Dysautonomia in Alzheimer's Disease


 and
and
Abstract
:1. Introduction
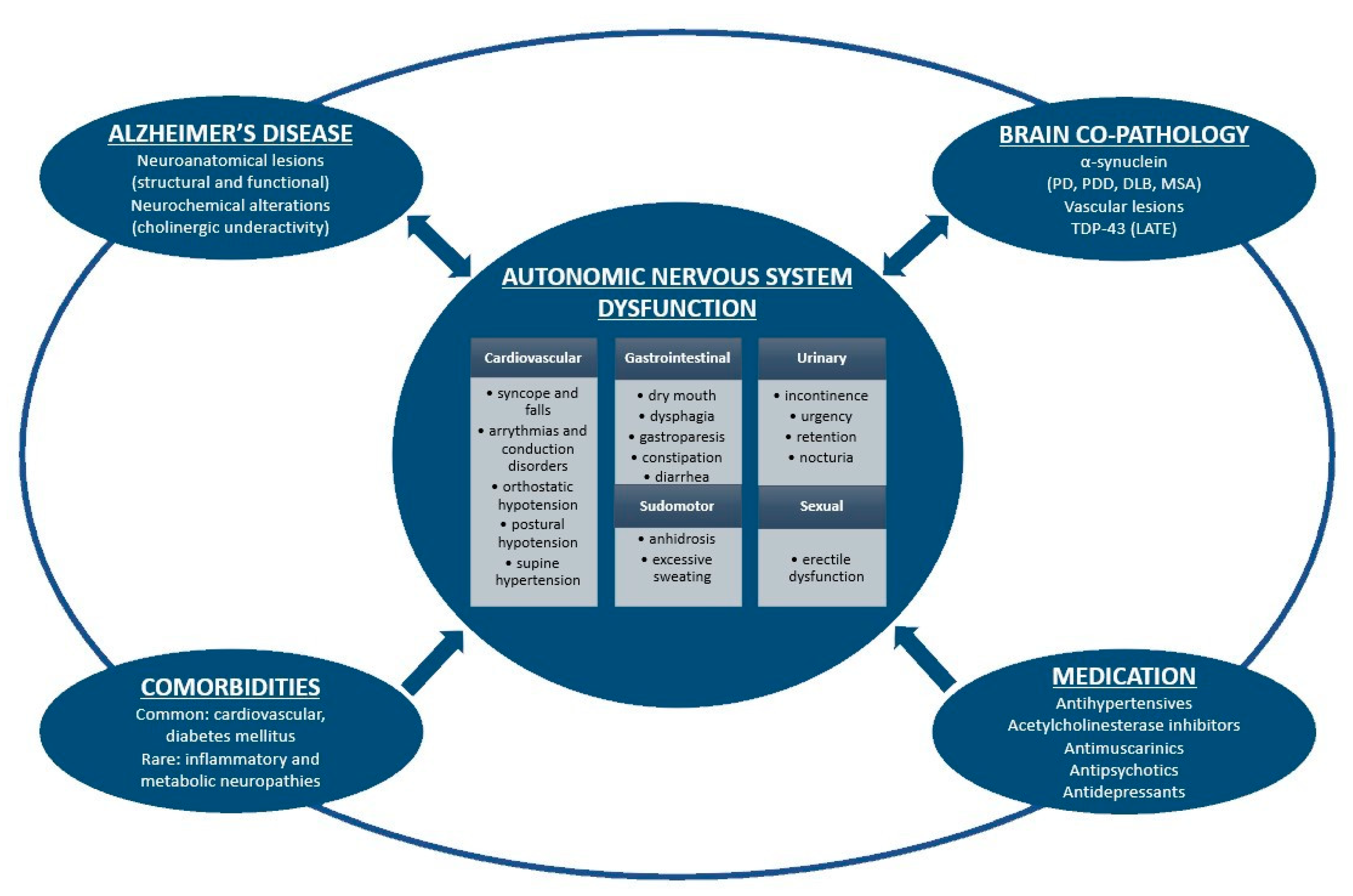
2. Dysautonomia in Alzheimer's Disease
3. Dysautonomia as a Result of Alzheimer's Disease Pathology
4. Dysautonomia as a Result of Brain Co-Pathologies
5. Dysautonomia as a Result of Comorbidities and Medication
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Alzheimer's Association. 2019 Alzheimer's disease facts and figures. Alzheimer's Dement. 2019, 15, 321–387. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Staging of Alzheimer's disease-related neurofibrillary changes. Neurobiol. Aging 1995, 16, 271–278. [Google Scholar] [CrossRef]
- Engelhardt, E.; Laks, J. Alzheimer disease neuropathology: Understanding autonomic dysfunction. Dement. Neuropsychol. 2008, 2, 183–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toledo, M.A.d.V.; Junqueira, L.F., Jr. Cardiac sympathovagal modulation evaluated by short-term heart interval variability is subtly impaired in Alzheimer's disease. Geriatr. Gerontol. Int. 2008, 8, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Katzman, R.; Hill, L.R.; Yu, E.S.; Wang, Z.Y.; Booth, A.; Salmon, D.P.; Liu, W.T.; Qu, G.Y.; Zhang, M. The malignancy of dementia. Predictors of mortality in clinically diagnosed dementia in a population survey of Shanghai, China. Arch. Neurol. 1994, 51, 1220–1225. [Google Scholar] [CrossRef]
- Burns, A.; Jacoby, R.; Luthert, P.; Levy, R. Cause of death in Alzheimer's disease. Age Aging 1990, 19, 341–344. [Google Scholar] [CrossRef]
- Manabe, T.; Mizukami, K.; Akatsu, H.; Hashizume, Y.; Ohkubo, T.; Kudo, K.; Hizawa, N. Factors associated with pneumonia-caused death in older adults with autopsy-confirmed dementia. Intern. Med. 2017, 56, 907–914. [Google Scholar] [CrossRef] [Green Version]
- Chu, C.; Tranel, D.; Damasio, A.R.; Van Hoesen, G.W. The autonomic-related cortex: Pathology in Alzheimer's disease. Cereb. Cortex 1997, 7, 86–95. [Google Scholar] [CrossRef] [Green Version]
- Zakrzewska-Pniewska, B.; Gawel, M.; Szmidt-Salkowska, E.; Kepczynska, K.; Nojszewska, M. Clinical and Functional Assessment of Dysautonomia and Its Correlation in Alzheimer's Disease. Am. J. Alzheimer's Dis. Other Dement. 2012, 27, 592–599. [Google Scholar] [CrossRef]
- Beach, P.A.; Huck, J.T.; Zhu, D.C.; Bozoki, A.C. Altered behavioral and autonomic pain responses in Alzheimer's disease are associated with dysfunctional affective, self-reflective and salience network resting-state connectivity. Front. Aging Neurosci. 2017, 9, 297. [Google Scholar] [CrossRef] [Green Version]
- Femminella, G.D.; Rengo, G.; Komici, K.; Iacotucci, P. Autonomic dysfunction in Alzheimer's disease: Tools for assessment and review of the literature. J. Alzheimer's Dis. 2014, 42, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Collins, O.; Dillon, S.; Finucane, C.; Lawlor, B.; Kenny, R.A. Parasympathetic autonomic dysfunction is common in mild cognitive impairment. Neurobiol. Aging 2012, 33, 2324–2333. [Google Scholar] [CrossRef] [PubMed]
- Affoo, R.H.; Foley, N.; Rosenbek, J.; Shoemaker, J.K.; Martin, R.E. Swallowing dysfunction and autonomic nervous system dysfunction in Alzheimer's disease: A scoping review of the evidence. J. Am. Geriatr. Soc. 2013, 61, 2203–2213. [Google Scholar] [CrossRef] [PubMed]
- Min, M.; Shi, T.; Sun, C.; Liang, M.; Zhang, Y.; Wu, Y.; Sun, Y. The association between orthostatic hypotension and dementia: A meta-analysis of prospective cohort studies. Int. J. Geriatr. Psychiatry 2018, 33, 1541–1547. [Google Scholar] [CrossRef]
- Braak, E.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; Del Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404. [Google Scholar] [CrossRef] [Green Version]
- Saper, C.B.; German, D.C. Hypothalamic pathology in Alzheimer's disease. Neurosci. Lett. 1987, 74, 364–370. [Google Scholar] [CrossRef]
- Giménez-Amaya, J.M.; McFarland, N.R.; Heras, S.D.L.; Haber, J.E. Organization of thalamic projections to the ventral striatum in the primate. J. Comp. Neurol. 1995, 354, 127–149. [Google Scholar] [CrossRef]
- Larner, A.J. The cerebellum in Alzheimer's disease. Dement. Geriatr. Cogn. Disord. 1997, 8, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E.; Bohl, J.; Lang, W. Alzheimer's disease: Amyloid plaques in the cerebellum. J. Neurol. Sci. 1989, 93, 277–287. [Google Scholar] [CrossRef]
- Parvizi, J.; Van Hoesen, G.W.; Damasio, A. The selective vulnerability of brainstem nuclei to Alzheimer's disease. Ann. Neurol. 2001, 49, 53–66. [Google Scholar] [CrossRef]
- Aletrino, M.A.; Vogels, O.J.; Van Domburg, P.H.; Ten Donkelaar, H.J. Cell loss in the nucleus raphes dorsalis in Alzheimer's disease. Neurobiol. Aging 1992, 13, 461–468. [Google Scholar] [CrossRef]
- Li, H.; Jia, X.; Qi, Z.; Fan, X.; Ma, T.; Ni, H.; Li, C.-S.R.; Li, K. Altered functional connectivity of the basal nucleus of Meynert in mild cognitive impairment: A resting-state fMRI study. Front. Aging Neurosci. 2017, 9, 127. [Google Scholar] [CrossRef]
- Grothe, M.; Zaborszky, L.; Atienza, M.; Gil-Neciga, E.; Rodriguez-Romero, R.; Teipel, S.J.; Amunts, K.; Suarez-Gonzalez, A.; Cantero, J.L. Reduction of basal forebrain cholinergic system parallels cognitive impairment in patients at high risk of developing Alzheimer's disease. Cereb. Cortex 2010, 20, 1685–1695. [Google Scholar] [CrossRef] [Green Version]
- Szili-Török, T.; Kálmán, J.; Paprika, D.; Dibó, G.; Rózsa, Z.; Rudas, L. Depressed baroreflex sensitivity in patients with Alzheimer's and Parkinson's disease. Neurobiol. Aging 2001, 22, 435–438. [Google Scholar] [CrossRef]
- Jengeleski, C.A.; Powers, R.E.; O'Connor, D.T.; Price, D.L. Noradrenergic innervation of human pineal gland: Abnormalities in aging and Alzheimer's disease. Brain Res. 1989, 481, 378–382. [Google Scholar] [CrossRef]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer's disease. Alzheimer's Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Head, E.; Schmitt, F.A.; Davis, P.R.; Neltner, J.H.; Jicha, G.A.; Abner, E.L.; Smith, C.D.; Van Eldik, L.J.; Kryscio, R.J.; et al. Alzheimer's disease is not “brain aging”: Neuropathological, genetic, and epidemiological human studies. Acta Neuropathol. 2011, 121, 571–587. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.L.; Lee, E.B.; Xie, S.X.; Rennert, L.; Suh, E.; Bredenberg, C.; Caswell, C.; Van Deerlin, V.M.; Yan, N.; Yousef, A.; et al. Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain 2018, 141, 2181–2193. [Google Scholar] [CrossRef] [Green Version]
- Boyle, P.A.; Yu, L.; Leurgans, S.E.; Wilson, R.S.; Brookmeyer, R.; Schneider, J.A.; Bennett, D. Attributable risk of Alzheimer's dementia attributed to age-related neuropathologies. Ann. Neurol. 2018, 85, 114–124. [Google Scholar] [CrossRef]
- Ferreira, D.; Nordberg, A.; Westman, E. Biological subtypes of Alzheimer disease. Neurol. 2020, 94, 436–448. [Google Scholar] [CrossRef] [Green Version]
- McKeith, I.G.; Boeve, B.F.; Dickson, D.W.; Halliday, G.M.; Taylor, J.P.; Weintraub, D.; Aarsland, D.; Galvin, J.; Attems, J.; Ballard, C.; et al. Diagnosis and management of dementia with Lewy bodies. Neurology 2017, 89, 88–100. [Google Scholar] [CrossRef] [Green Version]
- Irwin, D.J.; Grossman, M.; Weintraub, D.; Hurtig, H.I.; Duda, J.E.; Xie, S.X.; Lee, E.B.; Van Deerlin, V.M.; Lopez, O.L.; Kofler, J.K.; et al. Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: A retrospective analysis. Lancet Neurol. 2017, 16, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Smirnov, D.S.; Galasko, D.; Edland, S.D.; Filoteo, J.V.; Hansen, L.A.; Salmon, D.P. Cognitive decline profiles differ in Parkinson disease dementia and dementia with Lewy bodies. Neurology 2020, 94, e2076–e2087. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Dickson, D.W.; Trojanowski, J.Q.; Jack, C.R.; Boyle, P.A.; Arfanakis, K.; Rademakers, R.; Alafuzoff, I.; Attems, J.; Brayne, C.; et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): Consensus working group report. Brain 2019, 142, 1503–1527. [Google Scholar] [CrossRef] [Green Version]
- Josephs, K.A.; Whitwell, J.L.; Tosakulwong, N.; Weigand, S.D.; Murray, M.E.; Liesinger, A.M.; Petrucelli, L.; Senjem, M.L.; Ivnik, R.J.; Parisi, J.E.; et al. TAR DNA-binding protein 43 and pathological subtype of Alzheimer's disease impact clinical features. Ann. Neurol. 2015, 78, 697–709. [Google Scholar] [CrossRef]
- Hase, Y.; Polvikoski, T.M.; Firbank, M.J.; Craggs, L.J.L.; Hawthorne, E.; Platten, C.; Stevenson, W.; Deramecourt, V.; Ballard, C.; Kenny, R.A.; et al. Small vessel disease pathological changes in neurodegenerative and vascular dementias concomitant with autonomic dysfunction. Brain Pathol. 2019, 30, 191–202. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R.; Kawas, C.H.; Klunk, W.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer's disease: Recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimer's Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Eldholm, R.S.; Persson, K.; Barca, M.L.; Knapskog, A.B.; Cavallin, L.; Engedal, K.; Selbaek, G.; Skovlund, E.; Saltvedt, I. Association between vascular comorbidity and progression of Alzheimer's disease: A two-year observational study in Norwegian memory clinics. BMC Geriatr. 2018, 18, 120. [Google Scholar] [CrossRef]
- Wang, J.-H.; Wu, Y.-J.; Tee, B.L.; Lo, R.Y. Medical comorbidity in Alzheimer's disease: A nested case-control study. J. Alzheimer's Dis. 2018, 63, 773–781. [Google Scholar] [CrossRef]
- Testa, G.; Ceccofiglio, A.; Mussi, C.; Bellelli, G.; Nicosia, F.; Bo, M.; Riccio, D.; Curcio, F.; Martone, A.M.; Noro, G.; et al. Hypotensive drugs and syncope due to orthostatic hypotension in older adults with dementia (syncope and dementia study). J. Am. Geriatr. Soc. 2018, 66, 1532–1537. [Google Scholar] [CrossRef] [PubMed]
- Brignole, M.; Moya, A.; De Lange, F.J.; Deharo, J.-C.; Elliott, P.M.; Fanciulli, A.; Fedorowski, A.; Furlan, R.; Kenny, R.A.; Martín, A.; et al. 2018 ESC Guidelines for the diagnosis and management of syncope. Eur. Heart J. 2018, 39, 1883–1948. [Google Scholar] [CrossRef] [PubMed]
- Biaggioni, I. Orthostatic hypotension in the hypertensive patient. Am. J. Hypertens. 2018, 31, 1255–1259. [Google Scholar] [CrossRef] [Green Version]
- Freidenberg, D.L.; Shaffer, L.E.; Macalester, S.; Fannin, E.A. Orthostatic hypotension in patients with dementia. Cogn. Behav. Neurol. 2013, 26, 105–120. [Google Scholar] [CrossRef] [PubMed]
- Töyry, J.P.; Niskanen, L.K.; Mäntysaari, M.J.; Länsimies, E.A.; Uusitupa, M.I. Occurrence, predictors, and clinical significance of autonomic neuropathy in NIDDM. Ten-year follow-up from the diagnosis. Diabetes 1996, 45, 308–315. [Google Scholar] [CrossRef]
- Freeman, R. Diabetic autonomic neuropathy. Handb. Clin. Neurol. 2014, 126, 63–79. [Google Scholar] [CrossRef]
- Isik, A.T.; Bozoglu, E.; Naharci, M.I.; Kilic, S. Evaluation of the effects of galantamine on cardiac function in elderly patients with Alzheimer's disease. Am. J. Geriatr. Pharmacother. 2010, 8, 454–459. [Google Scholar] [CrossRef]
- van Beek, A.H.; Sijbesma, J.C.; Rikkert, M.G.M.O.; Claassen, J.A.H.R. Galantamine does not cause aggravated orthostatic hypotension in people with Alzheimer's disease. J. Am. Geriatr. Soc. 2010, 58, 409–410. [Google Scholar] [CrossRef] [Green Version]
- Isik, A.T.; Soysal, P.; Usarel, C. Effects of acetylcholinesterase inhibitors on balance and gait functions and orthostatic hypotension in elderly patients with Alzheimer Disease. Am. J. Alzheimer's Dis. Other Dement. 2016, 31, 580–584. [Google Scholar] [CrossRef]
- Gill, S.S.; Anderson, G.M.; Fischer, H.D.; Bell, C.M.; Li, P.; Normand, S.-L.T.; Rochon, P. Syncope and its consequences in patients with dementia receiving cholinesterase inhibitors. Arch. Intern. Med. 2009, 169, 867–873. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Brown, R.T.; Ding, E.L.; Kiel, D.P.; Berry, S.D. Dementia medications and risk of falls, syncope, and related adverse events: Meta-analysis of randomized controlled trials. J. Am. Geriatr. Soc. 2011, 59, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, R.K.; Farwell, W.; Cantor, M.D.; Lawler, E.V. Cholinesterase inhibitors and incidence of bradycardia in patients with dementia in the Veterans Affairs New England Healthcare System. J. Am. Geriatr. Soc. 2009, 57, 1997–2003. [Google Scholar] [CrossRef] [PubMed]
- Kröger, E.; Van Marum, R.; Souverein, P.; Carmichael, P.-H.; Egberts, T. Treatment with rivastigmine or galantamine and risk of urinary incontinence: Results from a Dutch database study. Pharmacoepidemiol. Drug Saf. 2015, 24, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Lampela, P.; Taipale, H.; Hartikainen, S. Use of cholinesterase inhibitors increases initiation of urinary anticholinergics in persons with Alzheimer's disease. J. Am. Geriatr. Soc. 2016, 64, 1510–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torvinen-Kiiskinen, S.; Taipale, H.; Tanskanen, A.; Tiihonen, J.; Hartikainen, S. Concomitant use of acetylcholine esterase inhibitors and urinary antispasmodics among Finnish community-dwelling persons with Alzheimer Disease. J. Clin. Psychopharmacol. 2014, 34, 722–727. [Google Scholar] [CrossRef]
- Chapple, C.R.; Khullar, V.; Gabriel, Z.; Muston, D.; Bitoun, C.E.; Weinstein, D. The effects of antimuscarinic treatments in overactive bladder: An update of a systematic review and meta-analysis. Eur. Urol. 2008, 54, 543–562. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.-F.; Tan, L.; Wang, H.-F.; Jiang, T.; Tan, M.-S.; Tan, L.; Xu, W.; Li, J.-Q.; Wang, J.; Lai, T.-J.; et al. The prevalence of neuropsychiatric symptoms in Alzheimer's disease: Systematic review and meta-analysis. J. Affect. Disord. 2016, 190, 264–271. [Google Scholar] [CrossRef]
- Alvares, G.; Quintana, D.S.; Hickie, I.B.; Guastella, A.J. Autonomic nervous system dysfunction in psychiatric disorders and the impact of psychotropic medications: A systematic review and meta-analysis. J. Psychiatry Neurosci. 2016, 41, 89–104. [Google Scholar] [CrossRef] [Green Version]
- Hattori, S.; Kishida, I.; Suda, A.; Miyauchi, M.; Shiraishi, Y.; Fujibayashi, M.; Tsujita, N.; Ishii, C.; Ishii, N.; Moritani, T.; et al. Effects of four atypical antipsychotics on autonomic nervous system activity in schizophrenia. Schizophr. Res. 2018, 193, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Howell, S.; Yarovova, E.; Khwanda, A.; Rosen, S.D. Cardiovascular effects of psychotic illnesses and antipsychotic therapy. Heart 2019, 105, 1852–1859. [Google Scholar] [CrossRef] [PubMed]
- GBD 2016 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1211–1259. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Antihypertensives | α-Blockers, β-Blockers, Central Sympatholytics, Nitrates, Diuretics | Bradycardia, Syncope, Orthostatic Hypotension |
|---|---|---|
| Acetylcholinesterase Inhibitors | donepezil, galantamine, rivastigmine | bradycardia, syncope, orthostatic hypotension |
| Antimuscarinic Agents | darifenacin, propoverine, solifenacin, tolderodine | dry mouth, constipation |
| trospium | constipation | |
| oxybutynin | dry mouth, urinary retention | |
| Typical Antipsychotics | haloperidol | cardiovascular events, sexual dysfunction |
| chlorpromazine | cardiovascular events, orthostatic hypotension, dry mouth, constipation, urinary retention | |
| thioridazine | orthostatic hypotension, dry mouth, constipation, urinary retention | |
| Atypical Antipsychotics | quetiapine, clozapine, olanzapine, risperidone, aripiprazole | cardiovascular events, dry mouth, constipation, urinary retention, sexual dysfunction |
| Antidepressants | tricyclic antidepressants | cardiovascular events, dry mouth, constipation, urinary retention |
| selective serotonin and serotonin-norepinephrine reuptake inhibitors | dry mouth, constipation, diarrhea, sexual dysfunction, excessive sweating |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tulbă, D.; Cozma, L.; Popescu, B.O.; Davidescu, E.I. Dysautonomia in Alzheimer's Disease. Medicina 2020, 56, 337. https://doi.org/10.3390/medicina56070337
Tulbă D, Cozma L, Popescu BO, Davidescu EI. Dysautonomia in Alzheimer's Disease. Medicina. 2020; 56(7):337. https://doi.org/10.3390/medicina56070337
Chicago/Turabian StyleTulbă, Delia, Liviu Cozma, Bogdan Ovidiu Popescu, and Eugenia Irene Davidescu. 2020. "Dysautonomia in Alzheimer's Disease" Medicina 56, no. 7: 337. https://doi.org/10.3390/medicina56070337
APA StyleTulbă, D., Cozma, L., Popescu, B. O., & Davidescu, E. I. (2020). Dysautonomia in Alzheimer's Disease. Medicina, 56(7), 337. https://doi.org/10.3390/medicina56070337