Modification of Marine Natural Product Ningalin B and SAR Study Lead to Potent P-Glycoprotein Inhibitors

Abstract
:
1. Introduction

2. Results and Discussion
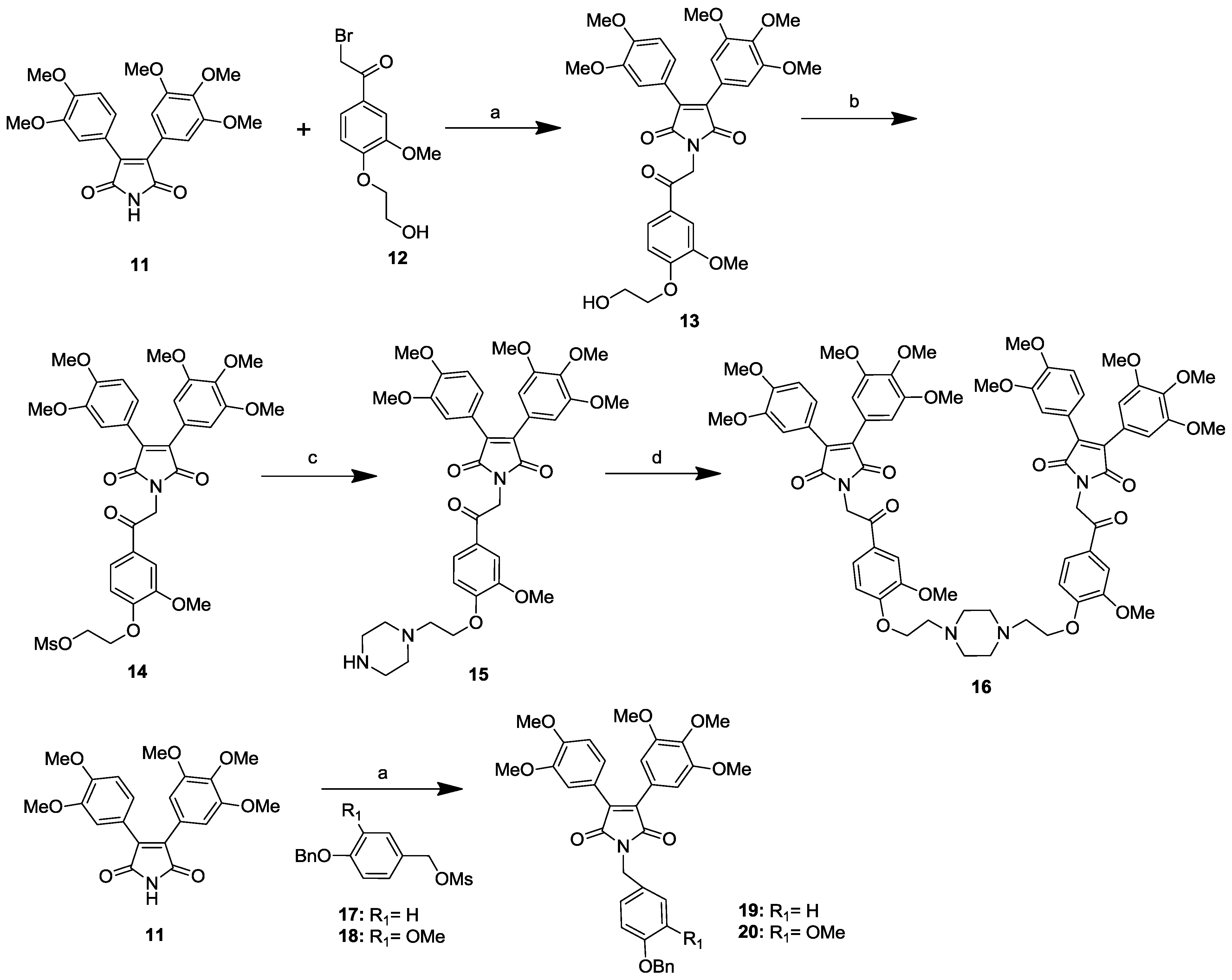
2.1. Synthesis of Permethyl Ningalin B Analogues

2.2. P-gp Modulating Activity of Permethyl Ningalin B Analogues


{kind=link}
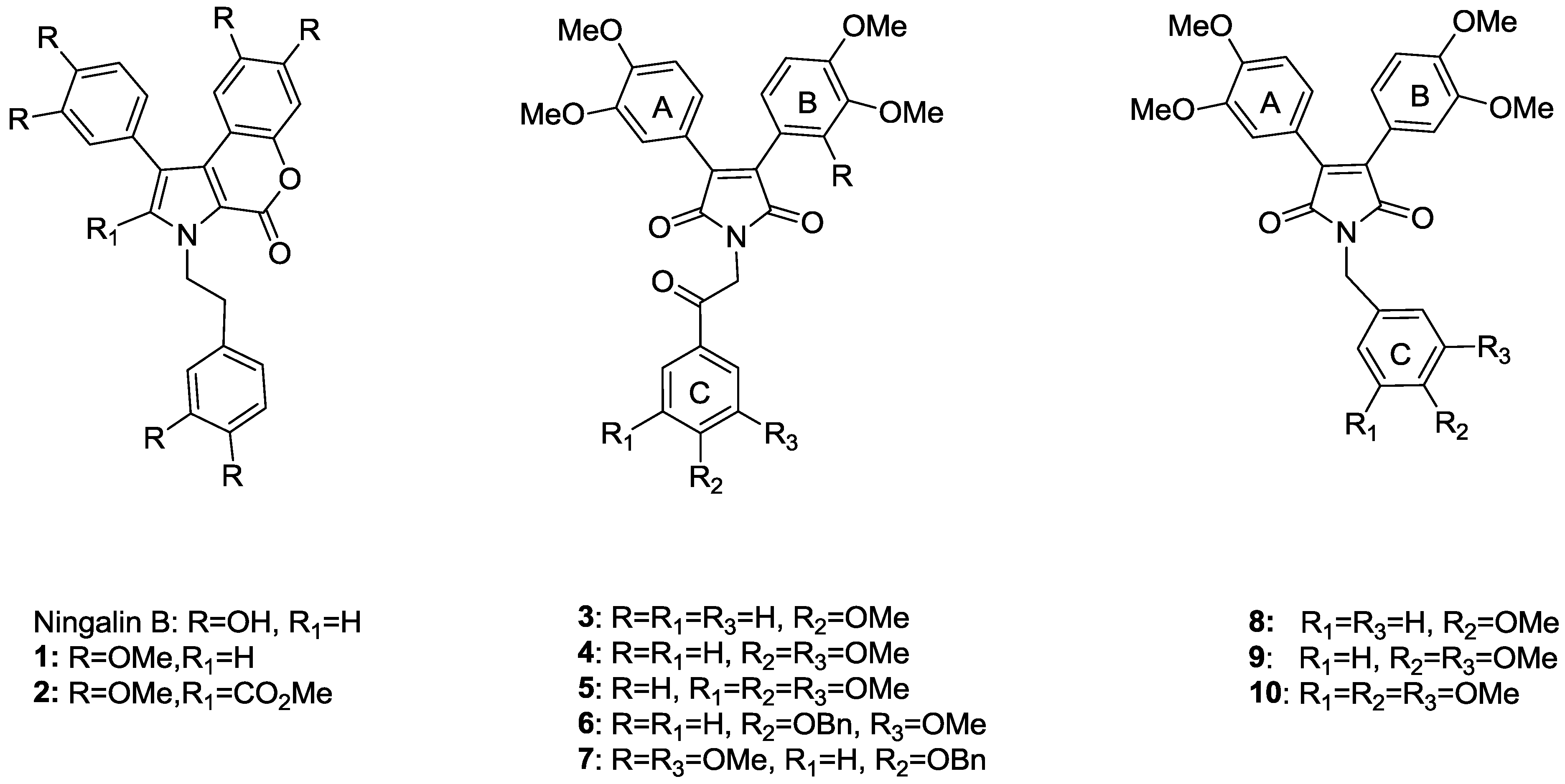{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Series | Cpds (1 μM) | No. of Methoxy Group on Acryl A Ring | No. of Methoxy Group on Acryl B Ring | Substituents at Acryl Ring C | Type of Linker | Cytotoxicity (IC50, μM) | LCC6MDR | K562/P-gp | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LCC6 | LCC6MDR | L929 | Mean IC50 of Paclitaxel (nM) | RF | Mean IC50 of Paclitaxel (nM) | RF | ||||||||||||||||
| 3 | di | di | mono-methoxy | carbonylmethylene | / | / | / | 9.9 a,b | / | / | ||||||||||||
| 4 | di | di | di-methoxy | carbonylmethylene | / | / | / | 8.2 a,b | / | / | ||||||||||||
| 5 | di | di | tri-methoxy | carbonylmethylene | / | / | / | 9.3 a,b | / | / | ||||||||||||
| Ι | 6 | di | di | mono-methoxy and mono-benzoloxy | carbonylmethylene | >100 b | >100 b | >100 b | 42.7 b | / | / | |||||||||||
| 7 | di | tri | mono-methoxy and mono-benzoloxy | carbonylmethylene | >100 b | >100 b | >100 b | 42.7 b | / | / | ||||||||||||
| 15 | di | tri | piperzine | carbonylmethylene | 31.2 | ± | 2.1 | 30.7 | ± | 0.6 | 44.6 | ± | 4.7 | 209.6 | ± | 22.1 | 0.7 | 539.1 | ± | 76.7 | 1.1 | |
| 16 | di | tri | piperzine | carbonylmethylene | >100 | >100 | >100 | 137.8 | ± | 7.7 | 1.0 | 458.4 | ± | 30.4 | 1.3 | |||||||
| 8 | di | di | mono-methoxy | methylene | / | / | / | 9.3 a,b | / | / | ||||||||||||
| 9 | di | di | di-methoxy | methylene | / | / | / | 12.4 a,b | / | / | ||||||||||||
| ΙΙ | 10 | di | di | tri-methoxy | methylene | / | / | / | 18.2 a,b | / | / | |||||||||||
| 19 | di | tri | mono-benzoloxy | methylene | >100 | >100 | >100 | 14.30 | ± | 2.3 | 10.1 | 4.3 | ± | 1.0 | 136.3 | |||||||
| 20 | di | tri | mono-methoxy and mono-benzoloxy | methylene | >100 | >100 | >100 | 11.00 | ± | 1.7 | 13.1 | 8.1 | ± | 2.0 | 72.3 | |||||||
| / | Verapamil | / | / | / | / | 63.9 | ± | 1.7 | 63.8 | ± | 0.1 | 89.2 | ± | 8.2 | 38.0 | ± | 7.0 | 3.8 | / | / | ||
| / | PSC833 | / | / | / | / | 14.6 | ± | 2.2 | 25.3 | ± | 4.3 | >100 | 1.8 | ± | 0.3 | 80.3 | 1.1 | ± | 0.5 | 520.9 | ||
| / | LCC6MDR c | / | / | / | / | 144.6 | ± | 9.4 | 1.0 | / | / | |||||||||||
| / | LCC6 c | / | / | / | / | 1.6 | ± | 0.3 | 90.4 | / | / | |||||||||||
| / | K562/P-gp | / | / | / | / | / | / | 586.0 | ± | 123.8 | 1.0 | |||||||||||
| / | K562 | / | / | / | / | / | / | 2.1 | ± | 0.0 | 279.0 | |||||||||||
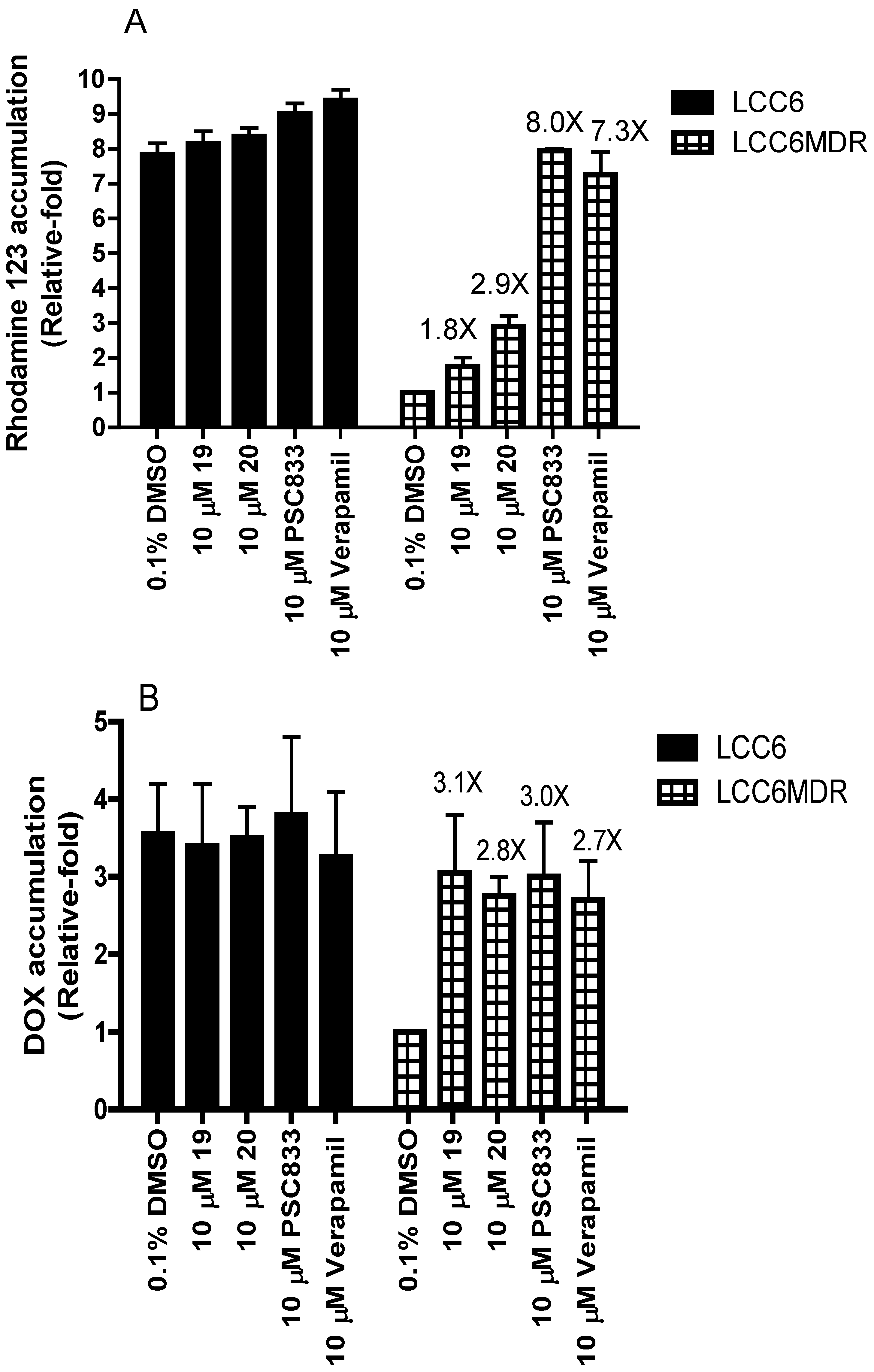
2.3. Effect of Permethyl Ningalin B Analogues on Intracellular Rhodamine 123 and DOX Accumulation

3. Experimental Section
3.1. Materials and Methods
3.2. Synthesis of Compounds 13–20
3.3. General Procedure for the Preparation of the Compounds 19, 20
3.4. Materials for Biological Studies
3.5. Cell Culture
3.6. Cell Proliferation Assay
3.7. Cytotoxicity Assay
3.8. Rhodamine 123 and DOX Accumulation Assay
3.9. Calculated Molecular Descriptors
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Amin, M.L. P-glycoprotein inhibition for optimal drug delivery. Drug Target Insights 2013, 7, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Baumert, C.; Hilgeroth, A. Recent advances in the development of P-gp inhibitors. Anticancer. Agents Med. Chem. 2009, 9, 415–436. [Google Scholar] [CrossRef] [PubMed]
- Eckford, P.D.; Sharom, F.J. ABC efflux pump-based resistance to chemotherapy drugs. Chem. Rev. 2009, 109, 2989–3011. [Google Scholar] [CrossRef] [PubMed]
- Kawase, M.; Motohashi, N. New multidrug resistance reversal agents. Curr. Drug Targets 2003, 4, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Robey, R.W.; Massey, P.R.; Amiri-Kordestani, L.; Bates, S.E. ABC transporters: Unvalidated therapeutic targets in cancer and the CNS. Anticancer Agents Med. Chem. 2010, 10, 625–633. [Google Scholar] [PubMed]
- Davis, R.A.; Carroll, A.R.; Pierens, G.K.; Quinn, R.J. New lamellarin alkaloids from the australian ascidian, didemnum chartaceum. J. Nat. Prod. 1999, 62, 419–424. [Google Scholar] [CrossRef]
- Fan, H.; Peng, J.; Hamann, M.T.; Hu, J.F. Lamellarins and related pyrrole-derived alkaloids from marine organisms. Chem. Rev. 2008, 108, 264–287. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.V.; Rao, M.R.; Rhodes, D.; Hansen, M.S.; Rubins, K.; Bushman, F.D.; Venkateswarlu, Y.; Faulkner, D.J. Lamellarin alpha 20-sulfate, an inhibitor of HIV-1 integrase active against HIV-1 virus in cell culture. J. Med. Chem. 1999, 42, 1901–1907. [Google Scholar] [CrossRef] [PubMed]
- Boger, D.L.; Soenen, D.R.; Boyce, C.W.; Hedrick, M.P.; Jin, Q. Total synthesis of ningalin B utilizing a heterocyclic azadiene Diels-Alder reaction and discovery of a new class of potent multidrug resistant (MDR) reversal agents. J. Org. Chem. 2000, 65, 2479–2483. [Google Scholar] [CrossRef] [PubMed]
- Bullington, J.L.; Wolff, R.R.; Jackson, P.F. Regioselective preparation of 2-substituted 3,4-diaryl pyrroles: A concise total synthesis of ningalin B. J. Org. Chem. 2002, 67, 9439–9442. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Fenical, W. Ningalins A-D: Novel aromatic alkaloids from a western australian ascidian of the genus didemnum. J. Org. Chem. 1997, 62, 3254–3262. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Li, Z.; Shen, S.; Zeng, Y.; Yang, Y.; Xu, M.; Bruhn, T.; Bruhn, H.; Morschhauser, J.; Bringmann, G.; Lin, W. Baculiferins A-O, O-sulfated pyrrole alkaloids with anti-HIV-1 activity, from the Chinese marine sponge Iotrochota baculifera. Bioorg. Med. Chem. 2010, 18, 5466–5474. [Google Scholar] [CrossRef] [PubMed]
- Rudi, A.; Evan, T.; Aknin, M.; Kashma, Y.N. Polycitone B and prepolycitrin A: Two novel alkaloids from the marine ascidian Polycitor africanus. J. Nat. Prod. 2000, 63, 832–833. [Google Scholar] [PubMed]
- Kreipl, A.T.; Reid, C.; Steglich, W. Total syntheses of the marine pyrrole alkaloids polycitone A and B. Org. Lett. 2002, 4, 3287–3288. [Google Scholar] [CrossRef] [PubMed]
- Bailly, C. Lamellarins, from A to Z: A family of anticancer marine pyrrole alkaloids. Curr. Med. Chem. Anticancer Agents 2004, 4, 363–378. [Google Scholar] [PubMed]
- Vervoort, H.C.; Fenical, W.; Keifer, P.A. A cyclized didemnimide alkaloid from the caribbean ascidian didemnumconchyliatum. J. Nat. Prod. 1999, 62, 389–391. [Google Scholar] [PubMed]
- Shen, S.; Liu, D.; Wei, C.; Proksch, P.; Lin, W. Purpuroines A-J, halogenated alkaloids from the sponge Iotrochota purpurea with antibiotic activity and regulation of tyrosine kinases. Bioorg. Med. Chem. 2012, 20, 6924–6928. [Google Scholar] [PubMed]
- Krishnaiah, P.; Reddy, V.L.; Venkataramana, G.; Ravinder, K.; Srinivasulu, M.; Raju, T.V.; Ravikumar, K.; Chandrasekar, D.; Ramakrishna, S.; Venkateswarlu, Y. New lamellarin alkaloids from the Indian ascidian Didemnum obscurum and their antioxidant properties. J. Nat. Prod. 2004, 67, 1168–1171. [Google Scholar] [CrossRef] [PubMed]
- Ballot, C.; Kluza, J.; Martoriati, A.; Nyman, U.; Formstecher, P.; Joseph, B.; Bailly, C.; Marchetti, P. Essential role of mitochondria in apoptosis of cancer cells induced by the marine alkaloid Lamellarin D. Mol. Cancer Ther. 2009, 8, 3307–3317. [Google Scholar] [CrossRef] [PubMed]
- Chittchang, M.; Batsomboon, P.; Ruchirawat, S.; Ploypradith, P. Cytotoxicities and structure-activity relationships of natural and unnatural lamellarins toward cancer cell lines. ChemMedChem 2009, 4, 457–465. [Google Scholar] [PubMed]
- Chou, T.C.; Guan, Y.; Soenen, D.R.; Danishefsky, S.J.; Boger, D.L. Potent reversal of multidrug resistance by ningalins and its use in drug combinations against human colon carcinoma xenograft in nude mice. Cancer Chemother. Pharmacol. 2005, 56, 379–390. [Google Scholar] [PubMed]
- Appendino, G.; Gibbons, S.; Giana, A.; Pagani, A.; Grassi, G.; Stavri, M.; Smith, E.; Rahman, M.M. Antibacterial cannabinoids from Cannabis sativa: A structure-activity study. J. Nat. Prod. 2008, 71, 1427–1430. [Google Scholar] [CrossRef] [PubMed]
- Bin, J.W.; Wong, I.L.; Hu, X.; Yu, Z.X.; Xing, L.F.; Jiang, T.; Chow, L.M.; Biao, W.S. Structure-activity relationship study of permethyl ningalin B analogues as P-glycoprotein chemosensitizers. J. Med. Chem. 2013, 56, 9057–9070. [Google Scholar] [PubMed]
- Zhang, P.Y.; Wong, I.L.; Yan, C.S.; Zhang, X.Y.; Jiang, T.; Chow, L.M.; Wan, S.B. Design and syntheses of permethyl ningalin B analogues: Potent multidrug resistance (MDR) reversal agents of cancer cells. J. Med. Chem. 2010, 53, 5108–5120. [Google Scholar] [PubMed]
- Venkata, M.; Reddy, M.V.R.; Faulkner, D.J.; Venkateswarlu, Y.; Rao, M.R. New Lamellarin alkaloids from an unidentified ascidian from the Arabian Sea. Tetrhedron 1997, 53, 3457–3466. [Google Scholar] [CrossRef]
- Chan, K.F.; Wong, I.L.; Kan, J.W.; Yan, C.S.; Chow, L.M.; Chan, T.H. Amine linked flavonoid dimers as modulators for P-glycoprotein-based multidrug resistance: Structure-activity relationship and mechanism of modulation. J. Med. Chem. 2012, 55, 1999–2014. [Google Scholar] [CrossRef]
- Chan, K.F.; Zhao, Y.; Burkett, B.A.; Wong, I.L.; Chow, L.M.; Chan, T.H. Flavonoid dimers as bivalent modulators for P-glycoprotein-based multidrug resistance: Synthetic apigenin homodimers linked with defined-length poly(ethylene glycol) spacers increase drug retention and enhance chemosensitivity in resistant cancer cells. J. Med. Chem. 2006, 49, 6742–6759. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.F.; Zhao, Y.; Chow, T.W.; Yan, C.S.; Ma, D.L.; Burkett, B.A.; Wong, I.L.; Chow, L.M.; Chan, T.H. Flavonoid dimers as bivalent modulators for p-glycoprotein-based multidrug resistance: Structure-activity relationships. ChemMedChem 2009, 4, 594–614. [Google Scholar] [CrossRef] [PubMed]
- Wong, I.L.; Chan, K.F.; Tsang, K.H.; Lam, C.Y.; Zhao, Y.; Chan, T.H.; Chow, L.M. Modulation of multidrug resistance protein 1 (MRP1/ABCC1)-mediated multidrug resistance by bivalent apigenin homodimers and their derivatives. J. Med. Chem. 2009, 52, 5311–5322. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, C.; Wong, I.L.K.; Jin, W.B.; Jiang, T.; Chow, L.M.C.; Wan, S.B. Modification of Marine Natural Product Ningalin B and SAR Study Lead to Potent P-Glycoprotein Inhibitors. Mar. Drugs 2014, 12, 5209-5221. https://doi.org/10.3390/md12105209
Yang C, Wong ILK, Jin WB, Jiang T, Chow LMC, Wan SB. Modification of Marine Natural Product Ningalin B and SAR Study Lead to Potent P-Glycoprotein Inhibitors. Marine Drugs. 2014; 12(10):5209-5221. https://doi.org/10.3390/md12105209
Chicago/Turabian StyleYang, Chao, Iris L. K. Wong, Wen Bin Jin, Tao Jiang, Larry M. C. Chow, and Sheng Biao Wan. 2014. "Modification of Marine Natural Product Ningalin B and SAR Study Lead to Potent P-Glycoprotein Inhibitors" Marine Drugs 12, no. 10: 5209-5221. https://doi.org/10.3390/md12105209
APA StyleYang, C., Wong, I. L. K., Jin, W. B., Jiang, T., Chow, L. M. C., & Wan, S. B. (2014). Modification of Marine Natural Product Ningalin B and SAR Study Lead to Potent P-Glycoprotein Inhibitors. Marine Drugs, 12(10), 5209-5221. https://doi.org/10.3390/md12105209