2.1. Discovery, Structural Determination and Biological Properties
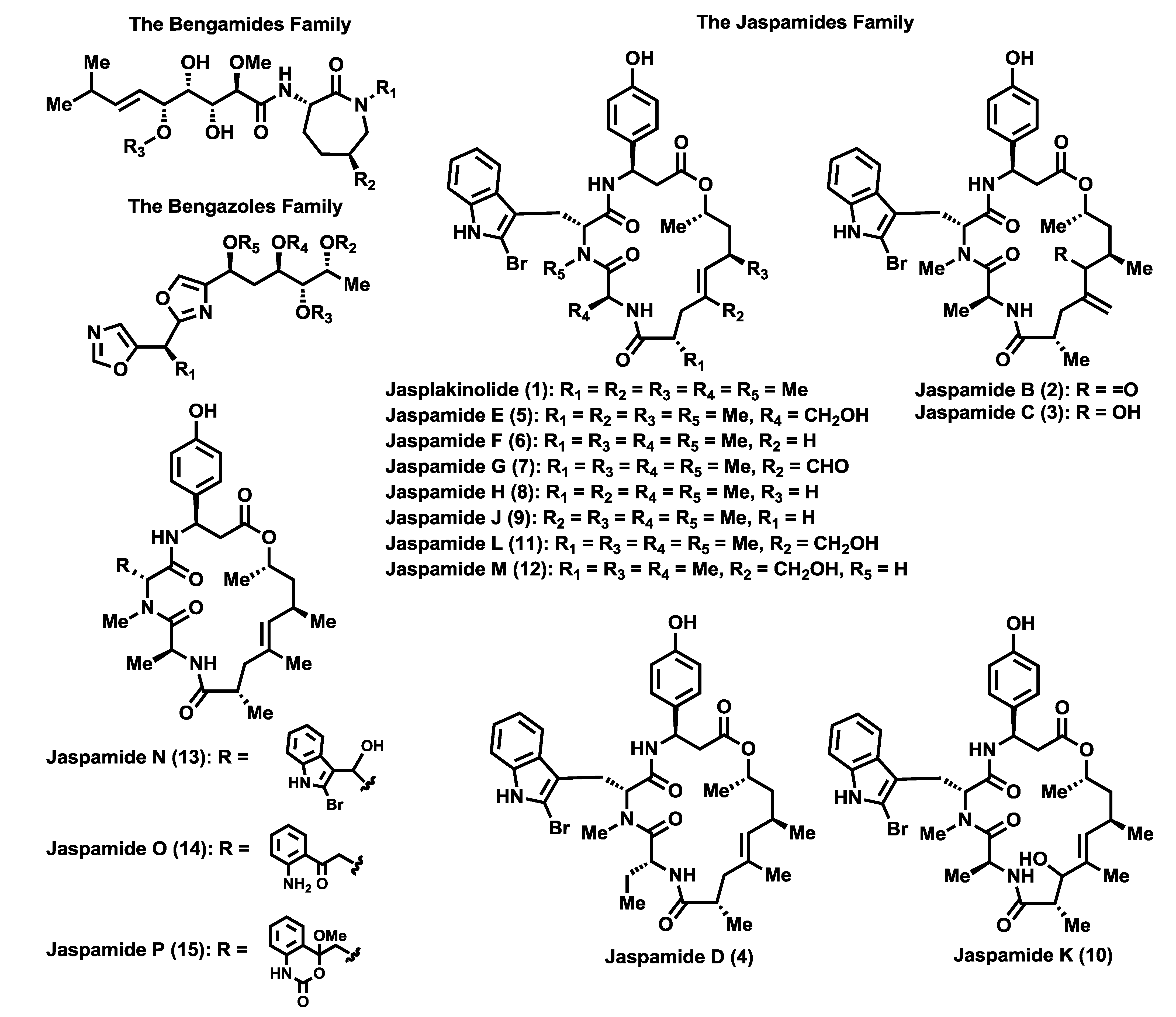
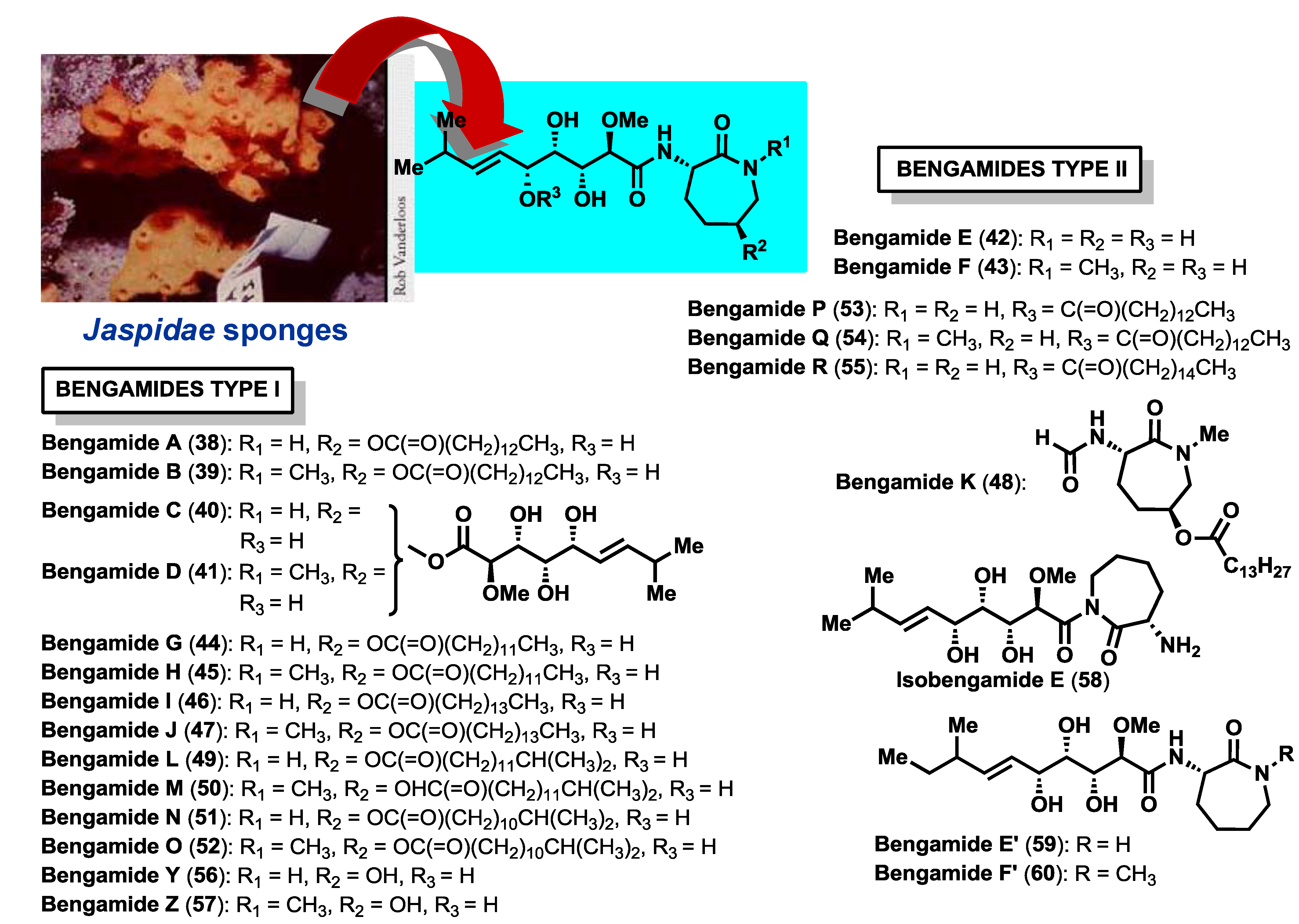
The bengamide family is comprised of a wide number of members (
Figure 3), with the bengamides A–F the first to be discovered and isolated between 1986 [
1] and 1989 [
10] by the research group of Professor Crews (University of California, Santa Cruz, CA, USA) from an undescribed specimen of an orange sponge belonging to the
Jaspidae family (family Choristida, order Astrophorida) that was collected in Benga Lagoon (Fiji Islands). The crude extract obtained from these sponges exhibited an impressive cytotoxicity profile against larynx epithelial carcinoma (1.0 μg/mL) and striking anthelmintic and antibacterial activities against the nematode
Nippostrongylus braziliensis and
Streptococcus pyrogenes [
1]. An extensive spectroscopic analysis led Crews and coworkers to establish the structures for the main components of this crude corresponding to bengamides A (
38) and B (
39). A third component, bengamide C (
40), was also identified in the crude extract, however, its structure determination was not possible at that time because it was not obtained in pure form. After two years the research group of Professor Crews was able to recollect huge amounts of this sponge and isolate new compounds related to the first bengamides, characterized as bengamides C–F (
40–
43), together with the related isomeric product isobengamide E (
58) and various oxazole derivatives, named bengazoles, which will be described in detail later in the review [
10,
34]. After this discovery, the extracts of the sponge
Jaspis carteri, collected in New Caledonia, showed a remarkable anticandidal activity and, after an expeditious purification process and structural analysis, the research group led by D’Auria isolated and identified bengamides A and B, together with a series of new members, including bengamides G–J (
44–
47) and a truncated derivative, bengamide K (
48) [
21]. Later, in 1999, Groweiss [
2] and co-workers isolated from
Jaspis sp. two new members of the bengamides; bengamide Y (
56), which lacks the fatty acid moiety and the
N-methyl group at position 15, and bengamide Z (
57), the nonacylated derivative of bengamide B. In the same year, Letourneux
et al. [
29] isolated from sponge
Pachastrissa sp. (family Calthropellidae, order Astrophorida) bengamide L (
49), together with novel members of the bengazoles. More recently, in 2001, more than 15 years after the discovery of the first bengamides, Crews
et al. [
20] described six new bengamides, bengamides M–R (
50–
55) as well as the known bengamides A (
38), B (
39), E (
42), F (
43), G (
44), H (
45), I (
46), L (
49), Y (
56) and Z (
57), from a collection of
Jaspis cf.
coriacea. The last contribution to this numerous family of natural products was generated also by Crews [
28] in 2012, who identified the bacteria
Myxococcus virescens as a new source of bengamide E (
42), and two new congeners, bengamides E′ (
59) and F′ (
60), isolated as an inseparable mixture of diastereomers, thus being the second example, in the peer-reviewed literature, of a sponge-derived natural product reported from a cultured microbial source (the first one being Makaluvamine A, isolated from the sponge
Zyzzya fuliginosa [
35] and myxomycetes
Didymium bahiense [
36]). It is also interesting to point out that bengamide E′ was initially synthesized in the laboratory prior to its isolation from a natural source [
37]. Currently, 23 members of the bengamide family have been described and their molecular structures determined by spectroscopic methods.
The bengamide structures contain a unique skeleton with a C-10 side chain possessing four contiguous hydroxyl groups as well as an E-olefin, that links to an aminocaprolactam moiety. According to the caprolactam ring, the bengamides have been classified into two structural classes: Type I that contains a hydroxylysine-derived caprolactam, bearing or not a lipidic chain (bengamides A–D, G–J, L–O, Y, Z) and Type II that contains a lysine-derived caprolactam (bengamides E, F, E′, F′, P–R).
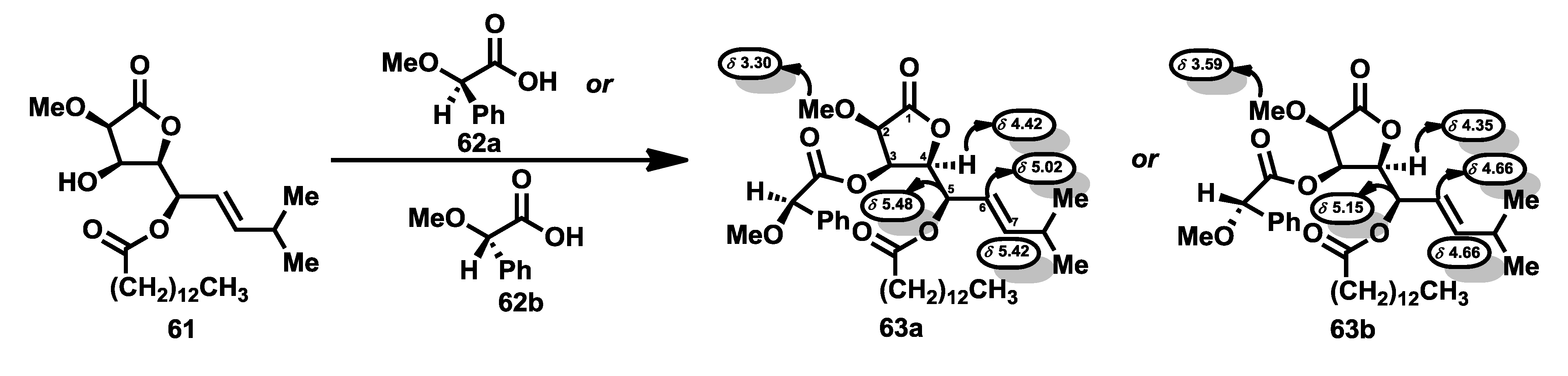
Structure determination of the bengamides was carried out by Crews
et al. [
38], as mentioned previously. Thus, the establishment of the absolute stereochemistry was possible by means of spectroscopic studies of the
1H-NMR of the (
R)- and (
S)-mandelate esters of lactone
61, a product obtained during the isolation process. Thus, these esters, compounds
63a and
63b, prepared by reaction of
61 with the O-methyl mandelic acids
62a and
62b respectively, showed significant differences in their respective
1H NMR spectra, particularly upfield shifts of H-4, H-5, H-6 and H-7 and downfield shifts for H-2 and the methoxyl group for the
S derivative
63b in comparison to the
R derivative
63a. The validity of the O-methylmandelate ester method [
39] was checked with the preparation of the corresponding esters of (−)-menthol, in which similar effects were observed in the chemical shifts of key protons (
Scheme 1). Subsequent total syntheses of bengamides A [
40], B [
41] and E [
42], as will be discussed in detail in
Section 2.3, confirmed the absolute configurations for the C10 side chain and for the hydroxylysine-derived caprolactam.
Figure 3.
Molecular structures of bengamides.
Figure 3.
Molecular structures of bengamides.
Scheme 1.
Chemical studies for structure determination.
Scheme 1.
Chemical studies for structure determination.
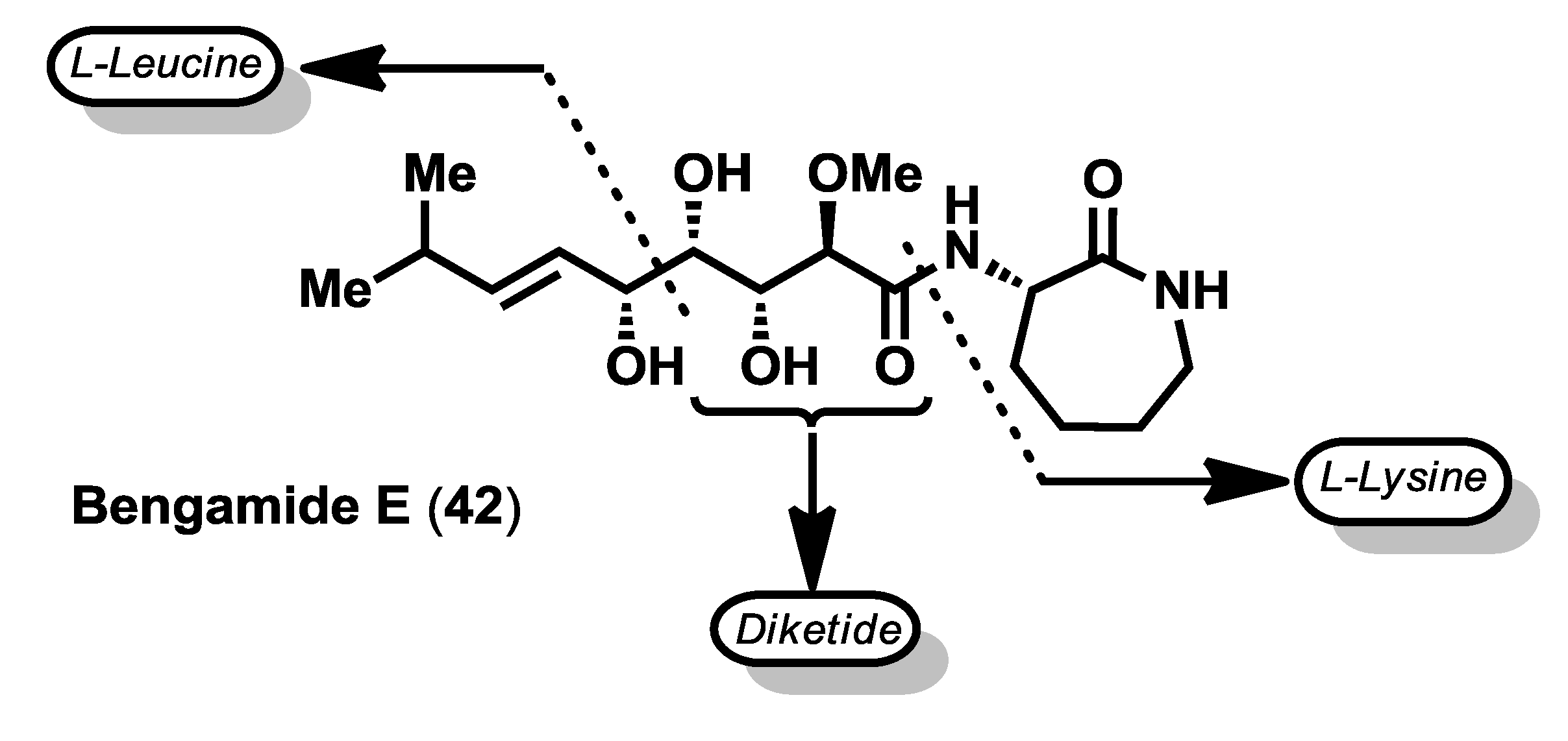
Interestingly, the biosynthetic origin of the bengamides seems to be the result of a symbiotic interaction between this class of sponges and bacteria, as revealed from their structures as the end-chain isopropyl group, characteristic of bacterial fatty acids [
43,
44]. Thus, the structure of the bengamides may arise from the linkage of a diketide with a six-carbon unit derived from
l-leucine to form the C10 side chain, which couples with cyclized
l-lysine (
Figure 4) [
10].
Figure 4.
Proposed biosynthetic origin of the bengamides.
Figure 4.
Proposed biosynthetic origin of the bengamides.
As indicated earlier, preliminary biological studies of the bengamides revealed
in vitro cytotoxicity towards larynx epithelial carcinoma at 1.0 µg/mL, together with activity against the bacteria
Streptococcus pyrogenes and the nematode
Nippostrongylus braziliensis [
1]. However, the bengamides proved inactive in assays against
Candida albicans and
Saccharomyces cerevisiae, in contrast to the bengazoles, which displayed potent antifungal activity [
29], as will be discussed later. Subsequent biological evaluations demonstrated the potent antiproliferative activities displayed by some members of the bengamides against different tumor cell lines, with IC
50 values ranging from 10 to 100 nM [
20]. Additionally, Crews
et al. [
28] recently discovered that the bengamides behave as immune modulating agents owing to their inhibition of NF-κB (nuclear factor kappa B) without accompanying cytotoxicity to RAW 264.7 macrophage immune cells, with the bengamides A (
38) and B (
39) as the most potent inhibitors. These studies suggest that the bengamides may serve as therapeutic leads for the treatment of diseases involving inflammation.
2.2. Antitumor Properties and Mechanism of Action
Intense cytotoxic studies of the antitumor activities of the bengamides, together with the results provided by the NCI-DTP database, has resulted in their identification as promising new anticancer agents [
20]. Thus, IC
50 values for natural bengamides A (
38), B (
39), E (
42), F (
43), M (
50), O (
52), P (
53) and Z (
57) were determined against MDA-MB-435 human mammary carcinoma with the best anti-proliferative
in vitro activities observed for bengamides having a fatty acid attached to the caprolactam ring (cases of bengamides A, B, M and O), which were >100-fold more potent than their nonlactam ester-bearing counterparts (bengamides E, F, P and Z) (
Table 2). However, despite the importance of a lipohilic ester on the caprolactam moiety for
in vitro potency,
in vivo studies revealed small differences in antitumor activity between all the bengamides, likely due to their poor water solubilities. On the other hand, it was demonstrated that bengamide B (
39) was converted to bengamide Z (
57) intracellularly, which suggests that this compound (
57) is actually responsible for the antiproliferative effects [
45]. Thus, the difference of activities observed
in vitro for bengamides B and Z likely arises from the poor cellular uptake of
57. Among all the natural bengamides, bengamide B (
39) displayed a unique profile in the NCI 60 cell line panel compared to standard antitumor agents, revealing arrest at both G1 and G2/M phases of the cell cycle by FACS (Fluorescence-activated cell sorter) analyses of transformed and non-transformed cells, respectively. Additional biological experiments led to the conclusion that the G1 arrest occurred at the G1/S restriction point and that the cells arrested in the G2/M phase of the cell cycle were not inhibited during mitosis but rather during cytokinesis [
20,
46,
47]. All these biological data suggested that the cytotoxicity exhibited by the bengamides was due to inhibition of a novel target [
48].
Table 2.
In vitro anti-proliferative activity of selected natural bengamides (IC50 [µM]) on MDA-MB-435 human mammary carcinoma cells.
Table 2.
In vitro anti-proliferative activity of selected natural bengamides (IC50 [µM]) on MDA-MB-435 human mammary carcinoma cells.
| Bengamide | MDA-MB-435 |
|---|
| A (38) | 0.001 ± 0.0006 |
| B (39) | 0.012 ± 0.003 |
| M (50) | 0.0101 ± 0.0021 |
| O (52) | 0.00029 ± 0.0005 |
| Z (57) | 2.9 ± 1.5 |
| E (42) | 3.3 ± 1.2 |
| F (43) | 2.9 ± 2.9 |
| P (53) | 1.2 ± 7.9 |
In 2003, Towbin
et al. [
48], in an effort to elucidate the mechanism of action of the bengamides, undertook extensive biological studies that allowed them to exclude many relevant biological targets involved in cancer, such as DNA, tubulins, actin, topoisomerases or proteases. On the other hand, proteomic studies performed on H1299 cells [
48] detected an alteration in a subset of proteins, the 14-3-3 protein isoforms, after treatment
in vitro with LAF389 (
64) (
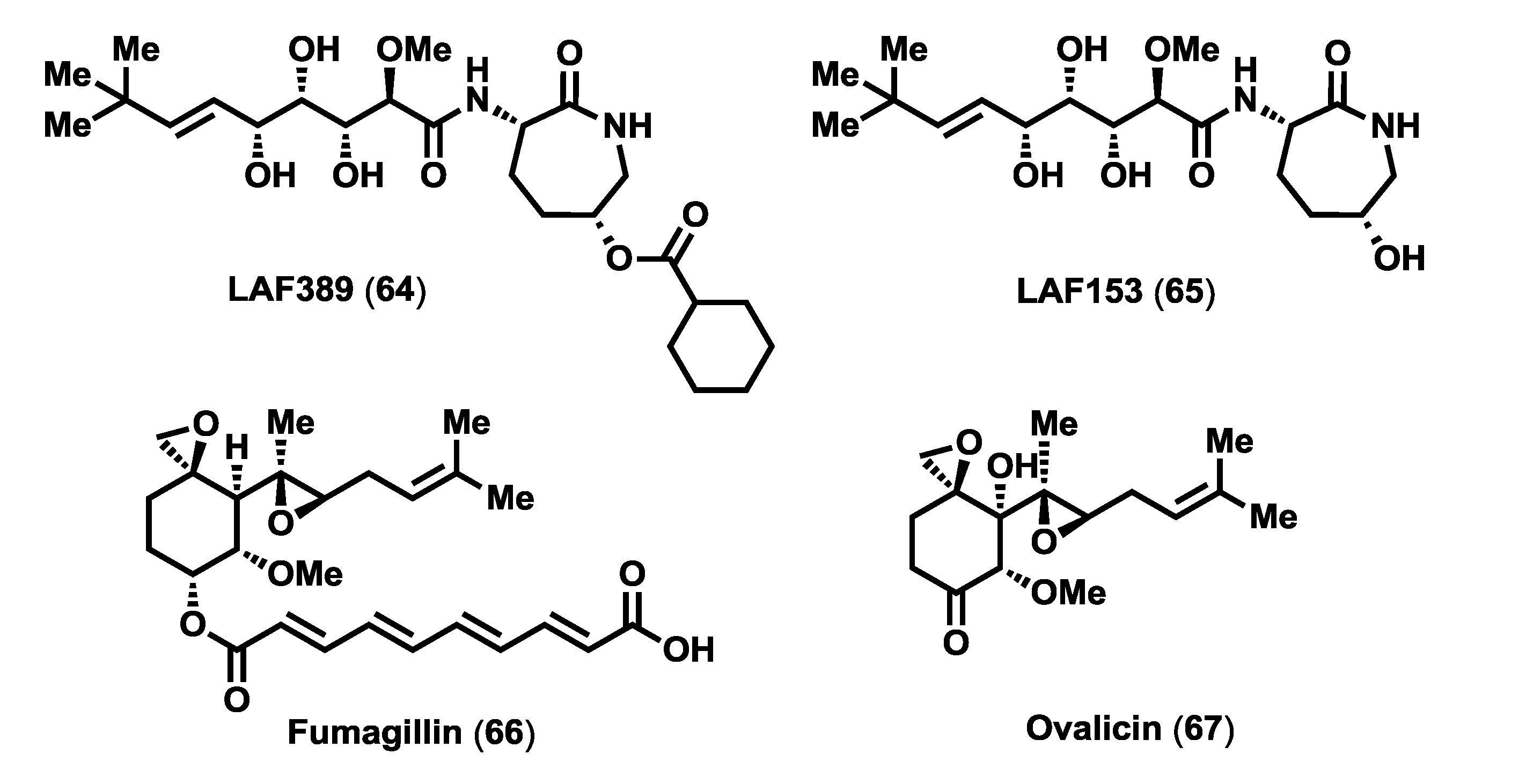
Figure 5), a more soluble synthetic analogue of bengamides, with striking inhibitory effects on tumor growth
in vitro and
in vivo [
49]. As a consequence of these results, a more detailed study focused on the 14-3-3 protein family (cytosolic adaptor proteins that modulate intracellular signaling, cell cycle control, transcriptional control and apoptosis [
50]) revealed a retention of the initiator methionine in the isoform 14-3-3 when treated with the bengamides, and consequently allowed to determine the direct targets of bengamides as both human methionine aminopeptidases (MetAP) isoforms, MetAP1 and MetAP2. This important finding prompted further investigations on the effect of LAF389 in endothelial cell proliferation and to compare it to fumagillin (
66), a well-known anti-angiogenic natural product that inhibits MetAP2 enzyme [
51,
52]. The result of this study revealed that LAF389 displayed less pronounced endothelial specificity. Consequently, whereas fumagillin (
66) and ovalicin (
67) express selective inhibition of endothelial cell proliferation, the bengamides lack such selectivity, inhibiting the proliferation of all cell types tested, both endothelial and epithelial cells [
48]. Nevertheless, additional biological studies with other bengamides revealed that bengamides M (
50) and O (
52) exhibited 10- to 20-fold selectivity toward MetAP1 which might imply a different antitumor activity profile (
Table 3) [
53,
54].
Table 3.
Inhibition of MetAP’s enzymatic activities by bengamides [IC50 (µM)].
Table 3.
Inhibition of MetAP’s enzymatic activities by bengamides [IC50 (µM)].
| Compound | MetAP1 | MetAP2 |
|---|
| Bengamide A (38) | 1.9 ± 0.2 | 10.5 ± 3.8 |
| Bengamide B (39) | 29.3 ± 10.4 | 17.9 ± 7.9 |
| Bengamide G (44) | 26.8 ± 18.3 | >50 |
| Bengamide L (49) | 37.1 ± 13.4 | >50 |
| Bengamide M (50) | 5.4 ± 2.3 | >50 |
| Bengamide N (51) | 40.2 ± 14.3 | >50 |
| Bengamide O (52) | 2.7 ± 0.4 | >50 |
| Fumagillin (66) | NA | 0.03 |
| Ovalicin (67) | NA | 0.0004 |
Figure 5.
Inhibitors of MetAPs: bengamide analogues LAF389 and LAF153, Fumagillin and Ovalicin.
Figure 5.
Inhibitors of MetAPs: bengamide analogues LAF389 and LAF153, Fumagillin and Ovalicin.
Having demonstrated that the methionine aminopeptidases were the direct biological targets for the bengamides, the next step in this research was to study their mode of action at the active site of these enzymes and to determine why this inhibition triggers an antiproliferative effect. To gain insight into the mode of action of the bengamides it is important to know the structure and function of the methionine aminopeptidases. These enzymes represent a unique class of metal dependent aminopeptidases that remove unblocked
N-terminal initiator methionine on either peptides or proteins [
55], both in a co-translational or post-translational mode [
56], suggesting a role in regulating processes rather than general protein degradation [
55]. The removal of the
N-terminal methionine by MetAPs is a critical step in the maturation of many proteins and is essential for further amino terminal modifications [
57]. Therefore, its inhibition has acquired a special importance since it has been demonstrated that MetAP2 is involved in the development of a certain number of tumors [
58]. There are two isoforms of MetAP, types I and II, differing from one another by particular differential sequences [
59,
60]. Thus, the type II enzymes present an α-helical domain of 60 residues inserted in a surface loop of the
C-terminal half of the molecule [
61]. The proximity of this domain to the active site suggests that it is the key for the differentiation in specificity of the two classes. Also, the modifications due to the presence of
N-terminal extensions further differentiate the enzymes [
55]. Moreover, type I is further divided into types Ia, Ic (procaryotes) and Ic (only in eukaryotes). The eukaryotic MetAPs are differentiated from their prokaryotic counterparts by an additional
N-terminal extension. The eukaryotic MetAP2 has two putative zinc finger motifs at the extreme
N-terminus and a highly charged
N-terminus with alternating polyacidic and polybasic stretches in a similarly sized segment. Although the catalytic domains of both MetAPs possess very similar structures, all the residues that form the methionine-binding pocket are different, but the shape of the pocket is conserved. Furthermore, MetAP2 also contains an inserted region contacting the catalytic domain in some of the same area covered by the connector of the type I [
62]. These data support the proposal that MetAP types I and II may present a common functional role. From the biomedical point of view, MetAP2 has attracted much more attention than MetAP1 due to the discovery of MetAP2 and not MetAP1 as the biological target of some of the anti-angiogenic compounds, such as the aforementioned fumagillin (
66) and ovalicin (
67) [
63], together with their synthetic analogues and other synthetic molecules [
64]. Taking into account that methionine aminopeptidases have been identified as antitumor targets for these anti-angiogenic agents, it would be interesting to investigate the role of these enzymes in angiogenesis. However, despite numerous studies stressing the role of aminopeptidases in the formation of new blood vessels, it is still unclear what role MetAP2 plays in regulating angiogenesis [
65].
The identification of MetAPs as the molecular targets for bengamides marked a turning point in the synthesis of potent and selective analogues that might assist not only to the generation of new anticancer leads but also to further investigate and unravel the role of MetAPs in cancer. As part of these investigations, Towbin
et al. [
48] were able to describe by X-ray studies the crystal structure of human MetAP2-bengamide complex, revealing that the real inhibitor was not LAF389 (
64) but its non-acylated derivative, LAF153 (
65) (
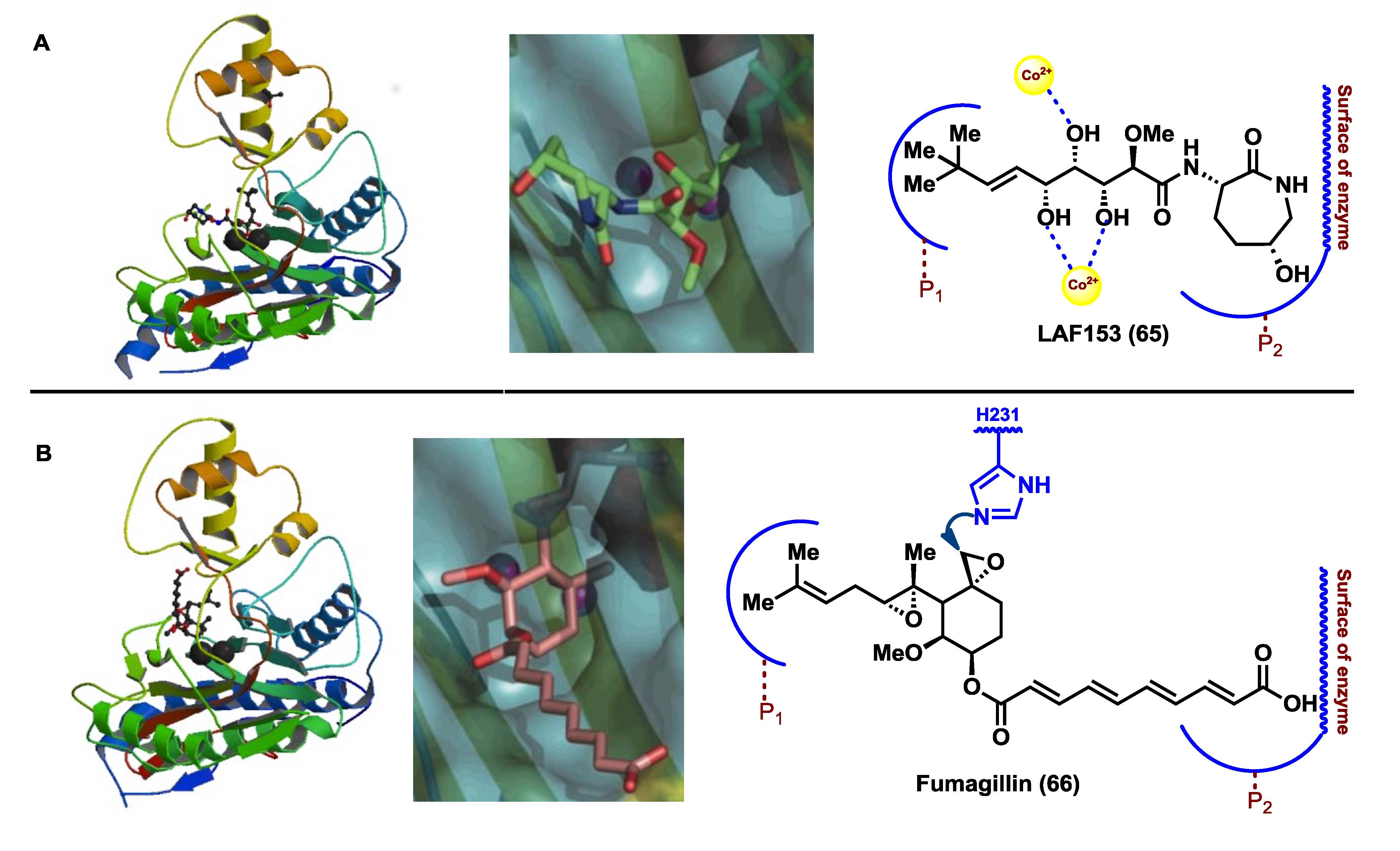
Figure 5). This enzyme-substrate complex structure proved the mode of interaction of these bioactive compounds at the active site of the methionine aminopeptidases. The X-ray structure revealed a critical dinuclear metal center placed as a deep invagination in the surface of the enzyme [
55]. On the other hand, the hydrophobic pocket P1, which contains the residues Phe-219, His-382 and Ala-414, in the innermost portion of the active-site, interacted with the terminal alkyl group of the olefin, while pocket P2, formed at the solvent-exposed surface by the residues of Leu-328, Phe-366 and His-231, hold the caprolactam ring. The coordination of the cobalt ions with the hydroxyl groups at C3, C4 and C5 occurred in a similar way to that observed for peptidic inhibitors of aminopeptidases [
66], forming two octahedral geometric centers (
Figure 6). Known MetAP2 inhibitors such as fumagilllin or ovalicin [
52] exhibit a mode of inhibition that differs from that demonstrated for the bengamides, due to a covalent bond formed by nucleophilic attack of the amine group of a histidine residue (His-231) to one of the oxirane rings contained in these molecules (
Figure 6). In contrast, the bengamides exert their inhibition as a result of multiple interactions. It should be noted that the mode of binding reported for LAF153 is similar to that of a bestatin-derived inhibitor of
Escherichia coli MetAP [
65].
More recently, in 2012, Ye
et al. [
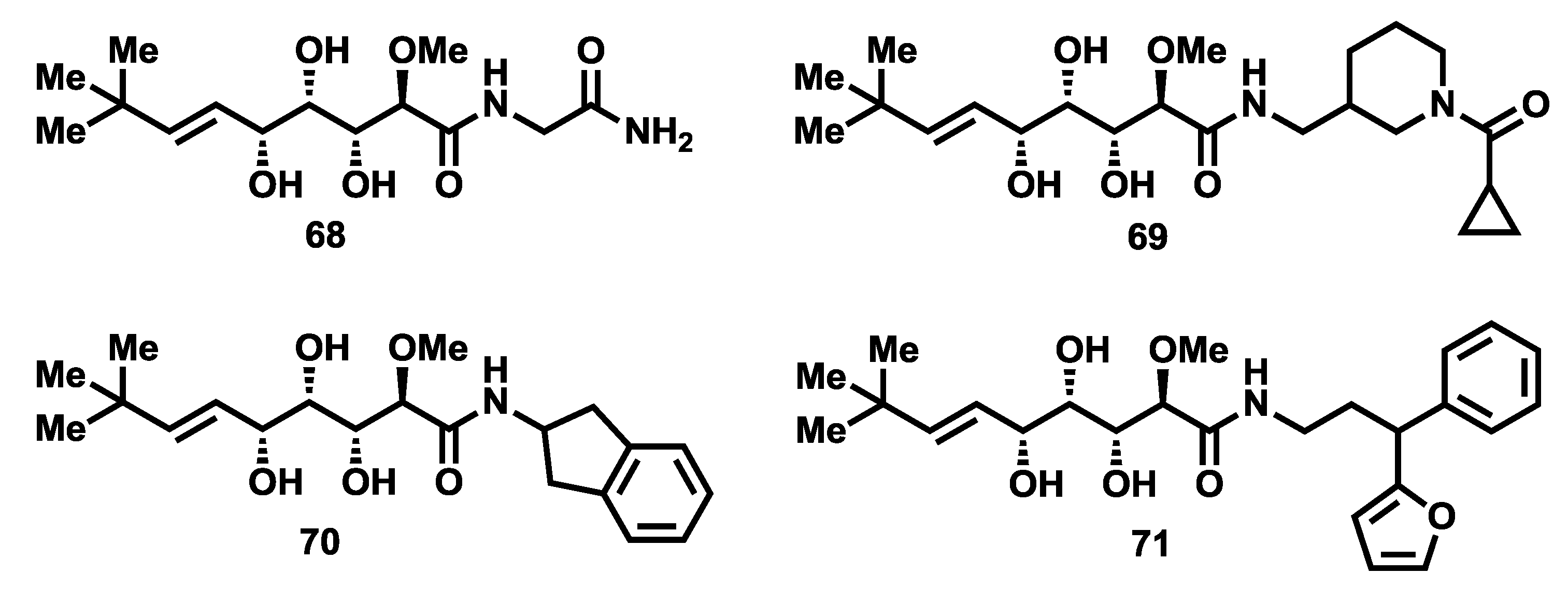
67] uncovered further evidence regarding the role of the bengamides as inhibitors of MetAPs. In particular, they reported the X-ray structures of four bengamide analogues (
68–
71,
Figure 7) in complex with
HsMetAP1 in the Mn (II) form, displaying a similar way of binding for these derivatives with respect to the natural bengamides except for the interaction at P2, due to the different amide structures. These four bengamide derivatives exhibited virtually similar IC
50 values in their inhibition of the Mn (II) form of
HsMetAP1. Since they structurally differ at the amide moiety, this study suggests that the major linkage and stronger interactions at the active site derived from the
tert-butylalkene and the triol fragments, although the modification of the caprolactam ring clearly affected the
in vitro and
in vivo activities, as reported earlier and will be described in
Section 2.4.
Figure 6.
Crystal structures of the MetAP2-inhibitor complexes for LAF153 (PDB-YQYZ) and Fumagillin (PDB-1BOA) and mode of interactions at the active site. Note: this figure is adapted with permissions from [
48]. Copyright © 2003, the American Society for Biochemistry and Molecular Biology.
Figure 6.
Crystal structures of the MetAP2-inhibitor complexes for LAF153 (PDB-YQYZ) and Fumagillin (PDB-1BOA) and mode of interactions at the active site. Note: this figure is adapted with permissions from [
48]. Copyright © 2003, the American Society for Biochemistry and Molecular Biology.
Figure 7.
Molecular structures of bengamide analogues 68–71.
Figure 7.
Molecular structures of bengamide analogues 68–71.
With regard to the role and involvement of
HsMetAP1 in cancer, Liu
et al. [
68] elucidated the physiological function of this enzyme in cell proliferation by studying the X-ray structure of the complex
HsMetAP1/pyridine-2-carboxylic acid inhibitors [
69,
70], demonstrating that MetAP1 plays an important role in G2/M phase of the cell cycle. Further biological investigations in this field were accomplished to elucidate the key protein or proteins that were affected by the inhibition of methionine aminopeptidases and, as a consequence, produced the antitumor response. In this research, these authors identified and validated the proto-oncogene
c-Src, involved in the development, growth, progression and metastasis of a number of human cancers [
71], as a substrate for both MetAP1 and MetAP2
in vivo and
in vitro. Thus, this research group showed that inhibition of MetAPs by the nonselective inhibitor bengamide A (
38) altered the subcellular distribution of
c-Src. This alteration significantly decreased its tyrosine kinase activity, and caused a remarkable delay in cell-cycle progression. Therefore, these results established a link between
c-Src and MetAP and suggested that inhibition of MetAPs could indirectly impair the functions of
c-Src and likely other oncogenes that are essential for tumor growth.
On the other hand, recent enzymatic studies has shown that depletion of MetAP2 by siRNA did not produce an inhibition of endothelial cell growth and, even more intriguing, these MetAP2-depleted endothelial cells remained responsive to inhibition by either bengamides or fumagillin [
72]. In view of the foregoing, this data seems to indicate that MetAP2 is not required for endothelial cell proliferation and that MetAP2 may not be the target for the anti-angiogenic effect of the bengamides and fumagillin despite all the biological studies and evidence, exposed before, clearly supported the methionine aminopeptidases as the biological targets of the bengamides. The activity of these compounds in MetAP2-depleted endothelial cells is particularly puzzling because both classes of inhibitors have been co-crystallized with human MetAP2 [
48,
63]. According to the authors, one possible explanation to explain these controversial results is that another member of the MetAP family exists in humans that could be expressed in endothelial cells but able to be inhibited by both bengamide and fumagillin. Nonetheless, the majority of experimental evidence supports the notion that MetAP2 is essential for endothelial cell viability [
73], although further biological studies are clearly required to gain insight into all these uncertainties.
Finally, the NF-κB (nuclear factor kappa B) inhibitory activity exhibited by the bengamides, recently discovered by Crews
et al. [
28], could be the responsible for their observed antitumor activities due to the close relationship between tumorigenesis and inflammation. In this biological study, qPCR analysis revealed that this inhibition affected the expression of the pro-inflammatory cytokines TNFα, IL6 and MCP-1. Thus, the effects of the bengamides on these key targets in the NF-κB pathway may contribute to their significant antitumor activity.
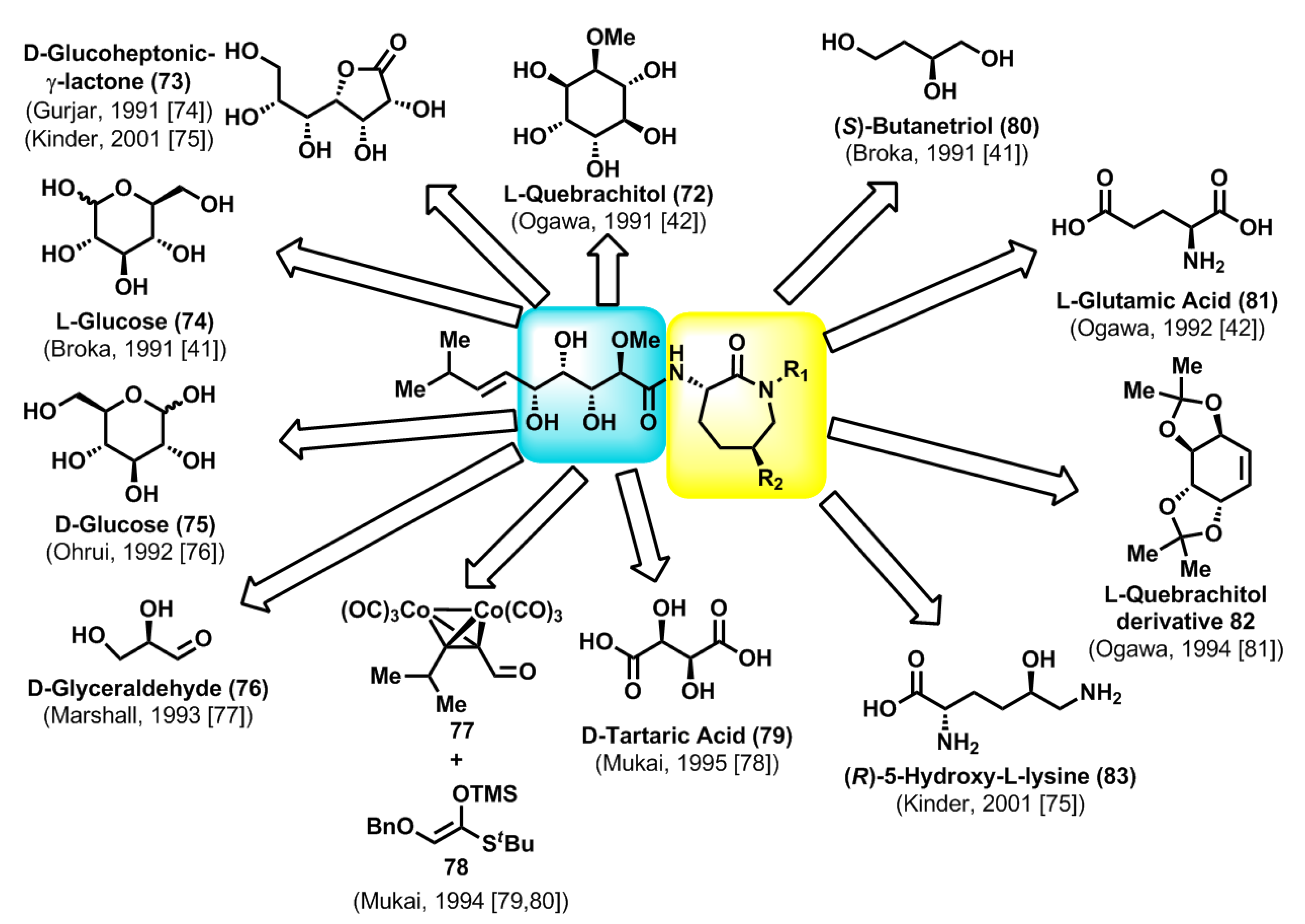
2.3. Chemical Synthesis of Natural Bengamides
The novel molecular structures of the bengamides, coupled with their antitumor properties, rapidly propelled them to the forefront of chemical research. Thus, a few years after the discovery of the first bengamides by Crews and co-workers, Ogawa [
42] reported the first total synthesis of bengamide E, which was accomplished starting from
l-quebrachitol (
72). The various total syntheses reported in the subsequent years after Ogawa’s first total synthesis employed monosaccharides such as
d-gluconolactone (
73) [
74,
75],
l- and
d-glucose (
74 and
75) [
41,
76],
d-glyceraldehyde (
76) [
77] or the
d-tartaric acid (
79) [
78] as suitable chiral starting materials, taking into account the polyhydroxylated nature of the side chain. In contrast, the first synthesis conducted by Mukai was based on Mukaiyama-type aldol reactions starting from the achiral precursor
77 and the silylenol ether
78 [
79,
80], although subsequent improvements by the same author of his first synthesis of bengamide E were carried out by use of
d-tartaric acid as starting material [
78]. On the other hand, whereas the synthesis of bengamide E did not require the preparation of the caprolactam moiety, the synthesis of other members, such as bengamides A or B, required the stereoselective construction of this caprolactam fragment. In this sense, (
S)-butanetriol (
80) [
41],
l-glutamic acid (
81) [
42],
l-quebrachitol derivative
82 [
81] or (
R)-5-hydroxy-
l-lysine (
83) [
75] were the starting chiral materials used for the syntheses of the caprolactam moieties (
Scheme 2). All the total syntheses reported in the literature during the period corresponding to 1991–2001 were reviewed by Kinder [
82] and, therefore, are not described in detail in the present article.
Scheme 2.
Total syntheses of bengamides in the period 1991–2001: general retrosynthetic analyses.
Scheme 2.
Total syntheses of bengamides in the period 1991–2001: general retrosynthetic analyses.
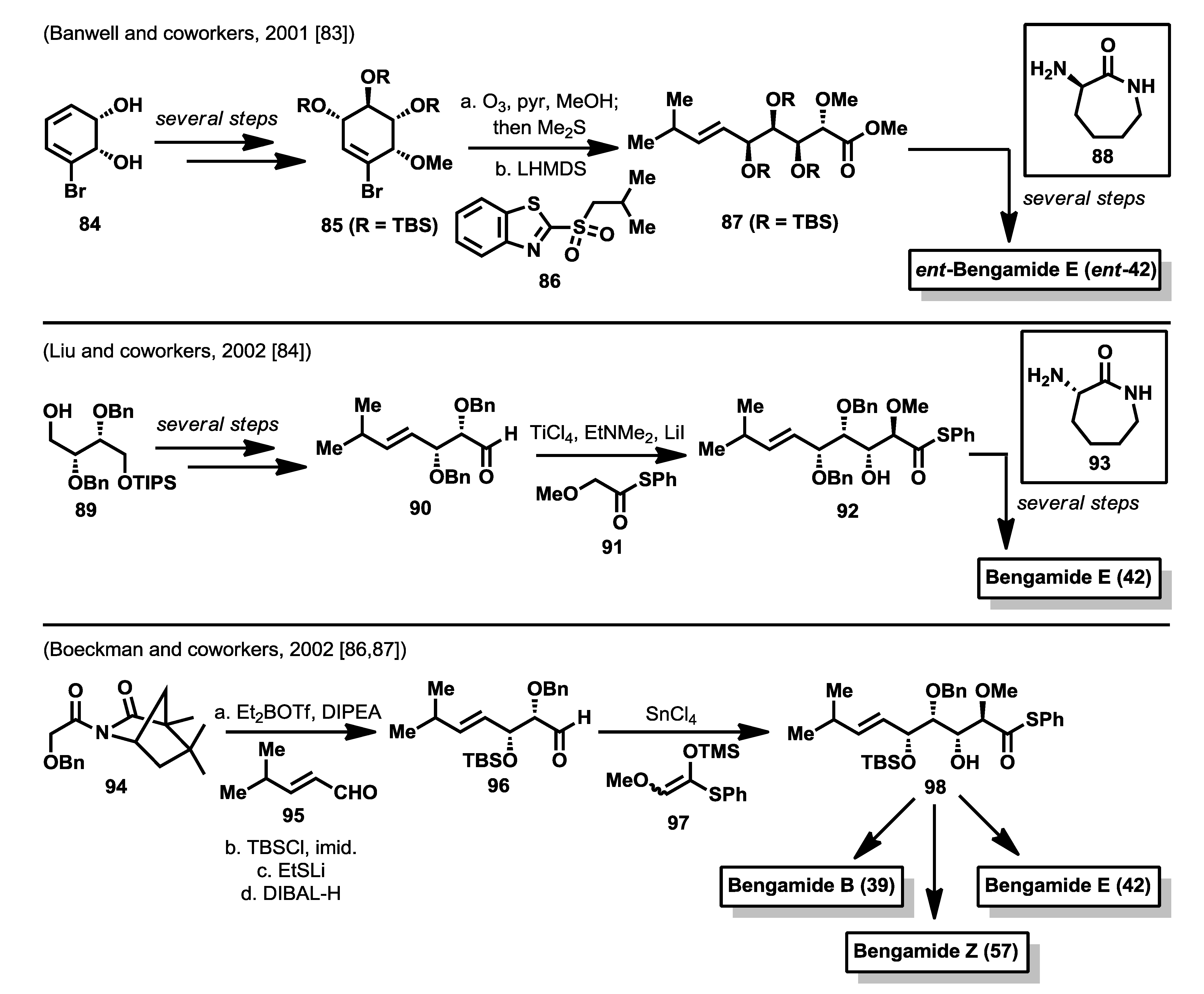
Soon after the synthesis by Kinder
et al. [
75] reported in 2001, Banwell and McRae reported a different synthesis of the enantiomer of bengamide E [
83] via a chemoenzymatic approach, in which starting from 1,2-dihydrocatechol
84, readily available by microbial oxidation of bromobenzene, compound
85 was prepared, after several steps involving oxirane ring formation, oxirane ring opening and protecting groups manipulations. Ozonolysis of this cyclohexene derivative
85, conducted in CH
2Cl
2-MeOH, afforded the corresponding aldehydic ester, which was exposed to the anion of sulfone
86 to yield
E alkene
87 almost exclusively. Ester
87 was then hydrolyzed and the resulting acid coupled with cyclo-
l-Lysine
88 to afford, after silyl ether cleavage, the enantiomer of bengamide E (
ent-
42). One year later, Liu and coworkers published the synthesis of bengamide E starting from alcohol
89, obtained from diisopropyl
d-tartrate and based on an aldol reaction strategy for the construction of the polyol system [
84]. Thus, after several steps in which the double bond was introduced via a Julia-Kocienski reaction, the aldehyde
90 resulting from
89 was subjected to an aldol reaction with thioester
91, under Annuziata conditions [
85], to obtain a 1:1 mixture of the
anti diastereoisomers. After separation of both isomers, the correct
anti diastereoisomer (compound
92) was coupled with cyclo-
l-lysine (
93), followed by Birch reduction of the benzyl ether protecting groups to obtain bengamide E (
42). As in the Mukai’s synthesis, the aldol approach to the bengamides is marked by the absence of stereoselectivity (
Scheme 3).
Scheme 3.
Total syntheses of bengamides by Banwell, Liu and Boeckman (2001–2002) [
83,
84,
86,
87].
Scheme 3.
Total syntheses of bengamides by Banwell, Liu and Boeckman (2001–2002) [
83,
84,
86,
87].
A new approach to the bengamides, based again on aldol reactions, was conducted by the Boeckman’s group who described the total synthesis of bengamides B (
39), E (
42) and Z (
57) [
86,
87]. For these synthesis, the C10 side chain was entirely constructed through sequential
syn and
anti asymmetric aldol reactions from the α,β-unsaturared aldehyde
95. The first aldol reaction was carried out with the chiral acetimide
94, which, after treatment with Et
2BOTf and DIPEA, was reacted with
95 to furnish the corresponding
syn aldol product in excellent diastereoselectivity (>24:1). The elaboration of this product for the second aldol reaction led to the aldehyde
96 which was subjected to the action of the silylenol ether
97 in the presence of SnCl
4 to obtain compound
98 in 73% yield and 11.5:1 diastereomeric ratio. This compound
98 represents the common intermediate for the syntheses of bengamides B, E and Z by direct coupling of the thioester
98 with the corresponding 2-aminocaprolactams. For the syntheses of the caprolactams contained in bengamides B and Z, the authors employed D-aspartic acid as the chiral starting material that allowed the construction of (
R)-2-bromobutane-1,4-diol as a key intermediate for their syntheses. However, for the bengamide B synthesis, the final benzyl ether deprotection, by the action of Na in NH
3, produced the cleavage of the fatty acid ester present in the caprolactam fragment, resulting in a need for a strategy modification to build the polyketide side chain. This modification consisted of the replacement of the benzyl ether group at C4 for a 2-naphtylmethyl group, which was cleaved at the end of the synthesis by sequential treatment with DDQ and PPTS that did not affect the integrity of the fatty acid ester moiety, allowing the completion of the synthesis (
Scheme 3).
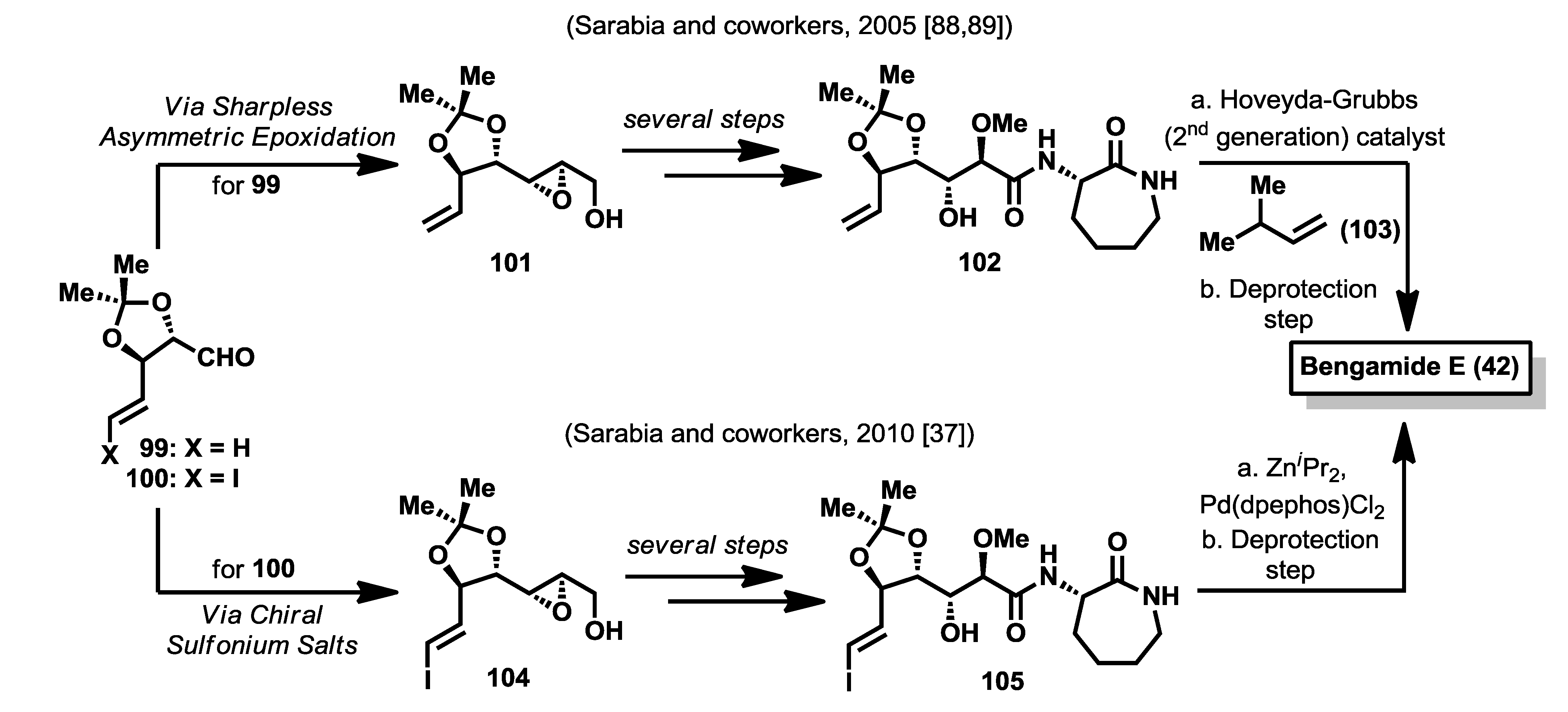
In 2005, Sarabia
et al. [
88,
89] reported a first synthesis of bengamide E based on three key steps that were: (1) a cross olefin metathesis to introduce the terminal alkyl group; (2) an epoxide ring opening reaction to construct the polyketide side chain; and (3) an amide bond formation to introduce the caprolactam moiety. Thus, starting from aldehyde
99 and, through the Sharpless methodology of asymmetric epoxidation, epoxy alcohol
101 was prepared in a complete stereoselective fashion. After a regioselective ring opening reaction of
101 with MeOH, chemoselective oxidation of the primary alcohol to the acid and coupling with
93 provided
102 which was subjected to a cross olefin metathesis with olefin
103, mediated by the 2nd generation Hoveyda-Grubbs catalyst, to afford the
E-substituted olefin in good yield and complete stereoselectivity. Bengamide E was finally synthesized after a deprotection step of the acetal group (
Scheme 4). In a second and improved synthesis by the same authors in 2010 [
37], and as an application of the group’s methodology in the field of amide-stabilized sulfur ylides [
90], aldehyde
100 was reacted with a chiral sulfonium salt to obtain in good yield and excellent diastereoselectivity (>98%) an epoxy amide that was reduced to epoxy alcohol
104. From this epoxy alcohol, the methodology proceeded in a similar manner as the previous synthesis, although the processes of ring opening reaction and the formation of the substituted olefin were remarkably improved with respect to the first synthesis. Thus, the ring opening reaction of
104 was carried out by treatment of MeOH in the presence of B(OMe)
3, as an extension of the methodology developed of Miyashita [
91]. After oxidation and coupling reactions of the ring opening product, the resulting vinyl iodide
105 was reacted with diisopropylzinc, catalyzed by palladium (0), to install the terminal olefinic substituent and, after a deprotection step, bengamide E was prepared in a shorter and more efficient route compared with the first one described in 2005 (See
Table 4). As will be discussed in the following section, one of the advantages of this latter synthetic strategy, as others described in the literature, is the versatility and convergency that allows one to generate bengamide analogues modified at various parts of the structure by suitable use of different reagents employed along the synthetic pathway previously delineated.
Scheme 4.
Total syntheses of bengamide E by Sarabia (2005 and 2010) [
37,
88,
89].
Scheme 4.
Total syntheses of bengamide E by Sarabia (2005 and 2010) [
37,
88,
89].
Table 4.
Synthesis of natural bengamides: A summary.
Table 4.
Synthesis of natural bengamides: A summary.
| Bengamide | Author/Year [Ref.] | Starting Material | Number of Steps 1 | Overall Yield 2 (%) |
|---|
| E (42) | Ogawa/1991 [42] | l-quebrachitol | 15 | 0.40% |
| E (42) | Broka/1991 [41] | l-glucose | 16 | 11.20% |
| B (39) | Broka/1991 [41] | l-glucose | 16 | 5.90% |
| E (42) | Ohrui/1992 [76] | d-glucose | 17 | 0.80% |
| A (38) | Ogawa/1992 [40] | l-quebrachitol | 16 | 0.24% |
| E (42) | Marshall/1993 [77] | d-mannose | 12 | 10.71% |
| E (42) | Mukai/1994 [79] | Isobutyraldehyde | 18 | 3.06% |
| B (39) | Ogawa/1994 [81] | l-quebrachitol | 16 | 0.09% |
| E (42) | Mukai/1995 [80] | Isobutyraldehyde | 18 | 4.70% |
| E (42) | Mukai/1995 [78] | DIDT 3 | 16 | 5.00% |
| B (39) | Kinder/2001 [75] | d-gluconolactone | 7 | 7.21% |
| E (42) | Kinder/2001 [75] | d-gluconolactone | 7 | 5.43% |
| ent-E (ent42) | Banwell/2001 [83] | Bromobenzene | 12 | 5.70% |
| E (42) | Liu/2002 [84] | DIDT 3 | 10 | 9.74% |
| B (39) | Boeckman/2002 [86,87] | Ethyl 2-hydroxyacetate | 12 | 10.86% |
| E (42) | Boeckman/2002 [86] | Isobutyraldehyde | 10 | 23.56% |
| Z (57) | Boeckman/2002 [86] | Isobutyraldehyde | 10 | 17.86% |
| E (42) | Sarabia/2005 [89] | Diethyl d-tartrate | 18 | 2.85% |
| E (42) | Sarabia/2010 [37] | Diethyl d-tartrate | 14 | 4.15% |
| E (42) | Li/2013 [92] | d-glucose | 16 | 4.00% |
| E (42) | Prasad/2013 [93] | Diethyl d-tartrate | 12 | 7.05% |
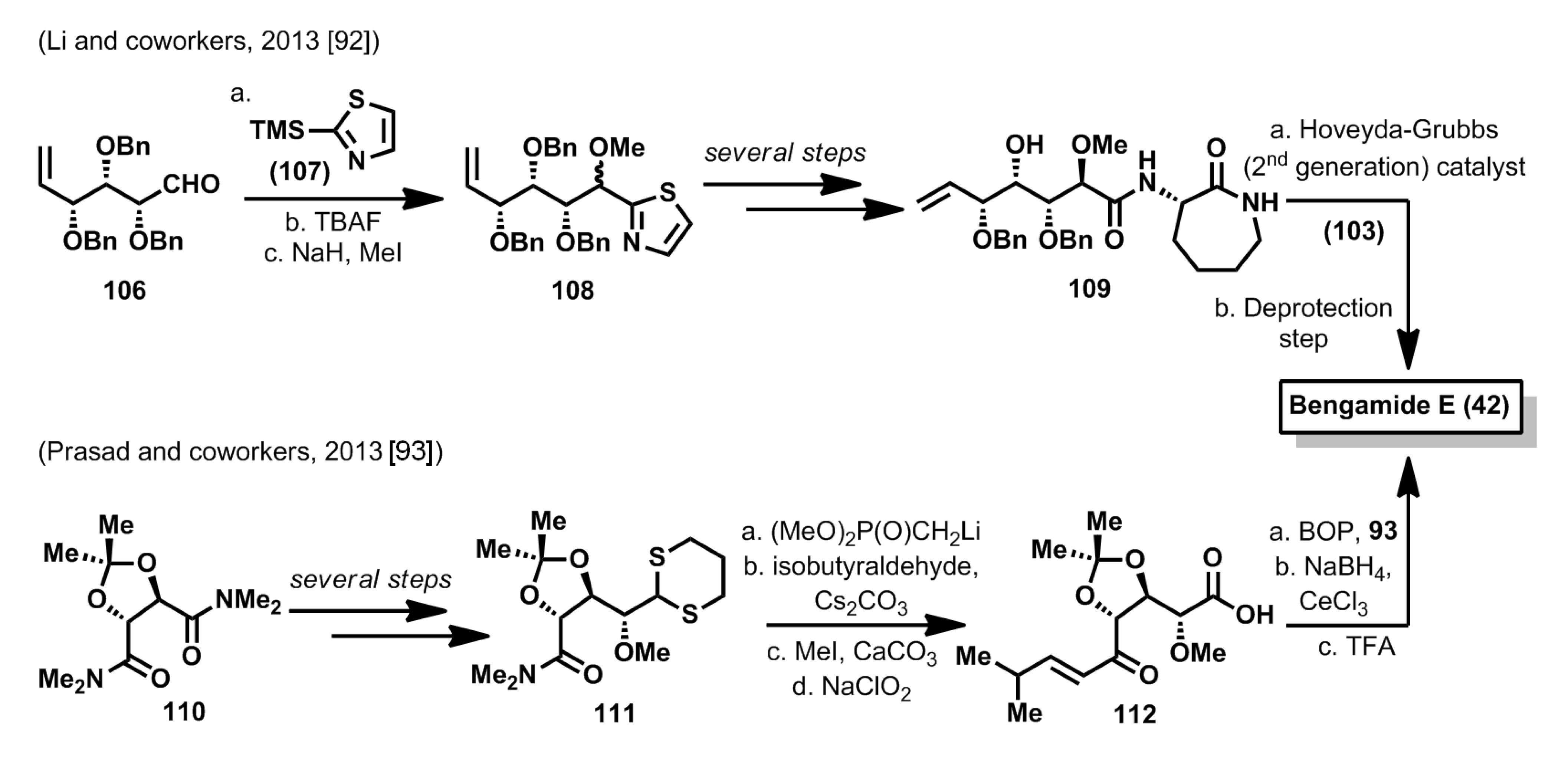
Two more total syntheses of bengamide E have been reported in the last year. In the first one, by Li
et al. [
92], bengamide E, together with some new analogues, were synthesized from hex-5-enal derivative
106, which was prepared from α-methyl-
d-glucopyranoside through a known synthetic sequence. Thus, by use of the Dondoni methodology [
94], aldehyde
106 was reacted with thiazol derivative
107 to afford a 1:1 diastereomeric mixture of compound
108, after TBAF treatment and methylation of the resulting alcohol. After subsequent transformation of the thiazol moiety into the acid and coupling with lactam
93, an olefin cross metathesis provided the bengamide E precursor, which was converted to the final product after deprotection steps (
Scheme 5). A second synthesis of bengamide E has been reported by Prasad
et al. [
93] starting from the bis(dimethylamide) of
d-tartaric acid
110, whose desymmetrization by reaction with the anion of 1,3-dithiane, led to compound
111, after a reduction and a methylation steps [
95]. After the synthesis of a β-ketophosphonate, a Horner-Wadsworth-Emmons olefination allowed the stereoselective introduction of the substituted olefinic fragment. Finally, dithiane oxidation that led to
112, followed by a caprolactam coupling, reduction of the ketone with NaBH
4/CeCl
3 and final acidic deprotection allowed the completion of the synthesis of bengamide E (
Scheme 5).
Scheme 5.
Total syntheses of bengamide E by Li and Prasad (2013) [
92,
93].
Scheme 5.
Total syntheses of bengamide E by Li and Prasad (2013) [
92,
93].
Throughout this section the total syntheses of natural bengamides has been reviewed. As a summary,
Table 4 comprises all the synthesis achieved up to now, indicating the number of steps and overall yields for comparative purposes. In this sense, it highlights the fact that over the years more efficient and shorter syntheses of the bengamides have been reported and has allowed preparation not only of natural bengamides in sufficient amounts for biological and clinical assays, but also the synthesis of analogues for identification of more potent bengamides from the biological point of view, as will be discussed further in the following section.
2.4. Synthesis of Bengamide Analogues and Biological Evaluation
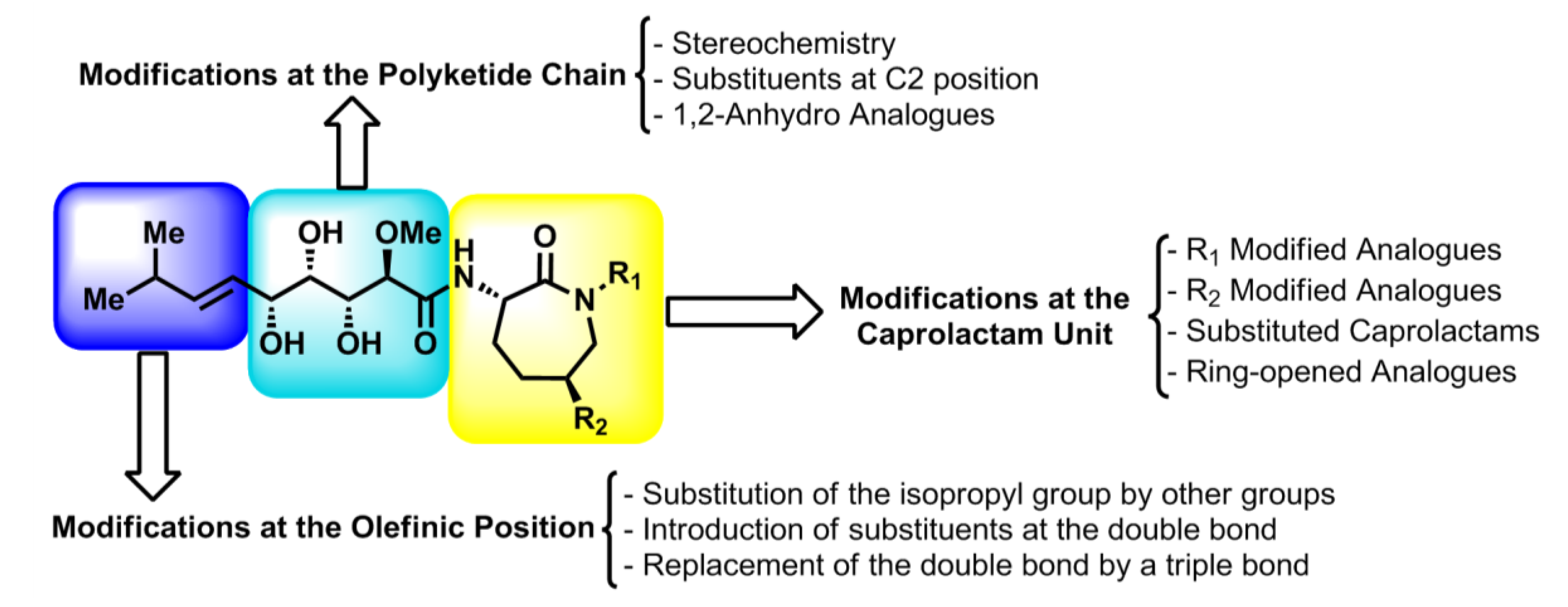
The structures of the bengamides are amenable to several modifications that could lead to the establishment of structure-activity relationships (SAR), which are essential for the design and synthesis of new potential drug candidates. Thus, configurational modifications of the different stereocenters located at the polyketide chain, changes of the substituent located at the terminal olefinic position or introduction of modified caprolactams have been reported by different authors (
Figure 8), contributing to the formulation of more potent bengamide derivatives that are detailed throughout this section.
Figure 8.
Designed bengamides: structural fragments amenable to modifications.
Figure 8.
Designed bengamides: structural fragments amenable to modifications.
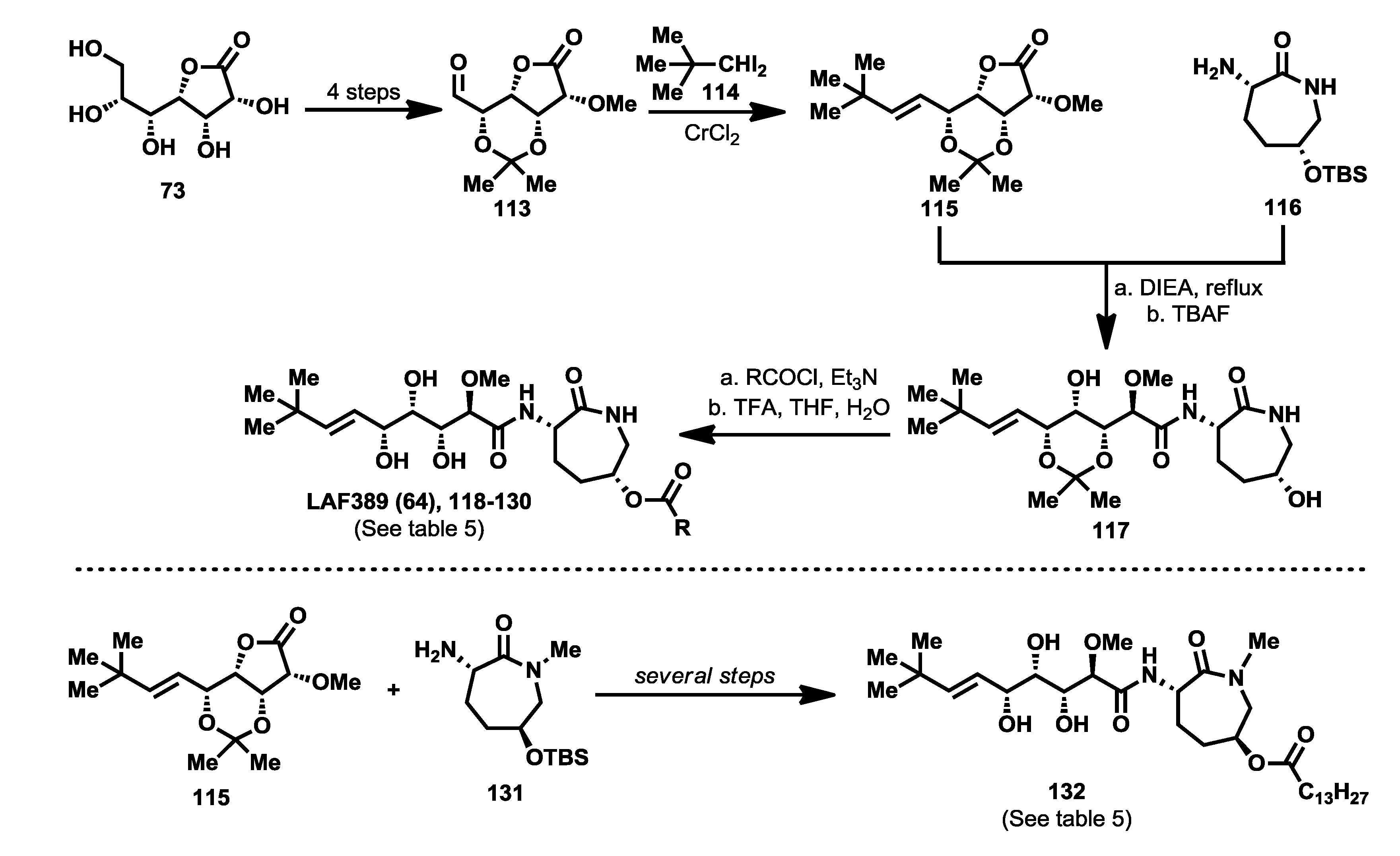
The first bengamide analogues were synthesized by Kinder’s group in 2001 [
46], consisting of ester-modified derivatives of bengamide B, that led to new bengamides with impressive
in vitro and
in vivo antitumor activities but with poor water solubility. In order to address this drawback, several key modifications were considered such as simplification of the lactam moiety and modification of the alkyl group at the olefinic position. The synthetic pathway used for the preparation of these analogues was based on the previous syntheses of bengamides B and E reported by the same author, starting from
d-gluconolactone
73 and replacing the terminal isopropyl group by a
tert-butyl [
75]. Thus, from aldehyde
113, readily prepared from
73, the introduction of the substituted
tert-butyl
E-olefin was achieved by means of a Takai-Utimoto olefination with 1,1-diiodoneopentane (
114) [
96] to obtain key lactone
115 in a reasonable good yield (63%). The reaction of this lactone with caprolactam
116, prepared from the commercially available (5
R)-5-hydroxy-
l-lysine, afforded bengamide derivative
117, which by reaction with either an acid chloride or a carboxylic acid, followed by acetal cleavage, provided an array of bengamide A analogues (
118–
130), including the aforementioned LAF389 (
64). Taking into account that all these analogues possess the opposite configuration at the C-5′ position of the caprolactam ring with respect to the natural bengamides, these authors prepared similarly the
tert-butyl analogue of bengamide B (
132) by reaction of lactone
115 with the amino caprolactam
131, which was prepared from
116 (
Scheme 6).
The cytotoxicity of these bengamide analogues was evaluated by measurement of their antiproliferative activities against MDA-MB-435 human breast carcinoma cells (
in vitro and
in vivo) (
Table 5), and as a result, some important conclusions related to the structure/activity relationship were established. Analogue
64, which replaces the myristate ester by a cyclohexane carboxylic ester and whose configuration at C5′ was inverted respected to bengamide B, was the most potent analogue of the series (both
in vitro and
in vivo). This result delivered interesting outcomes concerning both the configuration of the lactam at the 5′-position together with the increased water solubility due to the substitution of the myristate chain with fewer lipophilic groups. Additionally, compounds
64 and
124 were found to cause tumor regression with minor losses in body weight, with compound
64, corresponding to LAF389, being the most potent
in vivo, producing 29% tumor regression at 100 µmol/kg. Interestingly, the bengamide B analogue
132 displayed similar
in vitro and
in vivo potency as its natural counterpart, bengamide B [
46].
Scheme 6.
Syntheses of bengamide analogues by Kinder (2001) [
46].
Scheme 6.
Syntheses of bengamide analogues by Kinder (2001) [
46].
Table 5.
Cytotoxic activities (IC50, μM) of ester modified analogues of bengamides.
In light of these promising results, in particular for LAF389 (
64), Novartis developed in 2003 a large-scale and optimized synthesis of this analogue, which allowed its preparation on more than a 100 g scale for preclinical and clinical studies [
49]. Thus, LAF389, as a new potential antitumor lead, was launched to clinical trials. However, the lack of clinical activity and the finding of unpredictable cardiovascular toxicity caused the compound to be halted for further clinical investigation and future clinical assays were canceled [
97]. A possible reason for its failure may be due to its lack of selectivity in the inhibition of both MetAP1 and MetAP2, as well as its poor bioavailability, all this despite the potent cytotoxicity and significant
in vivo activity exhibited by LAF389. Nevertheless, these results prompted the search for new, more potent analogues with better selectivity and solubility properties.
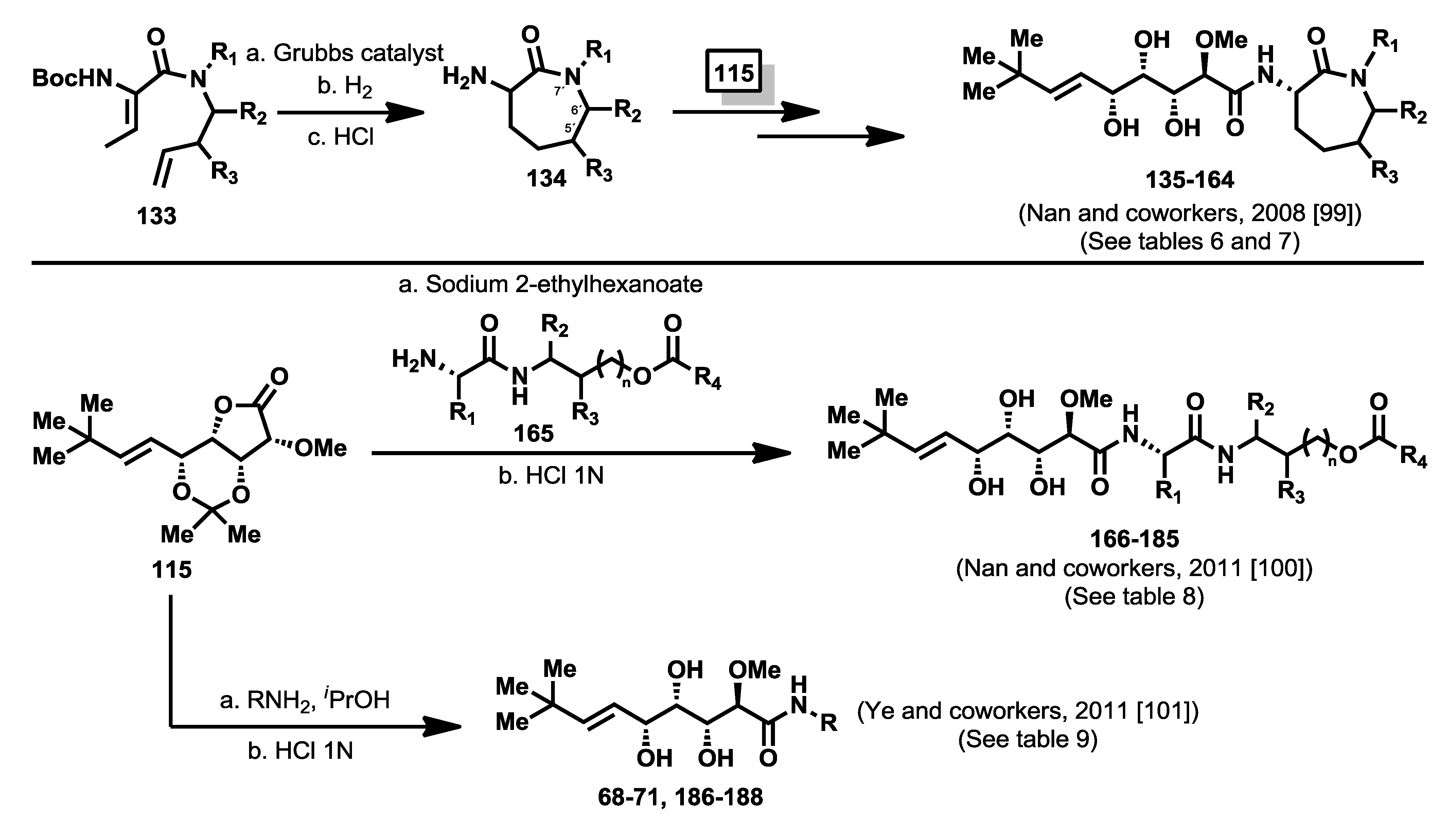
In 2008, Nan and co-workers, following the ring-closing metathesis methodology described previously by them for the synthesis of functionalized amino caprolactams [
98], described the synthesis of new bengamide analogues modified at the caprolactam moiety at positions 5′-, 6′-, and 7′- by reaction of lactone
115 with amino caprolactams type
134, obtained from the diene precursors
133, with an overall yield between 25% and 70% [
99] (
Scheme 7). The bengamide library (
135–
164) was evaluated against MDA-MB-435 human breast carcinoma cells (
Table 6 and
Table 7). The biological results showed that whereas modifications at position 5′ apparently did not seem to affect to the activity (which is in accordance with Novartis studies of modifications in this position), the 6′-substituted derivatives were well tolerated. Interestingly,
N-substituted bengamide analogues exhibited an increase in activity when the chain length of the R group was enlarged, with the best results provided for the two-carbon chain length (case of analogue
142 with a IC
50 of 17 nM). However, these effects were not clear when considering aryl containing analogues (cases of
144–
146), and in fact it decreased for simple
N-alkyl amines. This study resulted in the identification of a new potent analogue, the
N-acetyl derivative
142 [
99], which displayed greater activity (17 nM) and greater water solubility (10 mg/mL) than LAF389 (1.0 mg/mL) (
Table 6). As the data reflect in
Table 7, substitution at C5′ and C6′ positions led to less potent analogues than those showed in
Table 6 with the exception of the diastereomeric mixture of LAF385 (
64) and its C5′ epimer (
64′) whose cytotoxicity was similar to pure
64. A more profound modification of the caprolactam moiety led Nan and coworkers [
100] to the discovery in 2011 of a series of novel ring-opened bengamide analogues (
166–
185) in which the caprolactam ring was replaced by a linear peptidic chain (
Scheme 7). The biological evaluation of these new 20 analogues against MDA-MB-435 human breast carcinoma cells revealed compound
172, as one of the most potent antitumor compounds belonging to the bengamide family, with an IC
50 value of 4.0 nM and improved water solubility (1.0 mg/mL), when compared with the solubility of bengamide B (0.002 mg/mL) (
Table 8). This work has delivered relevant results that could be very useful for further SAR studies and novel modified analogues. In the same year, Ye
et al. [
101] outlined the design of a new type of bengamide derivatives displaying a strong inhibition of
Mycobacterium tuberculosis methionine aminpeptidases, but weak inhibition against human methionine aminopeptidases. While the triol moiety and the alkene substituent were kept to maintain the interaction with the two metal ions at P1, the caprolactam moiety was replaced with various amide moieties to study their interactions in P2 pocket. The syntheses of these analogues were performed by coupling of lactone
115 with several amines to obtain, after a deprotection step, the analogues
68–
71 and
186–
188 (
Scheme 7). Their inhibitory activities were tested toward
MtMetAP1a and
MtMetAP1c activated by the metal ions Co
2+, Mn
2+, Ni
2+ and Fe
2+. All compounds were almost inactive against the Co
2+ or Fe
2+ forms of
MtMetAP1c, and completely inactive against the Ni
2+ one, proving the selectivity for the Mn
2+ form in the low μM range. Additionally, compound
188 exhibited the best antitubercular activity against both replicating and non replicating
Mycobacterium tuberculosis, although their activities against human MetAPs were not completely suppressed according to the evaluation against human K562 cells that showed significant activity for some of these bengamide analogues (
Table 9).
Scheme 7.
Syntheses of caprolactam-modified bengamide analogues [
99,
100,
101].
Scheme 7.
Syntheses of caprolactam-modified bengamide analogues [
99,
100,
101].
Table 6.
Cytotoxic activities (IC
50, μM) of caprolactam-modified analogues of bengamides by Nan
et al. (2008) [
99].
Table 7.
Cytotoxic activities (IC
50, μM) of caprolactam-modified analogues of bengamides by Nan
et al. (2008) [
99].
Table 8.
Cytotoxic activities (IC
50, nM) of ring-opened analogues of bengamides by Nan
et al. (2011) [
100].
Table 9.
Activities (IC
50, μM) against
MtMetAPs and k56 cells of amide-modified analogues of bengamides by Ye
et al. (2011) [
101].
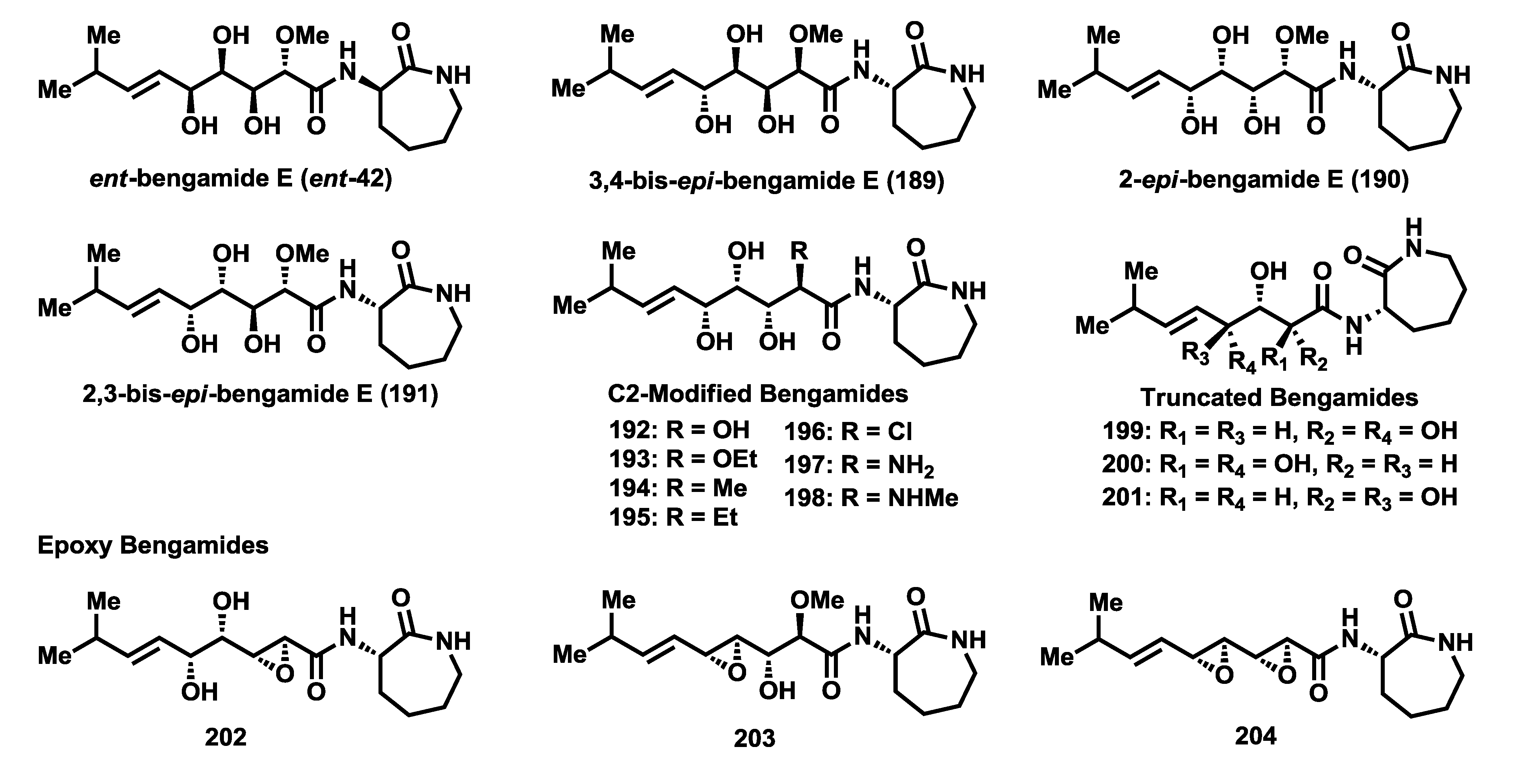
The essential role that the polyhydroxylated chain of the bengamides plays in their interaction with methionine aminopeptidases was demonstrated with the synthesis of various polyketide-modified analogues and their corresponding biological evaluations. The effect of the configuration of the polyketide chain on the cytotoxic potency was evaluated through the 3,4-
epi-, 2-
epi- and 2,3-
epi-bengamide E analogues (
189–
191) [
92,
102,
103] (
Figure 9). Together to all these stereo-isomers analogues of bengamide E, the enantiomer of the natural product (
ent-42) was prepared and biologically tested [
83]. It was also possible to assess the biological significance of the methoxyl group at C2 position through the synthesis of the C2-modified analogues
192–
198 [
89,
103]. Finally, the influence of the constitution of the polyketide chain on the biological activity was similarly studied through truncated analogues
199–
201 [
92] and the epoxy derivatives
202–
204 [
103], which were designed and prepared as potential fumagillin-bengamide hybrids. The biological results obtained by cytotoxic evaluations of all these analogues against a panel of different tumor cell lines revealed that this polyketide chain was essential to retain the biological activity of this class of compounds and that any change, configurational or constitutional, of this chain produced the complete loss of cytototoxic potency. Thus,
Table 10 displays only the most active analogues of this series. Consequently, it was clearly demonstrated that the polyol system was not amenable to modification, supporting the notion of the strong involvement of this polyketide chain in binding with the active site of the MetAP enzymes through metal ion coordination with the hydroxyl groups.
Figure 9.
Polyketide-modified bengamides.
Figure 9.
Polyketide-modified bengamides.
Table 10.
Cytotoxic activities (IC50, μM) against different tumor cells lines of selected polyketide-modified bengamides.
Table 10.
Cytotoxic activities (IC50, μM) against different tumor cells lines of selected polyketide-modified bengamides.
| Bengamide | Cell Line | IC50 | Bengamide | Cell Line | IC50 |
|---|
| 190 | HT29 | 85.2 ± 17.0 | 197 | HT29 | 38.5 ± 4.8 |
| MDA-MB-435 | 100.2 ± 13.3 | MDA-MB-435 | 70.7 ± 12.0 |
| HT1080 | 93.2 ± 8.1 | HT1080 | 30.2 ± 6.1 |
| HL60 | 60.3 ± 15.1 | HL60 | 25.3 ± 5.2 |
| BAEC | 69.7 ± 3.2 | BAEC | 28.9 ± 7.2 |
| 192 | HCT-116 | 25.49 | 198 | HT29 | 10.6 ± 1.2 |
| MDA-MB-435 | 11.36 | MDA-MB-435 | 12.5 ± 3.0 |
| MCF-7 | 8.35 | HT1080 | 5.8 ± 0.1 |
| - | - | HL60 | 9.2 ± 50.6 |
| - | - | BAEC | 4.1 ± 1.3 |
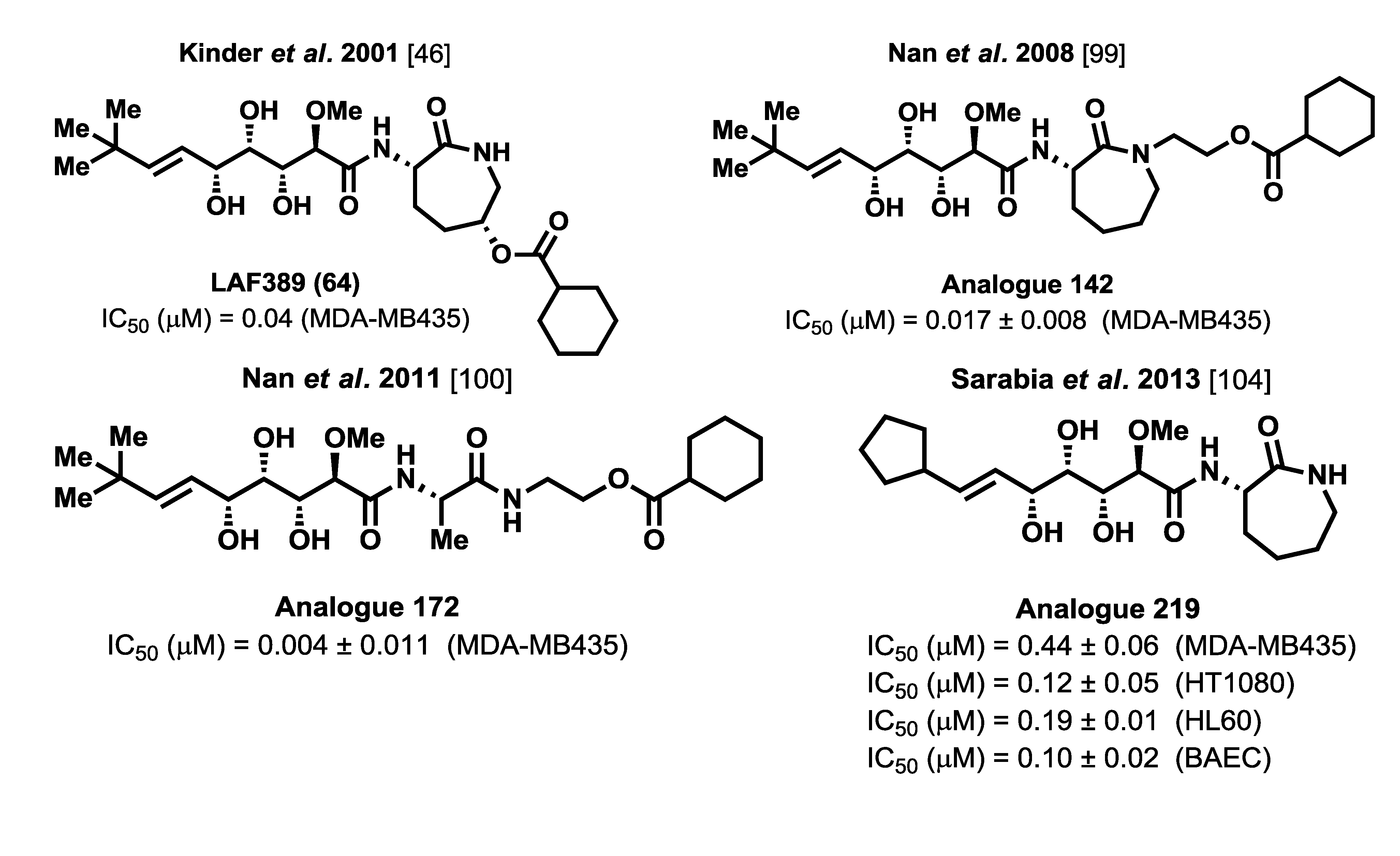
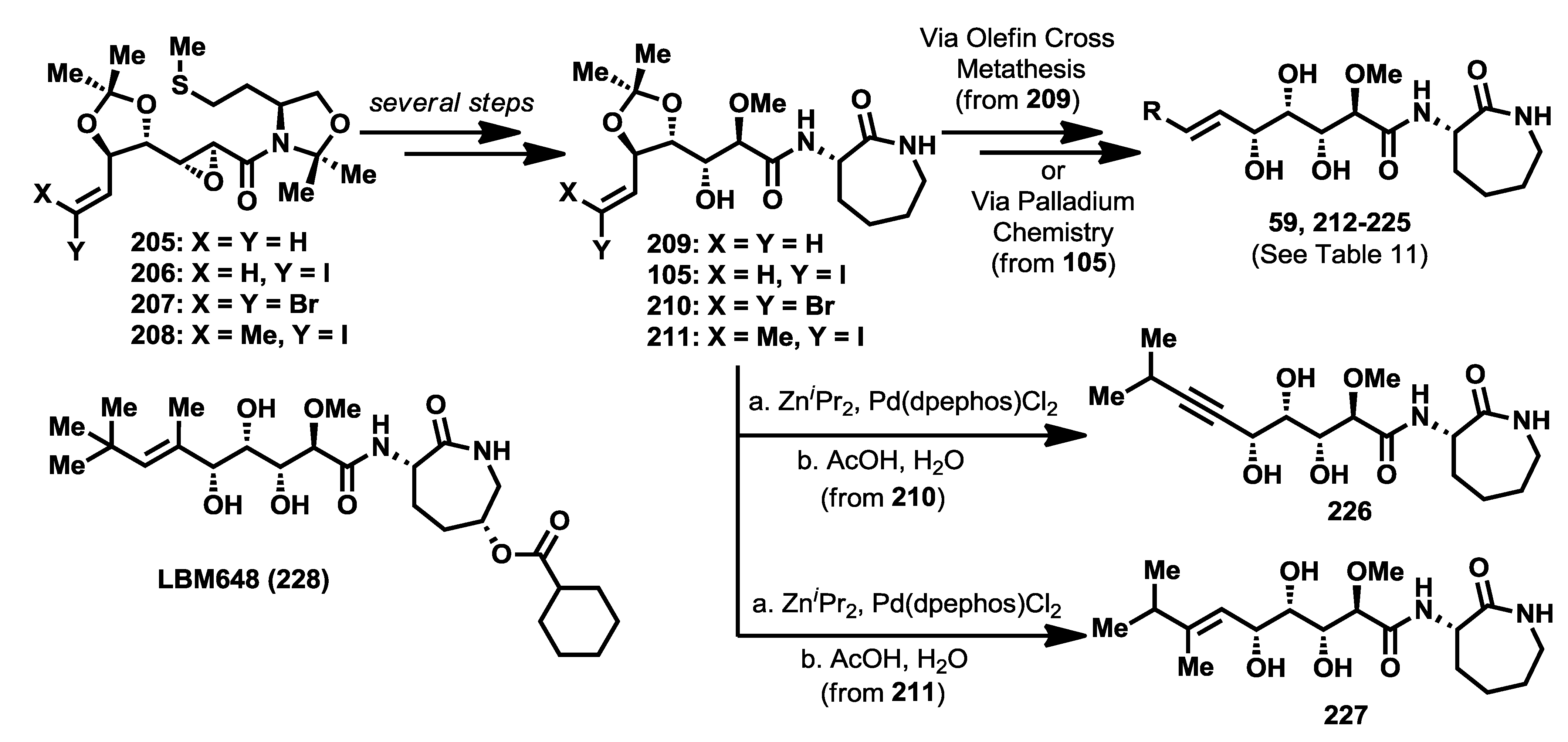
A final set of analogues, modified at the terminal olefinic position, was synthesized and biologically evaluated by Sarabia and co-workers [
104]. Apart from the bengamide analogue LAF389 (
64) and other related caprolactam-modified compounds, in which the isopropyl moiety was replaced with a
tert-butyl group, no other structural modifications were undertaken in this region of the molecule. Considering the binding mode of bengamides at the MetAP active site in which a hydrophobic interaction occurs at the P1 pocket, structural modifications in this region could reinforce this hydrophobic interaction and, therefore, have a positive effect upon the antitumor activity. To this aim, an array of bengamide analogues modified at this position was synthesized by use of an earlier described methodology based on chiral sulfur ylides [
90]. Thus, chiral epoxy amides
205–
208, efficiently prepared by this methodology, carried proper functionalization at the olefinic position for a subsequent introduction of different alkyl groups. These epoxy amides were transformed into the products
105 and
209–
211, which, through olefin cross metathesis or palladium-mediated couplings (Suzuki [
105], Sonogashira [
106] and Negishi [
107]), led to the preparation of bengamide analogues
212–
227. In addition to this array of bengamide E analogues, the olefin-modified analogue LBM648 (
228) was also prepared by a different research group [
72,
108] (
Scheme 8).
Scheme 8.
Synthesis of olefinic-modified bengamide analogues.
Scheme 8.
Synthesis of olefinic-modified bengamide analogues.
In order to evaluate the cytotoxic properties of this new library of bengamide E analogues, IC
50 values were determined first against HT29 human colon adenocarcinoma cell line and later against other additional cancer cell lines, namely MDA-MB-231 (human breast carcinoma), HT1080 (human fibrosarcoma) and HL60 (human promyyelocytic leukemia), as well as against a primary culture of non-transformed bovine aorta endothelial (BAE) cells (
Table 11), establishing a more complete and detailed antiproliferative profile for the most active members with fumagilin as a positive control. The data summarized in
Table 11 suggested that an increasing of the polyketide chain at the olefinic position resulted in a complete loss of cytotoxic activity. More exciting results were obtained for analogues bearing a cyclic group, emphasizing cyclopentyl analogue
219, which exhibited the best cytotoxic result among all the evaluated analogues, with a fourfold improvement in antitumor activity over bengamide E. It is interesting to highlight how essential the substitution at the olefinic position is for antitumor activity, as demonstrated by the methylene analogue
212, which was completely devoid of activity, as well as the importance of the molecular size at this position, proved with analogues
214–
216 and
221 which were similarly inactive.
In total, 111 analogues of the bengamides have been synthesized thus far and their biological activities evaluated. Among these analogues, the most potent and promising analogues are depicted in
Figure 10. Compilation of all the biological information obtained from the prepared analogues provides an extensive structure-activity relationship study that allows the establishment of a well-defined pharmacophore map for the bengamides and provides opportunities for the development of new generations of more active and promising analogues.
Table 11.
Cytotoxic activities (IC50, μM) against different tumor cells lines of bengamide E analogues modified at the terminal olefinic position.
Table 11.
Cytotoxic activities (IC50, μM) against different tumor cells lines of bengamide E analogues modified at the terminal olefinic position.
| Bengamide | R- | Cell Line | IC50 | Bengamide | R- | Cell Line | IC50 |
|---|
| 212 | H- | HT29 | >100 | 220 | Cyclohexyl | HT29 | 62.5 |
| 213 | Ph- | HT29 | 68.8 | 221 | E-ChxCH=CH- | HT29 | 100 |
| 214 | (CH3)2CHCH2- | HT29 | 38.5 | 222 | ![Marinedrugs 12 01580 i042]() | HT29 | 33.7 ± 3.0 |
| 215 | (CH3)3CCH2- | HT29 | 100 | 223 | TMSC≡C- | HT29 | 33.3 ± 10.0 |
| 216 | E-(CH3)3CH=CH- | HT29 | 61 | 224 | Cyclopropyl- | HT29 | 2.1 ± 0.4 |
| MDA-MB-435 | 3.7 ± 0.5 |
| HT1080 | 1.1 ± 0.5 |
| HL60 | 2.1 ± 0.4 |
| BAEC | 1.6 ± 0.5 |
| 217 | (CH3)3- | HT29 | 0.087 ± 0.2 | 225 | CH3C(=CH2)- | HT29 | 16.9 ± 3.7 |
| MDA-MB-435 | 2.2 ± 0.1 | MDA-MB-435 | 15.2 ± 2.5 |
| HT1080 | 0.25 ± 0.05 | HT1080 | 1.7 ± 0.7 |
| HL60 | 1.1 ± 0.2 | HL60 | 6.3 ± 0.2 |
| BAEC | 0.17 ± 0.01 | BAEC | 2.3 ± 0.2 |
| 218 | I- | HT29 | 14.8 ± 2.0 | 226 | See Scheme 8 | HT29 | 2.2 ± 0.4 |
| MDA-MB-435 | 14.2 ± 1.4 | MDA-MB-435 | 5.7 ± 2.0 |
| HT1080 | 3.6 ± 1.0 | HT1080 | 0.5 ± 0.1 |
| HL60 | 7.1 ± 0.5 | HL60 | 1.7 ± 0.1 |
| BAEC | 3.9 ± 0.4 | BAEC | 1.5 ± 0.2 |
| 219 | Cyclopentyl- | HT29 | 0.22 ± 0.05 | 227 | See Scheme 8 | HT29 | 6.6 ± 0.7 |
| MDA-MB-435 | 0.44 ± 0.06 | MDA-MB-435 | 11.1 ± 3.0 |
| HT1080 | 0.12 ± 0.05 | HT1080 | 1.21 ± 0.13 |
| HL60 | 0.19 ± 0.0 | HL60 | 5.5 ± 1.0 |
| BAEC | 0.10 ± 0.02 | BAEC | 2.4 ± 1.3 |
| 228 | See Scheme 8 | A549 | 0.74 ± 0.2 | - | - | - |
| HI299 | 0.99 ± 0.76 |
| HCT116 | 0.14 ± 0.15 |
| HUVEC | 0.14 ± 0.15 |
Figure 10.
Representative bengamide analogues with potent antitumor activities [
46,
99,
100,
104].
Figure 10.
Representative bengamide analogues with potent antitumor activities [
46,
99,
100,
104].






























































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
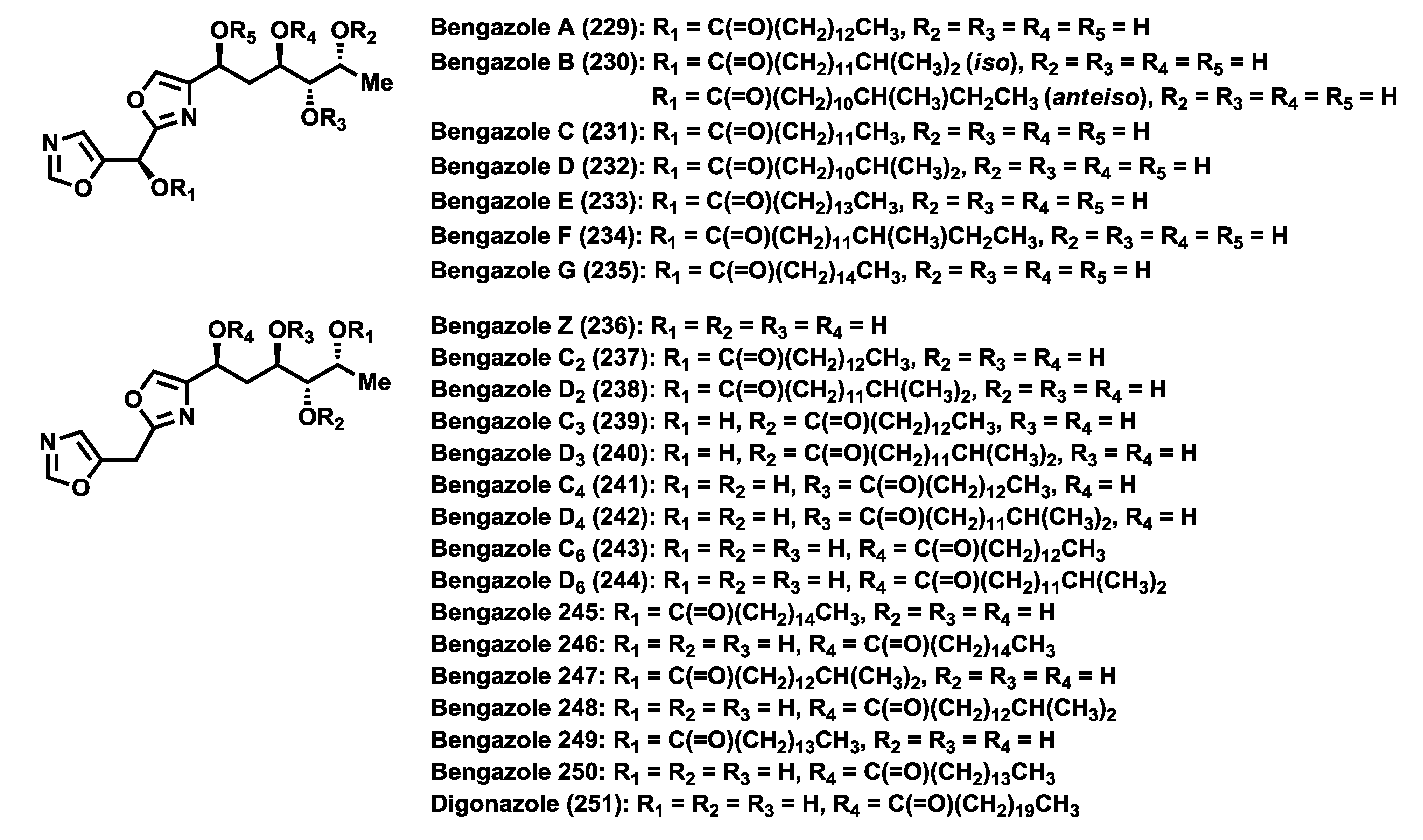{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





