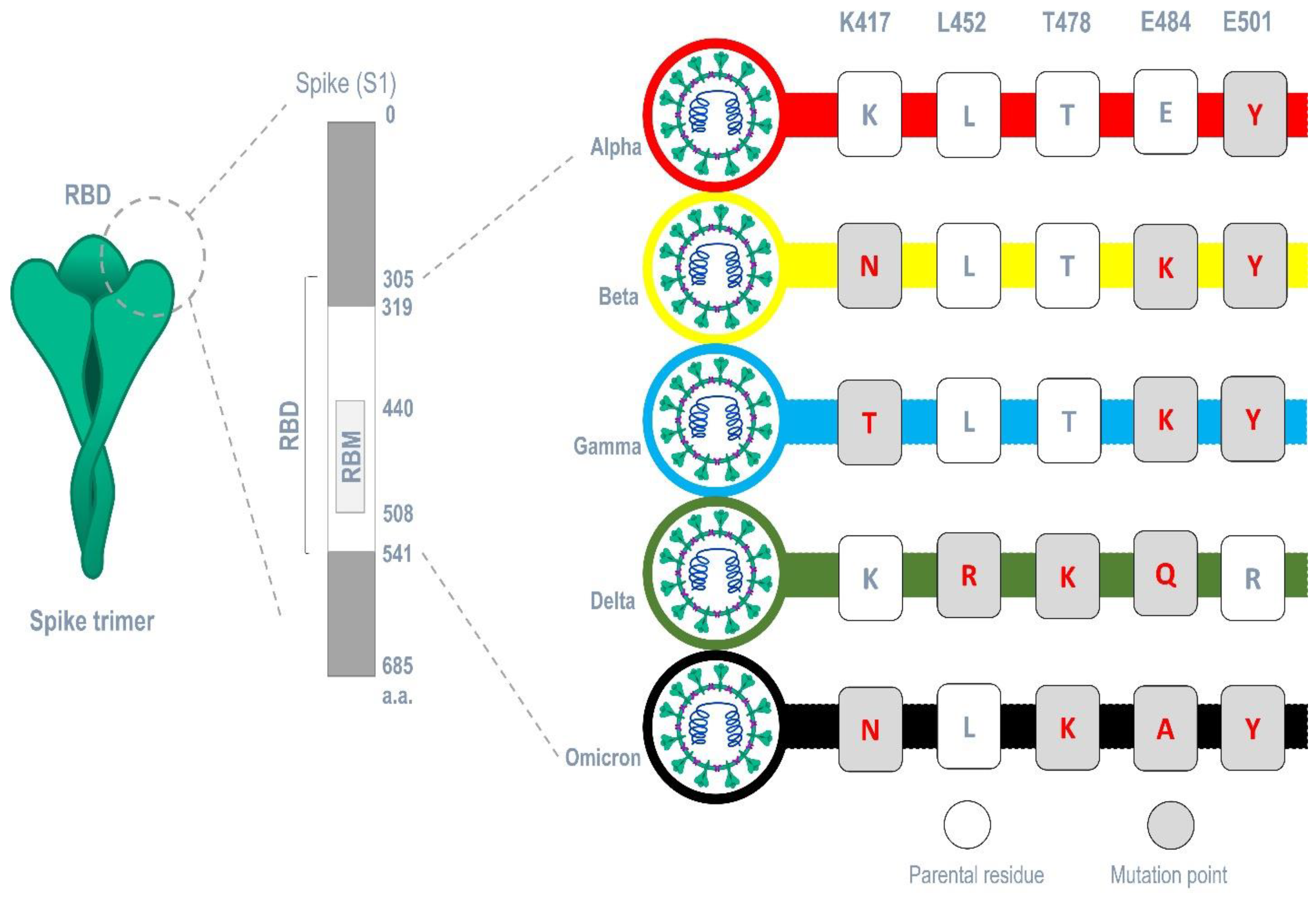
4.3. Interaction with Mutant SARS-CoV-2 Spike Protein’s RBD
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization (WHO). Health Emergency Dashboard: WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 2 January 2023).
- Weaver, A.K.; Head, J.R.; Gould, C.F.; Carlton, E.J.; Remais, J.V. Environmental Factors Influencing COVID-19 Incidence and Severity. Annu. Rev. Public Health 2022, 43, 271–291. [Google Scholar] [CrossRef] [PubMed]
- Redin, C.; Thorball, C.W.; Fellay, J. Host genomics of SARS-CoV-2 infection. Eur. J. Hum. Genet. 2022, 30, 908–914. [Google Scholar] [CrossRef] [PubMed]
- Jungreis, I.; Sealfon, R.; Kellis, M. SARS-CoV-2 gene content and COVID-19 mutation impact by comparing 44 Sarbecovirus genomes. Nat. Commun. 2021, 12, 1–20. [Google Scholar] [CrossRef]
- Prabhu, N.; Alonazi, M.A.; Algarni, H.A.; Issrani, R.; Alanazi, S.H.; Alruwaili, M.K.; Alanazi, G.R.; Iqbal, A.; Khattak, O. Knowledge, Attitude and Practice towards the COVID-19 Pandemic: A Cross-Sectional Survey Study among the General Public in the Kingdom of Saudi Arabia. Vaccines 2022, 10, 1945. [Google Scholar] [CrossRef]
- Konozy, E.H.E.; Osman, M.E.-F.M.; Ghartey-Kwansah, G.; Abushama, H.M. The striking mimics between COVID-19 and malaria: A review. Front. Immunol. 2022, 13, 957913. [Google Scholar] [CrossRef]
- Zhang, J.; Xiao, T.; Cai, Y.; Chen, B. Structure of SARS-CoV-2 spike protein. Curr. Opin. Virol. 2021, 50, 173–182. [Google Scholar] [CrossRef]
- Huang, H.-C.; Lai, Y.-J.; Liao, C.-C.; Yang, W.-F.; Huang, K.-B.; Lee, I.-J.; Chou, W.-C.; Wang, S.-H.; Wang, L.-H.; Hsu, J.-M. Targeting conserved N-glycosylation blocks SARS-CoV-2 variant infection in vitro. EBioMedicine 2021, 74, 103712. [Google Scholar] [CrossRef]
- Gong, Y.; Qin, S.; Dai, L.; Tian, Z. The glycosylation in SARS-CoV-2 and its receptor ACE2. Signal Transduct. Target. Ther. 2021, 6, 1–24. [Google Scholar] [CrossRef]
- Li, C.; Zhou, H.; Guo, L.; Xie, D.; He, H.; Zhang, H.; Liu, Y.; Peng, L.; Zheng, L.; Lu, W. Potential inhibitors for blocking the interaction of the coronavirus SARS-CoV-2 spike protein and its host cell receptor ACE2. J. Transl. Med. 2022, 20, 1–11. [Google Scholar] [CrossRef]
- Wang, G.; Yang, M.-L.; Duan, Z.-L.; Liu, F.-L.; Jin, L.; Long, C.-B.; Zhang, M.; Tang, X.-P.; Xu, L.; Li, Y.-C. Dalbavancin binds ACE2 to block its interaction with SARS-CoV-2 spike protein and is effective in inhibiting SARS-CoV-2 infection in animal models. Cell Res. 2021, 31, 17–24. [Google Scholar] [CrossRef]
- Peumans, W.J.; Van Damme, E.J. Lectins as plant defense proteins. Plant Physiol. 1995, 109, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Konozy, E.H.E.; Osman, M.E.-F.M.; Dirar, A.I.; Ghartey-Kwansah, G. Plant lectins: A new antimicrobial frontier. Biomed. Pharmacother. 2022, 155, 113735. [Google Scholar] [CrossRef] [PubMed]
- Konozy, E.H.E.; Osman, M.E.-F.M. Plant lectin: A promising future anti-tumor drug. Biochimie 2022, 202, 136–145. [Google Scholar] [CrossRef]
- Konozy, E.H.E.; Osman, M.E.-F.M.; Dirar, A.I. Plant lectins as potent Anti-coronaviruses, Anti-inflammatory, antinociceptive and antiulcer agents. Saudi J. Biol. Sci. 2022, 29, 103301. [Google Scholar] [CrossRef] [PubMed]
- Martinez, D.; Amaral, D.; Markovitz, D.; Pinto, L. The Use of Lectins as Tools to Combat SARS-CoV-2. Curr. Pharm. Des. 2021, 27, 4212–4222. [Google Scholar] [CrossRef] [PubMed]
- Lempp, F.A.; Soriaga, L.B.; Montiel-Ruiz, M.; Benigni, F.; Noack, J.; Park, Y.-J.; Bianchi, S.; Walls, A.C.; Bowen, J.E.; Zhou, J. Lectins enhance SARS-CoV-2 infection and influence neutralizing antibodies. Nature 2021, 598, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Barre, A.; Van Damme, E.J.; Simplicien, M.; Le Poder, S.; Klonjkowski, B.; Benoist, H.; Peyrade, D.; Rougé, P. Man-specific lectins from plants, fungi, algae and cyanobacteria, as potential blockers for SARS-CoV, MERS-CoV and SARS-CoV-2 (COVID-19) coronaviruses: Biomedical perspectives. Cells 2021, 10, 1619. [Google Scholar] [CrossRef]
- Sabzian-Molaei, F.; Nasiri Khalili, M.A.; Sabzian-Molaei, M.; Shahsavarani, H.; Fattah Pour, A.; Molaei Rad, A.; Hadi, A. Urtica dioica Agglutinin: A plant protein candidate for inhibition of SARS-COV-2 receptor-binding domain for control of Covid19 Infection. PLoS ONE 2022, 17, e0268156. [Google Scholar] [CrossRef]
- Setyawan, A.D. Traditionally utilization of Selaginella; field research and literature review. Nusant. Biosci. 2009, 13, 146–158. [Google Scholar] [CrossRef]
- Banks, J.A.; Nishiyama, T.; Hasebe, M.; Bowman, J.L.; Gribskov, M.; DePamphilis, C.; Albert, V.A.; Aono, N.; Aoyama, T.; Ambrose, B.A. The Selaginella genome identifies genetic changes associated with the evolution of vascular plants. Science 2011, 332, 960–963. [Google Scholar] [CrossRef] [Green Version]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, A.-Y.; Zhu, Q.-H.; Chen, X.; Luo, J.-C. GSDS: A gene structure display server. Yi Chuan Hered. 2007, 29, 1023–1026. [Google Scholar] [CrossRef]
- Qiao, X.; Li, Q.; Yin, H.; Qi, K.; Li, L.; Wang, R.; Zhang, S.; Paterson, A.H. Gene duplication and evolution in recurring polyploidization–diploidization cycles in plants. Genome Biol. 2019, 20, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547. [Google Scholar] [CrossRef]
- Mello, A.; Efroni, I.; Rahni, R.; Birnbaum, K.D. The Selaginella rhizophore has a unique transcriptional identity compared with root and shoot meristems. New Phytol. 2019, 222, 882–894. [Google Scholar] [CrossRef]
- Ge, S.X.; Son, E.W.; Yao, R. iDEP: An integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinform. 2018, 19, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Zhuang, Z.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server (2017 release). Nucleic Acids Res. 2018, 46, W49–W54. [Google Scholar] [CrossRef] [Green Version]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef] [Green Version]
- Krogh, A.; Larsson, B.; Von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.; Brunak, S. Prediction of glycosylation across the human proteome and the correlation to protein function. Pac. Symp. Biocomput. 2001, 37, 310–322. [Google Scholar]
- Steentoft, C.; Vakhrushev, S.Y.; Joshi, H.J.; Kong, Y.; Vester-Christensen, M.B.; Schjoldager, K.T.B.; Lavrsen, K.; Dabelsteen, S.; Pedersen, N.B.; Marcos-Silva, L. Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. EMBO J. 2013, 32, 1478–1488. [Google Scholar] [CrossRef] [Green Version]
- Horton, P.; Park, K.-J.; Obayashi, T.; Nakai, K. Protein subcellular localization prediction with WoLF PSORT. In Proceedings of the 4th Asia-Pacific Bioinformatics Conference, Taipei, Taiwan, 13–16 February 2006; pp. 39–48. [Google Scholar]
- Zhao, L.; Poschmann, G.; Waldera-Lupa, D.; Rafiee, N.; Kollmann, M.; Stühler, K. OutCyte: A novel tool for predicting unconventional protein secretion. Sci. Rep. 2019, 9, 19448. [Google Scholar] [CrossRef] [Green Version]
- Geourjon, C.; Deleage, G. SOPMA: Significant improvement in protein secondary structure prediction by c prediction from alignments and joint prediction. CABIOS 1995, 11, 681–684. [Google Scholar] [PubMed]
- Du, Z.; Su, H.; Wang, W.; Ye, L.; Wei, H.; Peng, Z.; Anishchenko, I.; Baker, D.; Yang, J. The trRosetta server for fast and accurate protein structure prediction. Nat. Protoc. 2021, 16, 5634–5651. [Google Scholar] [CrossRef] [PubMed]
- Lovell, S.C.; Davis, I.W.; Arendall III, W.B.; De Bakker, P.I.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins Struct. Funct. Bioinform. 2003, 50, 437–450. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e286. [Google Scholar] [CrossRef]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef] [PubMed]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017, 45, D200–D203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Zhang, D.; Zhou, P.; Li, B.; Huang, S.Y. HDOCK: A web server for protein-protein and protein-DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 2017, 45, W365–W373. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Weng, G.; Wang, E.; Wang, Z.; Liu, H.; Zhu, F.; Li, D.; Hou, T. HawkDock: A web server to predict and analyze the protein-protein complex based on computational docking and MM/GBSA. Nucleic Acids Res. 2019, 47, W322–W330. [Google Scholar] [CrossRef]
- Biovia, D.S. Discovery Studio Modeling Environment; Dassault Systèmes: San Diego, CA, USA, 2017. [Google Scholar]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.-Y. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 2020, 181, 894–904.e899. [Google Scholar] [CrossRef]
- Van Zundert, G.; Rodrigues, J.; Trellet, M.; Schmitz, C.; Kastritis, P.; Karaca, E.; Melquiond, A.; van Dijk, M.; De Vries, S.; Bonvin, A. The HADDOCK2. 2 web server: User-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Mitchell, J.C. KFC2: A knowledge-based hot spot prediction method based on interface solvation, atomic density, and plasticity features. Proteins Struct. Funct. Bioinform. 2011, 79, 2671–2683. [Google Scholar] [CrossRef]
- López-Blanco, J.R.; Aliaga, J.I.; Quintana-Ortí, E.S.; Chacón, P. iMODS: Internal coordinates normal mode analysis server. Nucleic Acids Res. 2014, 42, W271–W276. [Google Scholar] [CrossRef] [PubMed]
- Lopéz-Blanco, J.R.; Garzón, J.I.; Chacón, P. iMod: Multipurpose normal mode analysis in internal coordinates. Bioinformatics 2011, 27, 2843–2850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Cairns, QLD, Australia, June 28–1 July 2006; p. 84-es. [Google Scholar]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the OPLS force field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [Google Scholar] [CrossRef] [PubMed]
- Kezuka, Y.; Kojima, M.; Mizuno, R.; Suzuki, K.; Watanabe, T.; Nonaka, T. Structure of full-length class I chitinase from rice revealed by X-ray crystallography and small-angle X-ray scattering. Proteins Struct. Funct. Bioinform. 2010, 78, 2295–2305. [Google Scholar] [CrossRef]
- Morohashi, M.; Sakabe, K.; Sakabe, N.; Sasaki, K. Three-Dimensional Structure Analysis of Vigna Unguiculata Chitinase with Regulation Activity of the Yield Threshold of Cell Wall. Be Publ. 2013. Available online: https://doi.org/10.2210/pdb3w3e/pdb (accessed on 1 June 2023).
- Landim, P.G.C.; Correia, T.O.; Silva, F.D.; Nepomuceno, D.R.; Costa, H.P.; Pereira, H.M.; Lobo, M.D.; Moreno, F.B.; Brandão-Neto, J.; Medeiros, S.C. Production in Pichia pastoris, antifungal activity and crystal structure of a class I chitinase from cowpea (Vigna unguiculata): Insights into sugar binding mode and hydrolytic action. Biochimie 2017, 135, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Balu, K.E.; Ramya, K.; Radha, A.; Krishnasamy, G. Structure of intact chitinase with hevein domain from the plant Simarouba glauca, known for its traditional anti-inflammatory efficacy. Int. J. Biol. Macromol. 2020, 161, 1381–1392. [Google Scholar] [CrossRef]
- Hahn, M.; Hennig, M.; Schlesier, B.; Höhne, W. Structure of jack bean chitinase. Acta Crystallogr. Sect. D Biol. Crystallogr. 2000, 56, 1096–1099. [Google Scholar] [CrossRef]
- Kerff, F.; Amoroso, A.; Herman, R.; Sauvage, E.; Petrella, S.; Filée, P.; Charlier, P.; Joris, B.; Tabuchi, A.; Nikolaidis, N. Crystal structure and activity of Bacillus subtilis YoaJ (EXLX1), a bacterial expansin that promotes root colonization. Proc. Natl. Acad. Sci. USA 2008, 105, 16876–16881. [Google Scholar] [CrossRef]
- Yennawar, N.H.; Li, L.-C.; Dudzinski, D.M.; Tabuchi, A.; Cosgrove, D.J. Crystal structure and activities of EXPB1 (Zea m 1), a β-expansin and group-1 pollen allergen from maize. Proc. Natl. Acad. Sci. USA 2006, 103, 14664–14671. [Google Scholar] [CrossRef]
- Fedorov, A.A.; Ball, T.; Leistler, B.; Valenta, R.; Almo, S.C.; Burley, S.K. X-ray Crystal Structure of Phl p 1, a Major Timothy Grass Pollen Allergen. Be Publ. 2003. Available online: https://www.wwpdb.org/pdb?id=pdb_00001n10 (accessed on 1 June 2023).
- Yennawar, N.H.; Yennawar, H.P.; Georgelis, N.; Cosgrove, D.J. Crystal Structure of Wild Type and d78n Mutant Clavibacter Michiganensis Expansin, in Apo Form and in Complex with Oligosaccharides. Be Publ. 2013. Available online: https://doi.org/10.2210/pdb4JS7/pdb (accessed on 1 June 2023).
- Chakraborty, N.; Besra, A.; Basak, J. RETRACTED ARTICLE: Molecular Cloning of an Amino Acid Permease Gene and Structural Characterization of the Protein in Common Bean (Phaseolus vulgaris L.). Mol. Biotechnol. 2020, 62, 210–217. [Google Scholar] [CrossRef]
- Kumar, S.; Tsai, C.-J.; Nussinov, R. Factors enhancing protein thermostability. Protein Eng. 2000, 13, 179–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Wang, Y.; Liu, C.; Zhang, C.; Han, W.; Hong, X.; Wang, Y.; Hong, Q.; Wang, S.; Zhao, Q. Conformational dynamics of SARS-CoV-2 trimeric spike glycoprotein in complex with receptor ACE2 revealed by cryo-EM. Sci. Adv. 2021, 7, eabe5575. [Google Scholar] [CrossRef] [PubMed]
- Maffucci, I.; Contini, A. In silico drug repurposing for SARS-CoV-2 main proteinase and spike proteins. J. Proteome Res. 2020, 19, 4637–4648. [Google Scholar] [CrossRef]
- Lokhande, K.B.; Apte, G.R.; Shrivastava, A.; Singh, A.; Pal, J.K.; Swamy, K.V.; Gupta, R.K. Sensing the interactions between carbohydrate-binding agents and N-linked glycans of SARS-CoV-2 spike glycoprotein using molecular docking and simulation studies. J. Biomol. Struct. Dyn. 2022, 40, 3880–3898. [Google Scholar] [CrossRef] [PubMed]
- Shajahan, A.; Supekar, N.T.; Gleinich, A.S.; Azadi, P. Deducing the N-and O-glycosylation profile of the spike protein of novel coronavirus SARS-CoV-2. Glycobiology 2020, 30, 981–988. [Google Scholar] [CrossRef]
- Itakura, Y.; Nakamura-Tsuruta, S.; Kominami, J.; Tateno, H.; Hirabayashi, J. Sugar-binding profiles of chitin-binding lectins from the hevein family: A comprehensive study. Int. J. Mol. Sci. 2017, 18, 1160. [Google Scholar] [CrossRef] [Green Version]
- Scialo, F.; Daniele, A.; Amato, F.; Pastore, L.; Matera, M.G.; Cazzola, M.; Castaldo, G.; Bianco, A. ACE2: The Major Cell Entry Receptor for SARS-CoV-2. Lung 2020, 198, 867–877. [Google Scholar] [CrossRef]
- Khan, S.; Rizwan, M.; Zeb, A.; Eldeen, M.A.; Hassan, S.; Ur Rehman, A.; Eid, R.; Samir, A.; Zaki, M.; Albadrani, G.; et al. Identification of a Potential Vaccine against Treponema pallidum Using Subtractive Proteomics and Reverse-Vaccinology Approaches. Vaccines 2022, 11, 72. [Google Scholar] [CrossRef]
- Van Holle, S.; Van Damme, E.J.M. Messages From the Past: New Insights in Plant Lectin Evolution. Front. Plant Sci. 2019, 10, 36. [Google Scholar] [CrossRef] [Green Version]
- Slavokhotova, A.A.; Naumann, T.A.; Price, N.P.; Rogozhin, E.A.; Andreev, Y.A.; Vassilevski, A.A.; Odintsova, T.I. Novel mode of action of plant defense peptides–hevein-like antimicrobial peptides from wheat inhibit fungal metalloproteases. FEBS J. 2014, 281, 4754–4764. [Google Scholar] [CrossRef]
- Porto, W.F.; Souza, V.A.; Nolasco, D.O.; Franco, O.L. In silico identification of novel hevein-like peptide precursors. Peptides 2012, 38, 127–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khateeb, J.; Li, Y.; Zhang, H. Emerging SARS-CoV-2 variants of concern and potential intervention approaches. Crit. Care 2021, 25, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mannar, D.; Saville, J.W.; Zhu, X.; Srivastava, S.S.; Berezuk, A.M.; Zhou, S.; Tuttle, K.S.; Kim, A.; Li, W.; Dimitrov, D.S. Structural analysis of receptor binding domain mutations in SARS-CoV-2 variants of concern that modulate ACE2 and antibody binding. Cell Rep. 2021, 37, 110156. [Google Scholar] [CrossRef] [PubMed]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Consortium, C.-G.U. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Auth, J.; Fröba, M.; Große, M.; Rauch, P.; Ruetalo, N.; Schindler, M.; Morokutti-Kurz, M.; Graf, P.; Dolischka, A.; Prieschl-Grassauer, E. Lectin from Triticum vulgaris (WGA) inhibits infection with SARS-CoV-2 and its variants of concern alpha and beta. Int. J. Mol. Sci. 2021, 22, 10205. [Google Scholar] [CrossRef]
- Ahan, R.E.; Hanifehnezhad, A.; Kehribar, E.S.; Oguzoglu, T.C.; Foldes, K.; Ozcelik, C.E.; Filazi, N.; Ożtop, S.; Palaz, F.; Onder, S. A Highly Potent SARS-CoV-2 Blocking Lectin Protein. ACS Infect. Dis. 2022, 8, 1253–1264. [Google Scholar] [CrossRef]
- Sarkar, A.; Paul, S.; Singh, C.; Chowdhury, N.; Nag, P.; Das, S.; Kumar, S.; Sharma, A.; Das, D.K.; Dutta, D.; et al. A novel plant lectin, NTL-125, interferes with SARS-CoV-2 interaction with hACE2. Virus Res. 2022, 315, 198768. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Scaffold # | pI | MWt (KDa) | SP | TM | Targeting Class | N-Glycan | O-Glycan |
|---|---|---|---|---|---|---|---|---|
| Smo446851 | 79 | 6.43 | 34.364 | Sec/SPI | 0 | SP | −ve | +ve |
| Smo35272 | 30 | 8.62 | 15.603 | Other | 0 | IC | −ve | +ve |
| Smo125663 | 79 | 8.47 | 10.046 | Other | 0 | IC | −ve | +ve |
| Smo425957 | 79 | 8.02 | 12.554 | Sec/SPI | 1 | SP | −ve | −ve |
| Smo403798 | 1 | 8.6 | 13.395 | Sec/SPI | 1 | SP | +ve | −ve |
| Smo443112 | 30 | 6.79 | 34.635 | Sec/SPI | 0 | SP | −ve | +ve |
| Smo99416 | 21 | 5.79 | 10.038 | Sec/SPI | 0 | SP | −ve | −ve |
| Smo437354 | 0 | 5.75 | 20.944 | Sec/SPI | 0 | SP | +ve | +ve |
| Smo99732 | 22 | 8.09 | 6.57 | Sec/SPI | 0 | SP | −ve | −ve |
| Smo139127 | 757 | 8.07 | 6.204 | Other | 0 | IC | −ve | −ve |
| Type of Interactions | Interacting Residues of S Protein | Lectins | Interacting Residues of Lectins | Distance (Å) | Docking Score ● | Confidence Score * | MM/GBSA ● |
|---|---|---|---|---|---|---|---|
| H-bond | NAG 1307 | Smo446851 | Ala26 | 2.8 | −182.44 | 0.66 | −17.49 |
| NAG 1307 | Arg29 | 2.7 | |||||
| H-bond | Lys386 | Smo35272 | Gln110 | 2.2 | −163.59 | 0.568 | −50.07 |
| Thr385 | Gln94 | 2.0 | |||||
| Asn370 | Gln80 | 1.9 | |||||
| Ser366 | Gln80 | 2.2 | |||||
| Val320 | Arg122 | 2.4 | |||||
| H-bond | NAG 1321 | Smo125663 | Ser30 | 2.5 | −160.85 | 0.55 | −13.03 |
| H-bond | Asn370 | Smo425957 | Ser61 | 1.9 | −197.02 | 0.72 | −16.12 |
| Lys386 | Cys38 | 2.5 | |||||
| Lys386 | Pro36 | 2.4 | |||||
| Ser383 | Tyr58 | 3.1 | |||||
| Ser325 | Arg46 | 2.5 | |||||
| H-bond | Tyr369 | Smo403798 | Arg49 | 2.2 | −187.76 | 0.68 | −22.72 |
| H-bond | NAG 1321 | Smo99732 | Phe43 | 2.6 | −136.67 | 0.53 | −26.45 |
| Mutant | Lectin | # of H-Bonds | Interacting Residues | Docking Score ● | Confidence Score * | MM/GBSA ● | |
|---|---|---|---|---|---|---|---|
| S Protein | Lectins | ||||||
| Alpha | Smo446851 | 6 | Tyr351, Ser359, Asn360, NAG1306 | Ser6, Tyr38, Thr48, Thr49, Ala51 | −212.92 | 0.7788 | −23.64 |
| Smo125663 | 6 | Glu340, Thr345, Ser349, Ser359, Arg457, Arg466 | Tyr39, Gln22, Arg59, Asn6, Glu83, Ser44 | −196.60 | 0.7175 | −31.8 | |
| Smo99732 | 4 | Glu340, Arg357, Ser359 | Trp167, Gln195, Ser194 | −168.16 | 0.5895 | −9.22 | |
| Beta | Smo446851 | 7 | Asn331, Thr333, Thr581, NAG1306 | Ser108, Ser321, Asp234, Ser61, Pro33, Asn111 | −161.94 | 0.5594 | −6.31 |
| Smo125663 | 6 | Trp353, Arg355, Asn448, Arg466, Thr470 | Asn67, Ser30, Gly50, Tyr84 | −196.15 | 0.7157 | −21.49 | |
| Smo99732 | 7 | Tyr351, Ser359, Asn360, NAG1306 | Thr48, Ser6, Tyr38, Thr48, Thr49, Ala51 | −168.65 | 0.5922 | −8.67 | |
| Gamma | Smo446851 | 10 | Tyr369, Thr415, Phe456, Arg457, Ser459, Gln474, Thr478, Asn481, Tyr505 | Leu13, Ala225, Gln166, Gln195, Leu191, Gln165, Gln166, Asn202, Gln316 | −220.86 | 0.8049 | −35.31 |
| Smo125663 | 6 | Trp353, Arg355, Arg454, Arg466, Thr470 | Asn67, Ser42, GLy50, Tyr84 | −197.62 | 0.7216 | −15.04 | |
| Smo99732 | 5 | Phe347, Arg357, Asn450, Leu492 | Gln24, Ser6, Tyr38, Ala23, Thr48 | −218.65 | 0.7979 | −25.68 | |
| Delta | Smo446851 | 4 | Ser459, Asn481, Thr457, Gln755 | Gln195, Asn202, Asp48, Ser296 | −183.89 | 0.6632 | −6.6 |
| Smo125663 | 5 | Ala352, Asn450, Ser469, Thr470 | Arg21, Gln22, Tyr93, Arg91 | −202.50 | 0.7408 | 1.6 | |
| Smo99732 | 5 | Phe347, Ala352, Arg357, Asn450, Pro561 | Gln24, Lys20, Ser6, Tyr38, Lys2 | −208.37 | 0.7627 | −28.0 | |
| Omicron | Smo446851 | 6 | Tyr369, Lys378, Ser383, Ser459, Asn481, Gln755 | Leu13, Ala19, Ala17, Gln195, Asn202, Ser296 | −217.22 | 0.7932 | −16.22 |
| Smo125663 | 5 | Arg457, Arg466, Ile464, Glu516 | Glu83, Ser44, Gln36, Tyr84, Asn6 | −185.52 | 0.6705 | −22.37 | |
| Smo99732 | 7 | Phe347, Arg357, Ser359, Asn450, Leu492 | Gln24, Ser6, Tyr38, Ala23, Thr48 | −219.82 | 0.8016 | −25.94 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alsolami, A.; Dirar, A.I.; Konozy, E.H.E.; Osman, M.E.-F.M.; Ibrahim, M.A.; Alshammari, K.F.; Alshammari, F.; Alazmi, M.; Said, K.B. Genome-Wide Mining of Selaginella moellendorffii for Hevein-like Lectins and Their Potential Molecular Mimicry with SARS-CoV-2 Spike Glycoprotein. Curr. Issues Mol. Biol. 2023, 45, 5879-5901. https://doi.org/10.3390/cimb45070372
Alsolami A, Dirar AI, Konozy EHE, Osman ME-FM, Ibrahim MA, Alshammari KF, Alshammari F, Alazmi M, Said KB. Genome-Wide Mining of Selaginella moellendorffii for Hevein-like Lectins and Their Potential Molecular Mimicry with SARS-CoV-2 Spike Glycoprotein. Current Issues in Molecular Biology. 2023; 45(7):5879-5901. https://doi.org/10.3390/cimb45070372
Chicago/Turabian StyleAlsolami, Ahmed, Amina I. Dirar, Emadeldin Hassan E. Konozy, Makarim El-Fadil M. Osman, Mohanad A. Ibrahim, Khalid Farhan Alshammari, Fawwaz Alshammari, Meshari Alazmi, and Kamaleldin B. Said. 2023. "Genome-Wide Mining of Selaginella moellendorffii for Hevein-like Lectins and Their Potential Molecular Mimicry with SARS-CoV-2 Spike Glycoprotein" Current Issues in Molecular Biology 45, no. 7: 5879-5901. https://doi.org/10.3390/cimb45070372
APA StyleAlsolami, A., Dirar, A. I., Konozy, E. H. E., Osman, M. E. -F. M., Ibrahim, M. A., Alshammari, K. F., Alshammari, F., Alazmi, M., & Said, K. B. (2023). Genome-Wide Mining of Selaginella moellendorffii for Hevein-like Lectins and Their Potential Molecular Mimicry with SARS-CoV-2 Spike Glycoprotein. Current Issues in Molecular Biology, 45(7), 5879-5901. https://doi.org/10.3390/cimb45070372