
Structure Elucidation and in Vitro Toxicity of New Azaspiracids Isolated from the Marine Dinoflagellate Azadinium poporum

 , , ,
, , ,
Abstract
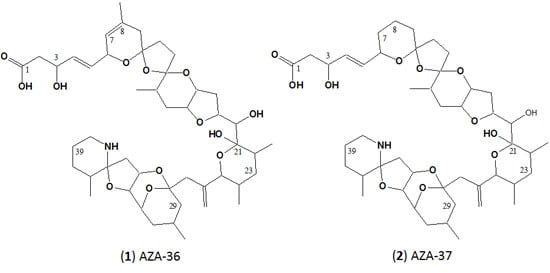
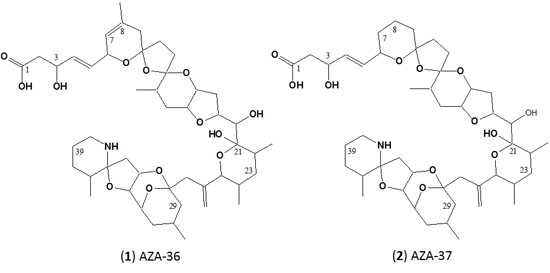
:
1. Introduction
2. Results
2.1. Purification of AZA-36 (1) and AZA-37 (2)

{kind=link}
{kind=link}
{kind=link}
| Step No | Step | AZA-36 [mg] | Weight [g] | Purity [%] † |
|---|---|---|---|---|
| HP20 resin extract | 1.30 | 8.30 | <0.1 | |
| 1 | Ethyl acetate partitioning | 1.20 | 0.74 | 0.2 |
| 2 | Silica gel | 1.05 | 0.18 | 0.6 |
| 3 | Flash (phenyl-hexyl) | 0.92 | <0.01 | 40.0 |
| 4 | Prep HPLC (C18) | 0.89 | - | > 95 |
| % Recovery (steps 1−4) | 69 |
| Step No | Step | AZA-37 [mg] | Weight [g] | Purity [%]† |
|---|---|---|---|---|
| HP20 resin extract | 1.70 | 1.03 | 0.2 | |
| 1 | Silica gel | 1.60 | 0.25 | 0.6 |
| 2 | Flash (phenyl-hexyl) | 1.57 | 0.01 | 20.0 |
| 3 | Prep HPLC (C18) | 1.28 | - | >95 |
| % Recovery (steps 1−3) | 75 |
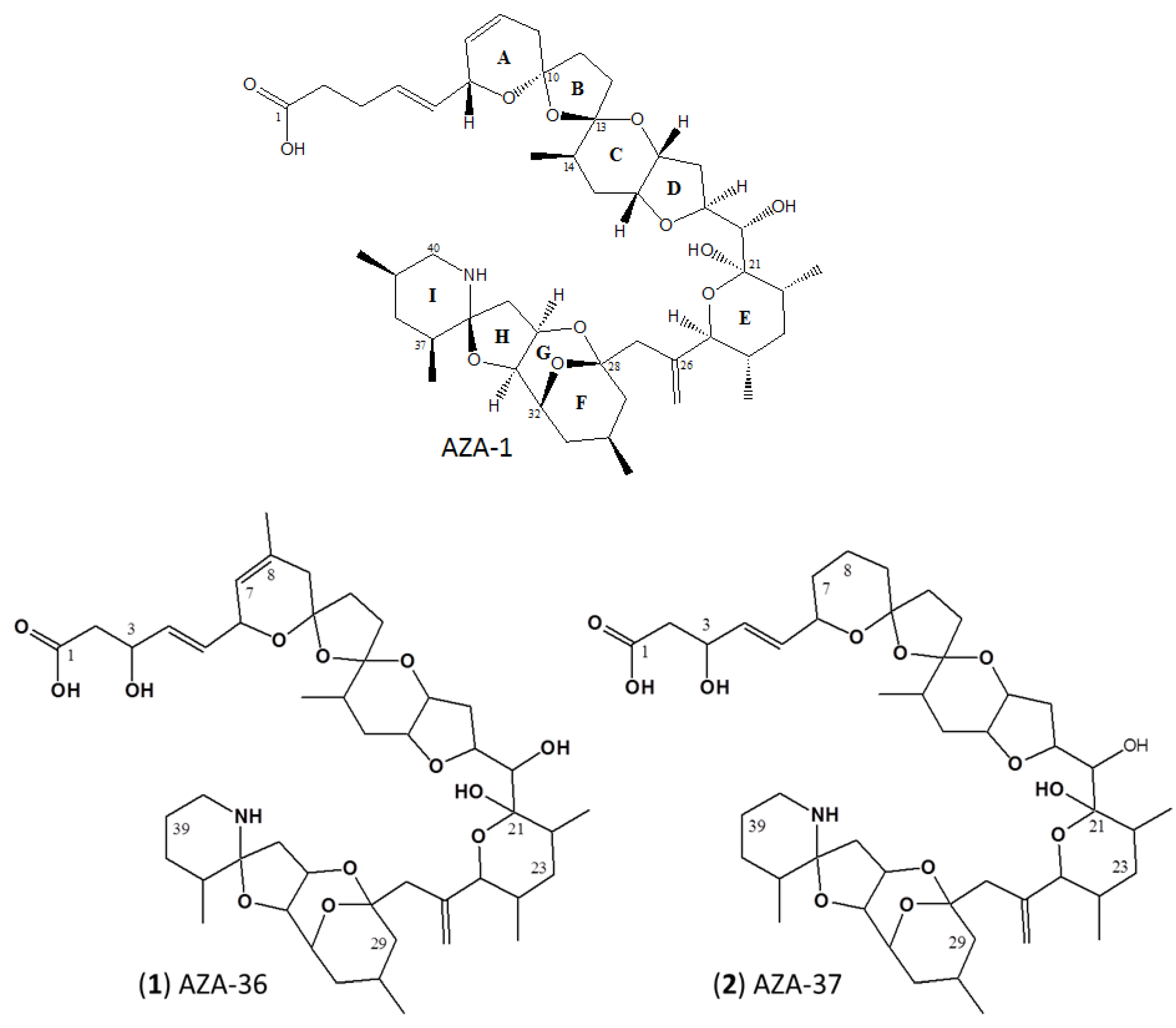
2.2. Structure Elucidation of AZA-36 (1) and AZA-37 (2)
| Atom No. | AZA-36 | AZA-37 | ||||
|---|---|---|---|---|---|---|
| δC | δH | δC | δH | |||
| 1 | 180.3 a | - | - | 180.3 a | - | - |
| 2 | 46.0 | 2.34 | 2.39 | 46.1 | 2.33 | - |
| 3 | 71.4 | 4.43 | - | 71.4 | 4.39 | - |
| 4 | 135.5 | 5.75 | - | 134.6 | 5.70 | - |
| 5 | 132.1 | 5.64 | - | 133.1 | 5.65 | - |
| 6 | 72.8 | 4.79 | - | 73.3 | 4.35 | - |
| 7 | 123.4 | 5.36 | - | 38.4 | 1.43 | 1.87 |
| 8 | 132.1 | - | - | 22.2 | 1.70 | 1.77 |
| 9 | 41.1 | 2.00 | 2.44 | 36.6 | 1.70 | 1.83 |
| 10 | 108.3 | - | - | 109.1 | - | - |
| 11 | 34.0 | 1.71 | 2.33 | 33.9 | 1.69 | 2.33 |
| 12 | 38.3 | 1.99 | 2.18 | 32.8 | 1.83 | 2.03 |
| 13 | 112.2 | - | - | 111.8 | - | - |
| 14 | 32.1 | 2.02 | - | 31.8 | 2.01 | - |
| 15 | 33.3 | 1.76 | 1.86 | 33.5 | 1.76 | 1.87 |
| 16 | 78.9 | 3.91 | - | 78.9 | 3.94 | - |
| 17 | 74.2 | 4.23 | - | 74.3 | 4.29 | - |
| 18 | 37.5 | 1.99 | 2.06 | 37.6 | 2.00 | 2.07 |
| 19 | 79.9 | 4.43 | - | 79.9 | 4.44 | - |
| 20 | 77.5 | 3.90 | - | 77.4 | 3.93 | - |
| 21 | 101.1 | - | - | 101.0 | - | - |
| 22 | 37.8 | 2.06 | - | 37.6 | 2.07 | - |
| 23 | 39.1 | 1.44 | 1.44 | 39.1 | 1.43 | 1.43 |
| 24 | 43.0 | 1.35 | - | 43.0 | 1.35 | - |
| 25 | 80.4 | 4.00 | - | 80.3 | 4.00 | - |
| 26 | 149.0 | - | - | 149.0 | - | - |
| 27 | 50.4 | 2.26 | 2.43 | 50.4 | 2.25 | 2.42 |
| 28 | 99.4 | - | - | 99.4 | - | - |
| 29 | 45.0 | 1.37 | 2.05 | 45.0 | 1.36 | 2.05 |
| 30 | 27.2 | 2.23 | - | 27.2 | 2.23 | - |
| 31 | 36.2 | 1.52 | 1.83 | 36.1 | 1.52 | 1.84 |
| 32 | 73.7 | 4.37 | - | 73.7 | 4.37 | - |
| 33 | 82.1 | 4.05 | - | 82.1 | 4.05 | - |
| 34 | 75.7 | 4.99 | - | 75.7 | 5.00 | - |
| 35 | 42.8 | 2.49 | 2.61 | 42.7 | 2.49 | 2.60 |
| 36 | 98.0 | - | - | 98.0 | - | - |
| 37 | 37.5 | 1.99 | - | 36.7 | 1.98 | - |
| 38 | 29.8 | 1.61 | 1.68 | 29.7 | 1.63 | 1.67 |
| 39 | 23.8 | 1.70 | 1.81 | 23.8 | 1.70 | - |
| 40 | 41.3 | 2.98 | 3.17 | 41.2 | 2.99 | 3.17 |
| 41 | 17.5 | 0.94 | - | 17.5 | 0.90 | - |
| 42 | 17.2 | 0.92 | - | 17.2 | 0.92 | - |
| 43 | 18.9 | 0.84 | - | 18.9 | 0.84 | - |
| 44 | 117.8 | 5.16 | 5.33 | 117.8 | 5.15 | 5.33 |
| 45 | 24.3 | 0.96 | - | 24.3 | 0.96 | - |
| 46 | 16.4 | 0.98 | - | 16.4 | 0.97 | - |
| 47 | 23.0 | 1.70 | - | - | - | - |

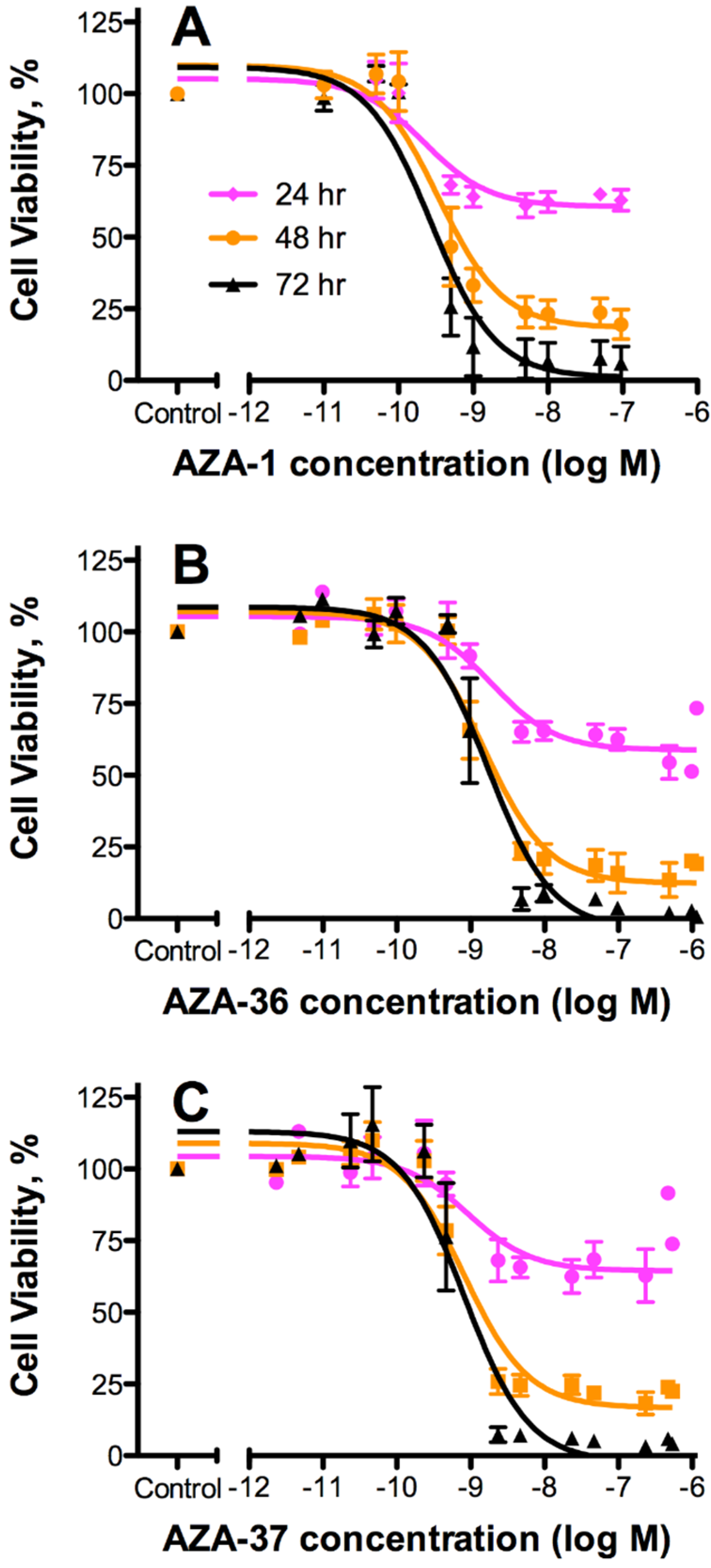
2.3. Cytotoxicity

| AZA Analogue | 24 h | 48 h | 72 h | Mean EC50 [nM] | Rel. Pot. | |||
|---|---|---|---|---|---|---|---|---|
| EC50 [nM] | 95% CI | EC50 [nM] | 95% CI | EC50 [nM] | 95% CI | |||
| AZA-1 | 0.22 | 0.10–0.49 | 0.34 | 0.18–0.65 | 0.27 | 0.14–0.52 | 0.28 | 1 |
| AZA-36* (1) | 1.9 | 0.89–4.1 | 1.5 | 0.92–2.4 | 1.7 | 0.92–3.2 | 1.70 | 0.16 |
| AZA-37 (2) | 0.91 | 0.27–3.1 | 0.82 | 0.51–1.3 | 0.82 | 0.46–1.5 | 0.85 | 0.33 |
3. Discussion
3.1. Structure Elucidation of AZA-36 (1) and AZA-37 (2)
3.2. Cytotoxicity
4. Experimental Section
4.1. Reagents
4.2. Cell Culture and Extraction
4.3. Purification of AZA-36 and AZA-37
4.4. LC-MS/MS Analysis
4.4.1. Method A
4.4.2. Method B
4.4.3. Method C
4.4.4. Method D
4.5. NMR Analysis
4.6. Toxicity Assays
4.6.1. Cell Culturing
4.6.2. Cytotoxicity Assay
5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- McMahon, T.; Silke, J. West coast of Ireland; winter toxicity of unknown aetiology in mussels. Harmful Algae News 1996, 14, 2. [Google Scholar]
- Satake, M.; Ofuji, K.; Naoki, H.; James, K.J.; Furey, A.; McMahon, T.; Silke, J.; Yasumoto, T. Azaspiracid, a new marine toxin having unique spiro ring assemblies, isolated from Irish mussels, Mytilus edulis. J. Am. Chem. Soc. 1998, 120, 9967–9968. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Frederick, M.O.; Petrovic, G.; Cole, K.P.; Loizidou, E.Z. Total synthesis and confirmation of the revised structures of azaspiracid-2 and azaspiracid-3. Angew. Chem. Int. Ed. 2006, 45, 2609–2615. [Google Scholar] [CrossRef] [PubMed]
- James, K.J.; Furey, A.; Satake, M.; Yasumoto, T. Azaspiracid Poisoning (AZP): A New Shellfish Toxic Syndrome in Europe. In Harmful Algal Blooms, Proceedings of the 9 International Conference on Harmful Algal Blooms, Hobart, Australia, 7–11 February 2000; Hallegraeff, G.M., Blackburn, S.I., Bolch, C.J.S., Lewis, R.J., Eds.; International Oceanogrphic Commission of UNESCO: Paris, France, 2000; pp. 250–253. [Google Scholar]
- Lehane, M.; Braña-Magdalena, A.; Moroney, C.; Furey, A.; James, K.J. Liquid chromatography with electrospray ion trap mass spectrometry for the determination of five azaspiracids in shellfish. J. Chromatogr. A 2002, 950, 139–147. [Google Scholar] [CrossRef]
- Vale, P.; Bire, R.; Hess, P. Confirmation by LC-MS/MS of azaspiracids in shellfish from the Portuguese north-western coast. Toxicon 2008, 51, 1449–1456. [Google Scholar] [CrossRef] [PubMed]
- Taleb, H.; Vale, P.; Amanhir, R.; Benhadouch, A.; Sagou, R.; Chafik, A. First detection of azaspiracids in mussels in North West Africa. J. Shellfish Res. 2006, 25, 1067–1070. [Google Scholar] [CrossRef]
- Álvarez, G.; Uribe, E.; Ávalos, P.; Mariño, C.; Blanco, J. First identification of azaspiracid and spirolides in Mesodesma donacium and Mulinia edulis from Northern Chile. Toxicon 2010, 55, 638–641. [Google Scholar] [CrossRef] [PubMed]
- Ueoka, R.; Ito, A.; Izumikawa, M.; Maeda, S.; Takagi, M.; Shin-ya, K.; Yoshida, M.; Matsunaga, S.; van Soest, R.W.M. Isolation of azaspiracid-2 from a marine sponge Echinoclathria sp. as a potent cytotoxin. Toxicon 2009, 53, 680–684. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Tan, Z.; Zhou, D.; Guo, M.; Xing, L.; Yang, S. Determination of azaspiracid-1 in shellfish by liquid chromatography with tandem mass spectrometry. Chin. J. Chromatogr. 2010, 28, 363–367. [Google Scholar] [CrossRef]
- Vershinin, A.; Moruchkov, A.; Morton, S.L.; Leighfield, T.A.; Quilliam, M.A.; Ramsdell, J.S. Phytoplankton composition of the Kandalaksha Gulf, Russian White Sea: Dinophysis and lipophilic toxins in the blue mussel (Mytilus edulis). Harmful Algae 2006, 5, 558–564. [Google Scholar] [CrossRef]
- Rehmann, N.; Hess, P.; Quilliam, M.A. Discovery of new analogs of the marine biotoxin azaspiracid in blue mussels Mytilus edulis by ultra-performance liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2008, 22, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Ofuji, K.; Satake, M.; McMahon, T.; Silke, J.; James, K.J.; Naoki, H.; Oshima, Y.; Yasumoto, T. Two analogs of azaspiracid isolated from mussels, Mytilus edulis, involved in human intoxication in Ireland. Nat. Toxins 1999, 7, 99–102. [Google Scholar] [CrossRef]
- Ofuji, K.; Satake, M.; McMahon, T.; James, K.J.; Naoki, H.; Oshima, Y.; Yasumoto, T. Structures of azaspiracid analogs, azaspiracid-4 and azaspiracid-5, causative toxins of azaspiracid poisoning in Europe. Biosci. Biotechnol. Biochem. 2001, 65, 740–742. [Google Scholar] [CrossRef] [PubMed]
- James, K.J.; Sierra, M.D.; Lehane, M.; Braña Magdalena, A.; Furey, A. Detection of five new hydroxyl analogues of azaspiracids in shellfish using multiple tandem mass spectrometry. Toxicon 2003, 41, 277–283. [Google Scholar] [CrossRef]
- Kilcoyne, J.; Keogh, A.; Clancy, G.; LeBlanc, P.; Burton, I.W.; Quilliam, M.A.; Hess, P.; Miles, C.O. Improved isolation procedure for azaspiracids from shellfish, structural elucidation of azaspiracid-6, and stability studies. J. Agric. Food Chem. 2012, 60, 2447–2455. [Google Scholar] [CrossRef] [PubMed]
- Krock, B.; Tillmann, U.; John, U.; Cembella, A.D. Characterization of azaspiracids in plankton size-fractions and isolation of an azaspiracid-producing dinoflagellate from the North Sea. Harmful Algae 2009, 8, 254–263. [Google Scholar] [CrossRef]
- Tillmann, U.; Elbrächter, M.; Krock, B.; John, U.; Cembella, A.D. Azadinium spinosum gen. et sp. nov. (Dinophyceae) identified as a primary producer of azaspiracid toxins. Eur. J. Phycol. 2009, 44, 63–79. [Google Scholar] [CrossRef]
- Jauffrais, T.; Kilcoyne, J.; Séchet, V.; Herrenknecht, C.; Truquet, P.; Hervé, F.; Bérard, J.B.; Nulty, C.; Taylor, S.; Tillmann, U.; et al. Production and isolation of azaspiracid-1 and-2 from Azadinium spinosum culture in pilot scale photobioreactors. Mar. Drugs 2012, 10, 1360–1382. [Google Scholar] [CrossRef] [PubMed]
- Kilcoyne, J.; Nulty, C.; Jauffrais, T.; McCarron, P.; Herve, F.; Foley, B.; Rise, F.; Crain, S.; Wilkins, A.L.; Twiner, M.J.; et al. Isolation, structure elucidation, relative LC-MS response, and in vitro toxicity of azaspiracids from the dinoflagellate Azadinium spinosum. J. Nat. Prod. 2014, 77, 2465–2474. [Google Scholar] [CrossRef] [PubMed]
- Tillmann, U.; Elbrächter, M.; John, U.; Krock, B.; Cembella, A.D. Azadinium obesum (Dinophyceae), a new nontoxic species in the genus that can produce azaspiracid toxins. Phycologia 2010, 49, 169–182. [Google Scholar] [CrossRef]
- Tillmann, U.; Elbrächter, M.; John, U.; Krock, B. A new non-toxic species in the dinoflagellate genus Azadinium: A. poporum sp. nov. Eur. J. Phycol. 2011, 46, 74–87. [Google Scholar] [CrossRef]
- Tillmann, U.; Soehner, S.; Nézan, E.; Krock, B. First record of the genus Azadinium (Dinophyceae) from the Shetland Islands, including the description of Azadinium polongum sp. nov. Harmful Algae 2012, 20, 142–155. [Google Scholar] [CrossRef]
- Akselman, R.; Negri, R.M. Blooms of Azadinium cf. spinosum Elbrächter et Tillmann (Dinophyceae) in northern shelf waters of Argentina, Southwestern Atlantic. Harmful Algae 2012, 19, 30–38. [Google Scholar] [CrossRef]
- Percopo, I.; Siano, R.; Rossi, R.; Soprano, V.; Sarno, D.; Zingone, A. A new potentially toxic Azadinium species (Dinophyceae) from the Mediterranean Sea, A. dexteroporum sp. nov. J. Phycol. 2013, 49, 950–966. [Google Scholar]
- Luo, Z.; Gu, H.; Krock, B.; Tillmann, U. Azadinium dalianense, a new dinoflagellate species from the Yellow Sea, China. Phycologia 2013, 52, 625–636. [Google Scholar] [CrossRef]
- Tillmann, U.; Gottschling, M.; Nézan, E.; Krock, B.; Bilien, G. Morphological and molecular characterization of three new Azadinium species (Amphidomataceae, Dinophyceae) from the Irminger Sea. Protist 2014, 165, 417–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potvin, É.; Jeong, H.J.; Kang, N.S.; Tillmann, U.; Krock, B. First report of the photosynthetic dinoflagellate genus Azadinium in the Pacific Ocean: Morphology and molecular characterization of Azadinium cf. poporum. J. Eukaryot. Microbiol. 2012, 59, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Luo, Z.; Krock, B.; Witt, M.; Tillmann, U. Morphology, phylogeny and azaspiracid profile of Azadinium poporum (Dinophyceae) from the China Sea. Harmful Algae 2013, 21–22, 64–75. [Google Scholar] [CrossRef]
- Krock, B.; Tillmann, U.; Voß, D.; Koch, B.P.; Salas, R.; Witt, M.; Potvin, É.; Jeong, H.J. New azaspiracids in Amphidomataceae (Dinophyceae). Toxicon 2012, 60, 830–839. [Google Scholar] [CrossRef] [PubMed]
- Twiner, M.J.; El-Ladki, R.; Kilcoyne, J.; Doucette, G.J. Comparative effects of the marine algal toxins azaspiracid-1,-2, and-3 on Jurkat T lymphocyte cells. Chem. Res. Toxicol. 2012, 25, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Kilcoyne, J.; Jauffrais, T.; Twiner, M.; Doucette, G.; Aasen Bunæs, J.A.; Sosa, S.; Krock, B.; Séchet, V.; Nulty, C.; Salas, R.; et al. Azaspiracids—Toxicological Evalution, Test Methods and Identification of the Source Organisms (ASTOX 2); Series, M.E.H., Ed.; Marine Institute: Galway, Ireland, 2014. [Google Scholar]
- Twiner, M.J.; Hess, P.; Bottein Dechraoui, M.-Y.; McMahon, T.; Ramsdell, J.S.; Samons, M.S.; Satake, M.; Yasumoto, T.; Doucette, G.J. Cytotoxic and cytosketetal effects of azaspiracid-1 on mammalian cell lines. Toxicon 2005, 45, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Twiner, M.J.; Rehmann, N.; Hess, P.; Doucette, G.J. Azaspiracid Shellfish Poisoning: A review on the chemistry, ecology, and toxicology with an emphasis on human health impacts. Mar. Drugs 2008, 6, 39–72. [Google Scholar] [CrossRef] [PubMed]
- Krock, B.; Tillmann, U.; Witt, M.; Gu, H. Azaspiracid variability of Azadinium poporum (Dinophyceae) from the China Sea. Harmful Algae 2014, 36, 22–28. [Google Scholar] [CrossRef] [Green Version]
- MacKenzie, L.; Beuzenberg, V.; Holland, P.; McNabb, P.; Selwood, A. Solid phase adsorption toxin tracking (SPATT): A new monitoring tool that simulates the biotoxin contamination of filter feeding bivalves. Toxicon 2004, 44, 901–918. [Google Scholar] [CrossRef] [PubMed]
- Rein, K.S.; Snyder, R.V. The biosynthesis of polyketide metabolites by dinoflagellates. In Advances in Applied Microbiology; Academic Press: San Diego, CA, USA, 2006; Volume 59, pp. 93–125. [Google Scholar]
- Kilcoyne, J.; McCarron, P.; Twiner, M.J.; Nulty, C.; Crain, S.; Wilkins, A.; Rise, F.; Quilliam, M.A.; Miles, C.O. Epimers of azaspiracids: isolation, structural elucidation, relative LC-MS response, and in vitro toxicity of 37-epi-azaspiracid-1. Chem. Res. Toxicol. 2014, 27, 587–600. [Google Scholar] [CrossRef] [PubMed]
- Guillard, R.R.L.; Ryther, J.H. Studies on marine planktonic diatoms. I. Cyclotella nana Hustedt and Detonula confervaceae (Cleve) Gran. Can. J. Microbiol. 1962, 8, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Potvin, E.; Hwang, Y.J.; Yoo, Y.D.; Kim, J.S.; Jeong, H.J. Feeding by heterotrophic protists and copepods on the photosynthetic dinoflagellate Azadinium cf. poporum from western Korean waters. Aquat. Microb. Ecol. 2013, 68, 143–158. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krock, B.; Tillmann, U.; Potvin, É.; Jeong, H.J.; Drebing, W.; Kilcoyne, J.; Al-Jorani, A.; Twiner, M.J.; Göthel, Q.; Köck, M. Structure Elucidation and in Vitro Toxicity of New Azaspiracids Isolated from the Marine Dinoflagellate Azadinium poporum. Mar. Drugs 2015, 13, 6687-6702. https://doi.org/10.3390/md13116687
Krock B, Tillmann U, Potvin É, Jeong HJ, Drebing W, Kilcoyne J, Al-Jorani A, Twiner MJ, Göthel Q, Köck M. Structure Elucidation and in Vitro Toxicity of New Azaspiracids Isolated from the Marine Dinoflagellate Azadinium poporum. Marine Drugs. 2015; 13(11):6687-6702. https://doi.org/10.3390/md13116687
Chicago/Turabian StyleKrock, Bernd, Urban Tillmann, Éric Potvin, Hae Jin Jeong, Wolfgang Drebing, Jane Kilcoyne, Ahmed Al-Jorani, Michael J. Twiner, Qun Göthel, and Matthias Köck. 2015. "Structure Elucidation and in Vitro Toxicity of New Azaspiracids Isolated from the Marine Dinoflagellate Azadinium poporum" Marine Drugs 13, no. 11: 6687-6702. https://doi.org/10.3390/md13116687
APA StyleKrock, B., Tillmann, U., Potvin, É., Jeong, H. J., Drebing, W., Kilcoyne, J., Al-Jorani, A., Twiner, M. J., Göthel, Q., & Köck, M. (2015). Structure Elucidation and in Vitro Toxicity of New Azaspiracids Isolated from the Marine Dinoflagellate Azadinium poporum. Marine Drugs, 13(11), 6687-6702. https://doi.org/10.3390/md13116687