1. Introduction
Consumption of carotenoids may reduce the risk of developing chronic diseases associated with oxidative stress. Since carotenoids accumulate in much larger concentrations in the gastro-intestinal (GI) tract than in plasma and tissues, and because the GI tract may undergo substantial oxidative stress in postprandial conditions [
1,
2,
3], part of this protection could take place prior to intestinal absorption [
4,
5,
6]. For instance, dietary iron is highly present in food (especially in red meat), both as heme and in the free form [
7,
8]. Iron is a potent initiator of lipid peroxidation, especially in acidic conditions [
9,
10]. Indeed, under gastric conditions, dietary lipid peroxidation may be quite rapid due to high dioxygen concentrations, moderate temperature (37 °C), a pH varying between 2 (empty stomach) and 6 (postprandial), and constant mixing [
5].
In solution and in organized lipid assemblies (e.g., micelles, liposomes, emulsions, and lipoproteins), carotenoids and other dietary antioxidants can interact. Previous studies have highlighted the possible regeneration of some antioxidants by others [
11,
12]. For instance, antioxidant synergism has been demonstrated using combinations of α-tocopherol, vitamin C, and β-carotene, as well as lutein [
13,
14,
15,
16,
17,
18,
19,
20]. The mechanisms proposed involve a transfer of electrons or hydrogen atoms between antioxidants partitioned between the aqueous and lipid phases via antioxidants located at the interface. A few studies have been conducted with combinations of carotenoids. For instance, in membranes, lycopene or β-carotene has been shown to significantly prolong the antioxidant activity of the xanthophyll zeaxanthin by reducing its radical, thereby restoring its active form [
21].
In this work, we investigated possible mechanism(s) underlying the high stability and antioxidant activity of novel carotenoids extracted from a marine bacterial strain.
Bacillus indicus HU36 was initially selected for its high production of carotenoids, the resistance of its spores to UV radiation [
22], and its probiotic properties [
23]. It was isolated from human feces and shown to synthesize yellow-orange pigments in variable proportions depending on whether the bacteria were present as vegetative cells or as spores [
22]. The most abundant pigments in a lipophilic extract of vegetative cells were found to be 1-(6-C
n:0)-glycosyl-apo-8′-lycopene esters, and in a lipophilic extract of spores were found to be methyl-1-(6-C
n:0)-glycosyl-apo-8′-lycopenoate esters. Other isoprenoids were also observed [
24]. In particular, menaquinone MQ-7, a form of vitamin K
2, was extracted with the carotenoids in a crude extract. Menaquinones are constituents of bacterial cytoplasmic membranes and play important roles in electron transport, oxidative phosphorylation, active transport, and endospore formation [
25]. As MQ-7 is potentially redox active, its influence on the stability and antioxidant activity of the bacterial carotenoids was tested. The common dietary carotenoids β-carotene and lycopene were also investigated for comparison.
Studies were carried out in micelle solutions [
26], used as a simple model to mimic the environmental conditions experienced by dietary carotenoids in the GI tract [
27,
28]. Peroxidation was initiated either by free ferrous iron or by metmyoglobin, and followed at pH 5.8 and 4.0, corresponding to the early phase and mid-phase of digestion, respectively. The degradation of carotenoids and the formation of lipid oxidation products were followed by UV-VIS spectroscopy.
In a first step, the effect of the bacterial vitamin menaquinone MQ-7 (
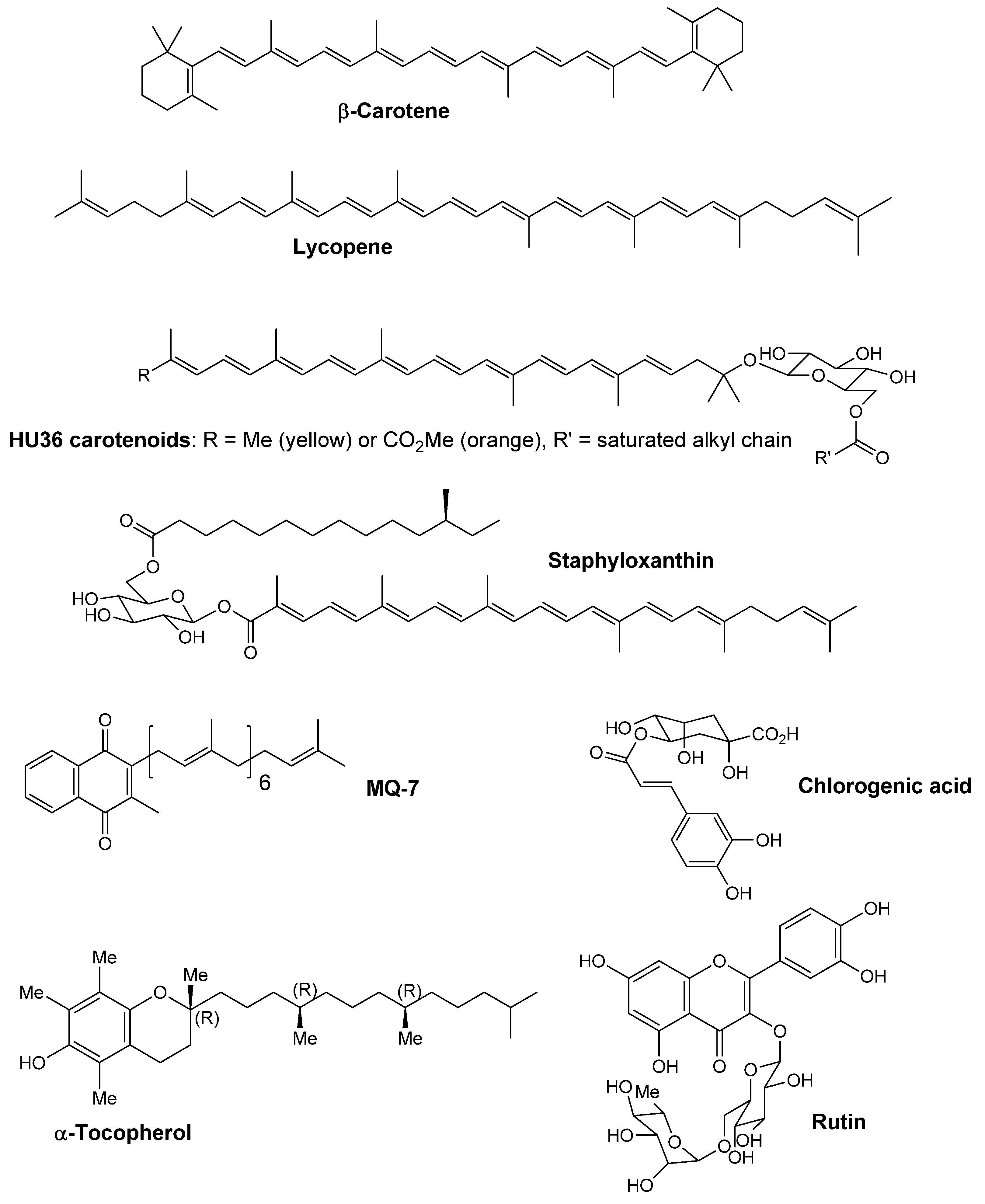
Scheme 1) on the stability and antioxidant properties of HU36 carotenoids was evaluated and compared with the effect on β-carotene. Then, combinations of carotenoids with similar structures (β-carotene and lycopene,
Scheme 1) were evaluated to determine possible antioxidant interactions as compared to the cocktail of bacterial carotenoids. Finally, purified HU36 carotenoids were combined with other antioxidants (
Scheme 1) in order to suggest formulations that could enhance their stability and antioxidant properties.
Scheme 1.
Chemical structures of studied molecules.
Scheme 1.
Chemical structures of studied molecules.
3. Discussion
The stability and lipid-protecting capacity of antioxidant mixtures were investigated in this work using the following combinations: mixtures of carotenoids, carotenoids + MQ-7, and carotenoids + phenolic antioxidants.
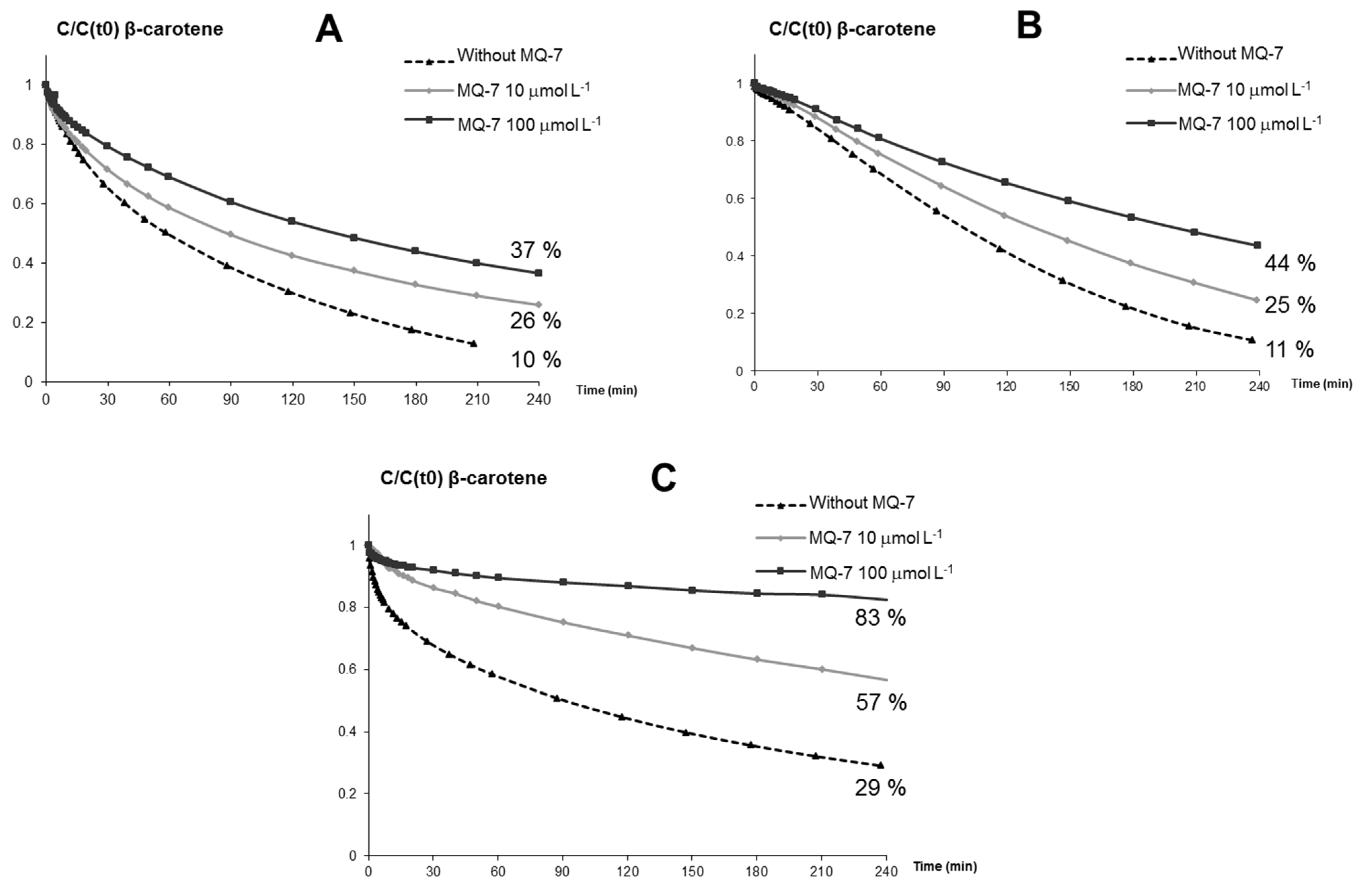
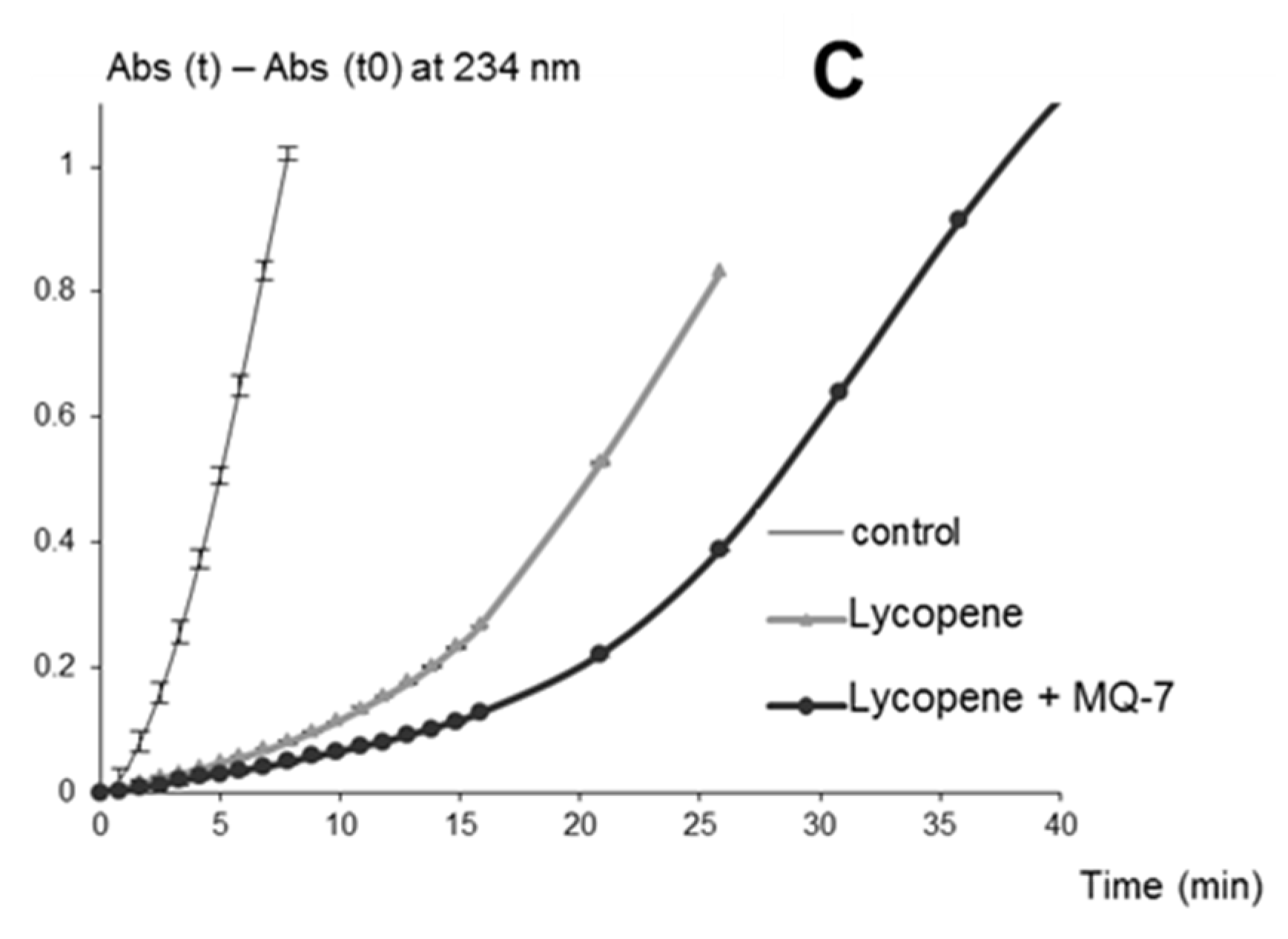
MQ-7 does not significantly inhibit iron-induced autoxidation of HU36 carotenoids (a weak protection was observed only with FeIII). However, the protective effect of MQ-7 is very significant with β-carotene, especially in the presence of heme iron. Thus, protection is very dependent on the form of iron and the type of carotenoid. Although a poor antioxidant compared to HU36 carotenoids and lycopene, MQ-7 in relatively high concentrations (10 molar equiv.) can efficiently increase the antioxidant activity of carotenoids. This effect is maximally observed in FeII-induced lipid peroxidation with the bacterial carotenoids, and in heme-induced peroxidation with lycopene. However, MQ-7 is not degraded in the presence of iron.
Although not an electron-donor by itself, MQ-7 (abbreviated as MQ below and in
Scheme 2) may be involved in redox cycling through its one-electron reduced (semiquinone, MQH
•) and two-electron reduced (hydroquinone, MQH
2) forms [
35,
36]. Such species could be generated by electron transfer from iron—carotenoid systems to MQ depending on the location and reducing capacity of carotenoids. MQH
• and MQH
2 could in turn effectively transfer electrons to lipid-derived radicals, carotenoid radical cations, and/or hypervalent heme-iron species involved in the initiation of lipid peroxidation.
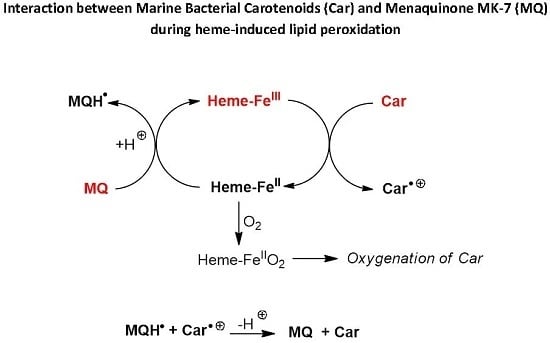
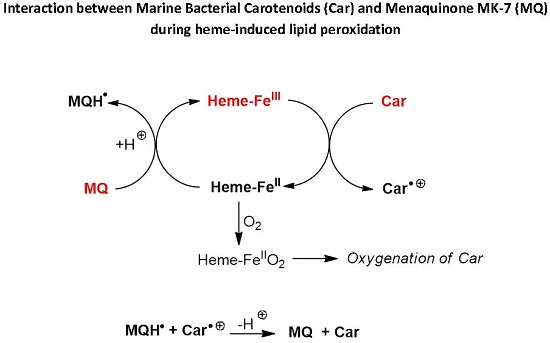
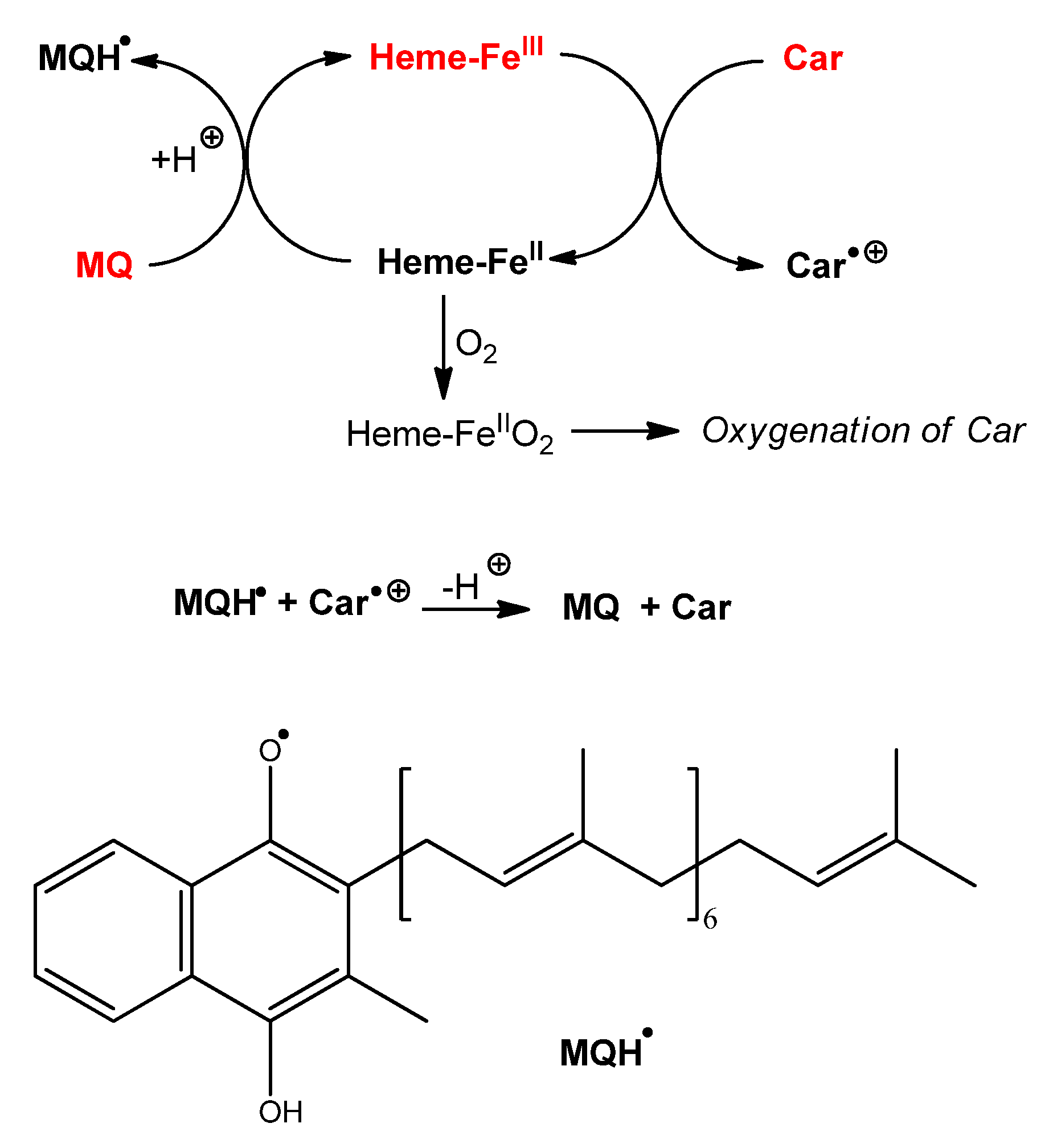
Scheme 2.
Proposed mechanisms of protection of carotenoids by menaquinone during heme-induced lipid peroxidation.
Scheme 2.
Proposed mechanisms of protection of carotenoids by menaquinone during heme-induced lipid peroxidation.
For instance, from the mechanism of heme-induced autoxidation of carotenoids previously proposed [
30], a mechanism for the protection of carotenoids by MQ-7 can be suggested (
Scheme 2). By sparing carotenoids, MQ-7 is also susceptible to increase their apparent antioxidant efficiency in inhibiting heme-induced lipid peroxidation. However, this mechanism is likely limited to carotenes (β-carotene, lycopene).
The observation that MQ-7 strongly improves the ability of HU36 carotenoids to inhibit Fe
II-induced lipid peroxidation is unexpected. MQ-7 does not react with iron and is a poor antioxidant by itself, which makes it unlikely that it could directly quench propagating lipid peroxyl radicals (LOO
•). The previously proposed mechanism for the inhibition of Fe
II-induced lipid peroxidation by carotenoids is reintroduced in
Scheme 3 [
31]. A possible interpretation of the effect of MQ-7 is that the initiating oxyl radical(s) (
i.e., LO
•, or other radicals derived from LO
•) could combine with MQ-7 to form a stabilized radical, which could then be rapidly reduced by the carotenoids. In other words, HU36 carotenoids would be able to interfere in the initiation process via MQ-7, in addition to their conventional action on the propagation step (LOO
• scavenging).
Scheme 3.
Proposed mechanism for the inhibition of Fe
II-induced lipid peroxidation (from [
31]) by carotenoids, and interaction of marine bacterial carotenoids with menaquinone MQ-7.
Scheme 3.
Proposed mechanism for the inhibition of Fe
II-induced lipid peroxidation (from [
31]) by carotenoids, and interaction of marine bacterial carotenoids with menaquinone MQ-7.
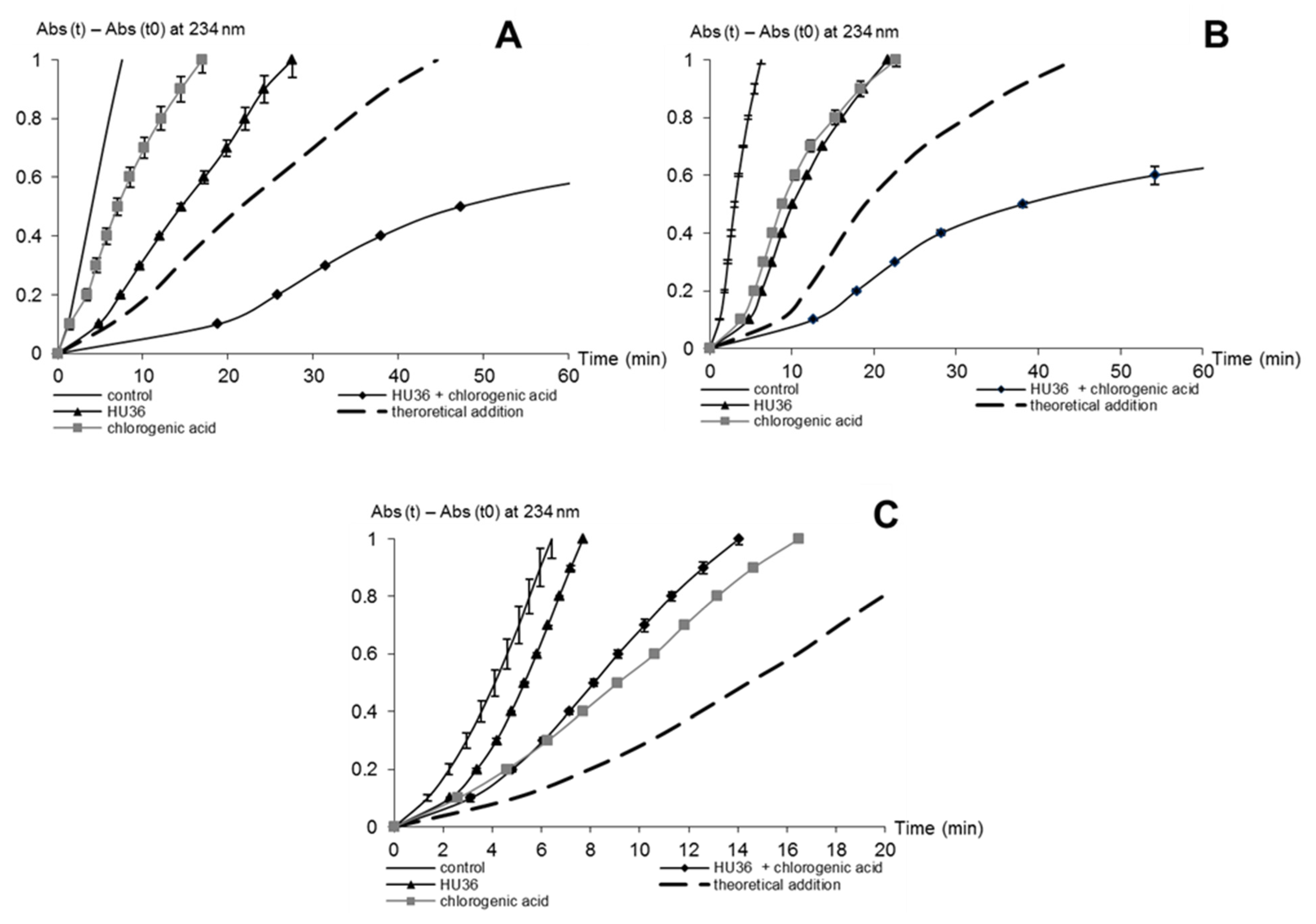
When used in combination, β-carotene and lycopene exhibit a slightly greater stability toward iron-induced autoxidation than the individual carotenoids in equivalent concentrations. It is possible that each carotenoid may be protected by the radical-scavenging activity of the other. However, HPLC analysis would be required to reveal whether one carotenoid is more significantly spared than the other. Stability of the cocktail of HU36 carotenoids might be reinforced by similar interactions.
A mixture of lycopene and β-carotene displays the same efficiency at inhibiting lipid peroxidation as that expected from the activity of the individual pigments (additivity). Thus, the antioxidant activity of HU36 carotenoids might also correspond to the additive contribution of the individual carotenoids in the cocktail.
Combinations of HU36 carotenoids and phenolic antioxidants displayed synergistic activities in the inhibition of linoleic acid peroxidation induced by heme iron, but not by free iron. Phenolic antioxidants may act in synergy with HU36 carotenoids, either by protecting them from oxidation during lipid peroxidation, or by acting through a complementary antioxidant mechanism. Simultaneous monitoring of carotenoid consumption showed that α-tocopherol and the polyphenols tested did not significantly protect the carotenoids in the presence of iron (data not shown). Thus, synergism cannot be explained by a recycling of the bacterial carotenoids by the phenolic antioxidants.
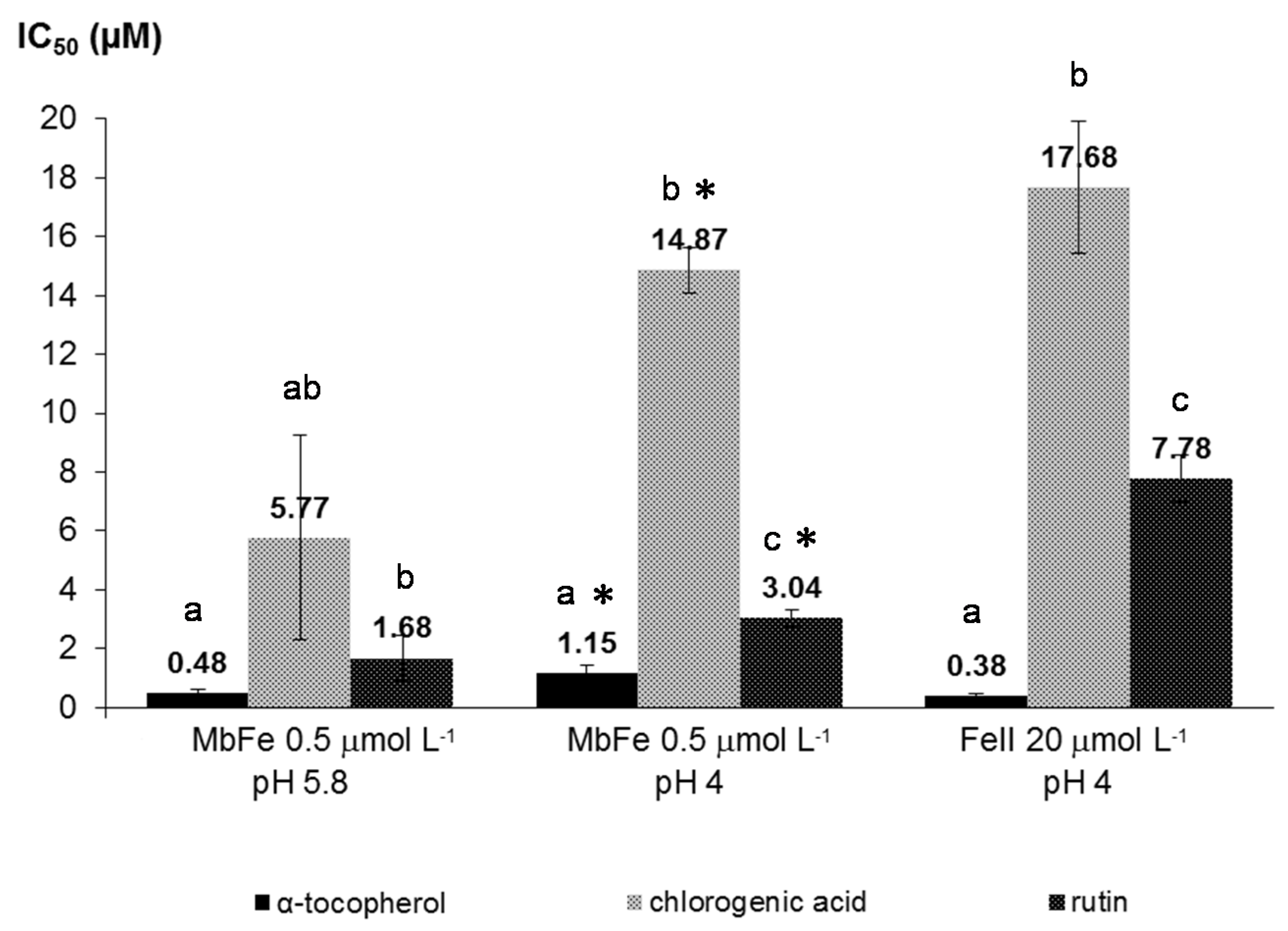
Complementary experiments have highlighted that chlorogenic acid and rutin rapidly reduce ferrylmyoglobin (Fe
IV), a potential initiator of lipid peroxidation, while HU36 carotenoids and α-tocopherol are essentially inactive. On the other hand, α-tocopherol and the bacterial carotenoids can act as chain-breaking antioxidants by direct scavenging of the propagating lipid peroxyl radicals in the lipid phase [
26]. Thus, complementary antioxidant mechanisms that may result in synergy can be suggested for the polyphenols and bacterial carotenoids (
Scheme 4). The heme cofactor of metmyoglobin was already proposed as a key component of synergism between α-tocopherol and quercetin [
12]. In this previously reported study, it was demonstrated that α-tocopherol, while unable to reduce the iron-oxo center of ferrylmyoglobin, can protect the porphyrin nucleus from oxidative degradation (as revealed by the decay of the Soret band). Moreover, quercetin, which can quickly reduce MbFe
IV=O, is partially spared when quercetin + α-tocopherol mixtures are used for the inhibition of lipid peroxidation (while α-tocopherol consumption is enhanced). Hence, the synergism observed was attributed to a regeneration of quercetin (and/or some of its oxidation products with a residual antioxidant activity) from its radical by α-tocopherol via electron transfer through the porphyrin nucleus. A similar mechanism may apply between polyphenols and the bacterial carotenoids.
Scheme 4.
Proposed mechanism for the complementary antioxidant activity of marine bacterial carotenoids and phenolic compounds during heme-induced lipid peroxidation.
Scheme 4.
Proposed mechanism for the complementary antioxidant activity of marine bacterial carotenoids and phenolic compounds during heme-induced lipid peroxidation.
4. Experimental Section
4.1. Chemicals
Natural (all-
E) lycopene from tomato oleoresin (C
40H
56, M = 536 g·mol
−1, >90%) was obtained from Conesa, Badajoz, Spain. Carotenoid extracts from
Bacillus strains HU36 were provided by members of the Colorspore consortium (Small Collaborative Project No. 207948, funded by FP7 and coordinated by the Royal Holloway University of London) [
37]. Synthetic type II (all-
E) β-carotene (>95%), α-tocopherol (95%), rutin (≥95%), chlorogenic acid (≥95%), menaquinone K2 (MK-4, ≥99.9%), polyoxyethyleneglycol 23 lauryl ether (Brij
®35), (9
Z,12
Z)-octadecadienoic acid (linoleic acid, >99%), FeSO
4·7H
2O (>99.5%), Fe(NO
3)
3·9H
2O (98%), myoglobin from equine heart (>90%, essentially salt-free), and ammonium formate (99.995%) were purchased from Sigma-Aldrich (St-Quentin-Fallavier, France). The buffers used in the experiments were a 0.02 mol·L
−1 acetate buffer (pH 4.0) and a 0.2 mol·L
−1 phosphate buffer (pH 5.8). Water was purified through a Millipore Q-Plus apparatus.
Acetic acid (100%), formic acid (>98%) and LC-MS grade methanol (MeOH) were purchased from Merck (Darmstadt, Germany). All other solvents were from Fisher-Scientific (Loughborough, UK). Dichloromethane (CH2Cl2) and acetonitrile (MeCN) were of UPLC grade, and ethylacetate (EtOAc) was of analytical grade.
4.2. Purification of the Bacterial Carotenoid Extracts
Fractionation of crude bacterial extracts by flash liquid chromatography. The crude bacterial extracts from HU36 were dissolved in CH2Cl2/MeOH (1/1, v:v), filtered through glass cotton to eliminate residues of the culture medium, and dried by evaporation under reduced pressure. For purification, about 250 mg of dry crude extract was dissolved in the smallest volume of MeOH and placed in the top a burette, (30 cm length, 2 cm diameter) filled with C-18 silica gel column 47 μm × 60 μm A.P.S. (DAVISIL® Chromatographic Silica Media for separation and purification applications, grade 633N, W.R. Grace Columbia, MD, USA) and containing a sintered glass filter. The C-18 silica gel was previously moistened with MeOH, 1% aq. CH3CO2H, and the column was rinsed with MeOH/H2O (80:20, v/v). The elution of the content of the bacterial extract was performed with a two solvent-gradient and the flow rate was maintained regular with a flow of argon. Solvent A was MeOH/H2O (80:20, v/v) and solvent B was EtOAc/CH2Cl2 (80:20, v/v). The gradient was: 250 mL A 100%, 75 mL A 90% B 10%, 75 mL A 85% B 15%, 75 mL A 80% B 20%, 100 mL A 75% B 25%, 250 mL A 65% B 35%, 75 mL A 50% B 50%, 50 mL de A 40% B 60%, 75 mL A 20% B 80%, 50 mL B 100%. The elution fractions were progressively collected in 25 mL glass tubes. A sample of each fraction was filtered through a 20 μm PTF filter and analyzed by UPLC-MS to ensure the absence of menaquinone in colored fractions and the presence of menaquinone in the last uncolored fraction. After each new purification, the C-18 silica gel was regenerated by several washings with Milli-Q water and MeOH/CH2Cl2 (1:1, v/v).
Structural analysis of the fractions. Reversed-phase ultra-fast liquid chromatography analyses were performed on a Waters Acquity™ Ultra Performance LC® system equipped with a diode array detector and coupled to an esquire HCT™ (high capacity trap) ultra-MS® mass spectrophotometer (Bruker-Daltonics). A Waters Acquity™ reversed-phase C18 column type HSS T3 (2.1 mm× 150 mm, 1.8 μm particle size) was used to separate fraction components. For the separation, gradient elution was performed at 25 °C, using a two solvent mobile phase at a flow rate of 0.5 mL·min−1. Eluent A contained 5 μmol·L−1 HCO2H and 10 mmol·L−1 HCO2NH4 in a mixture of MeCN/MeOH/H2O (60:20:20, v/v/v). Eluent B contained 5 μmol·L−1 HCO2H in EtOAc/CH2Cl2 (80:20, v/v). The gradient was as follows: 0% B held for 2 min, linearly increasing to 55% B over 14.5 min, holding at 55% B for 0.5 min, increasing to 60% B over 7 min, increasing to 70% B over 3 min, holding at 70% B for 1 min, returning to 0% B over 0.01 min, and holding at 0% B for 2 additional minutes (to rinse the system and return the column to the initial conditions before the following injection). Absorbance was followed by UV-VIS detection every 2 nm from 250 to 800 nm, with a time interval of 0.1 min. The effluent of the column was interfaced with the ion source of the MS using APCI (atmospheric pressure chemical ionization) alternating between positive and negative mode. The additional MS parameters were as follows: corona intensity = 1 μA, cone pressure = 40 psi, dry gas flow rate = 4 L·min−1 at a temperature of 300 °C. Spectra were acquired every 40 ms with a mass range of 50 to 1000 m/z. For optimal detection, windows were drawn to focus on target masses throughout the analysis. A m/z of 350 was targeted for t = 0–7 min, m/z 600 for t = 7–11.5 min, and m/z 750 for t = 11.5–26 min. In each fraction, m/z = 649.3 corresponding to MQ-7 was searched. Fractions were collected, gathered, and the total volume was measured and evaporated under reduced pressure. Finally, a pure bacterial carotenoid stock solution was obtained by dissolving the dry residue in MeOH/CH2Cl2 (1:1, v/v).
Quantification of purified bacterial carotenoids and the menaquinone content of the initial crude extract. Before evaporation, a sub-sample of the total volume of collected fractions was evaporated under reduced pressure, and dissolved it in the same volume of CH
2Cl
2 for spectrophotometric measurement. Absorbance was recorded at the respective λ
max for the particular parent carotenoid (previously described in the introduction) and the fraction concentration was calculated using the previously determined molar absorption coefficient (ε = 165 × 10
3 M
−1·cm
−1 at 454 nm) [
31]. Total extracted MQ-7 was quantified by UPLC-MS using an external calibration curve established with commercially available menaquinone.
4.3. Chemical Models of Antioxidant Activity in the Gastric Compartment
Stock solutions of antioxidants. Four stock carotenoid solutions (~1 mmol·L
−1) were used: a crude bacterial extract from HU36, purified bacterial carotenoids extract from HU36, (all-
E) lycopene, and (all-
E) β-carotene. Bacterial extracts were dissolved in CH
2Cl
2/MeOH (1:1, v/v) and lycopene and β-carotene in CH
2Cl
2. The concentrations of stock carotenoid solutions were calculated by spectrophotometric measurement. The molar absorption coefficients used were 128.5 × 10
3 L·mol
−1·cm
−1 at 460 nm for β-carotene, 178 × 10
3 L·mol
−1·cm
−1 at 482 nm for lycopene and 165 × 10
3 L·mol
−1·cm
−1 at 454 nm for HU36 carotenoids in CH
2Cl
2 [
38].
A 232 μmol·L−1 α-tocopherol solution, a 1 mmol L−1 chlorogenic acid solution, and a 350 μmol·L−1 rutin solution were also prepared by dissolving 1.00 mg, 3.54 mg, and 2.74 mg, respectively, in 10 mL MeOH.
Micelle solution. Both stability and antioxidant tests were performed using a model adapted from a previous study [
26]. Experiments were performed at 37 °C, pH 4 (0.2 mol·L
−1 acetate buffer) and pH 5.8 (0.2 mol·L
−1 phosphate buffer), mimicking the two stages of digestion in humans [
27]. The synthetic non-ionic hydroperoxide-free surfactant Brij
®35 (polyoxyethyleneglycol dodecyl ether) was selected for the preparation of carotenoid micelles in the aqueous medium [
39]. Brij
®35 was dissolved in CH
2Cl
2 to obtain a 40 mmol·L
−1 stock solution. Linoleic acid was added only for antioxidant tests (not for stability tests). A 28 mmol·L
−1 stock solution was prepared by dissolving 39.25 mg commercial linoleic acid in 5 mL CH
2Cl
2.
Aliquots of stock carotenoid solutions and/or other antioxidant solutions were dispersed with 2 mL stock surfactant solution and 0 or 250 μL linoleic acid stock solution. After evaporation of the solvent under reduced pressure and dilution of the dried viscous residue in 20 mL aqueous buffer, initial concentrations were 4 mmol·L−1 Brij®35 and 10 μmol·L−1 carotenoid for stability tests; 4 mmol·L−1 Brij®35, 0.7 mmol·L−1 linoleic acid, 0 to 8 μmol·L−1 carotenoid, and/or 0 to 100 μmol·L−1 of another antioxidant for the antioxidant activity experiments. Micelles were formed by stirring the solution on a magnetic stir plate until total homogenization. The micelle solution was perfectly transparent.
Iron solutions. Oxidation was initiated by FeII, FeIII, or metmyoglobin (MbFeIII, heme iron).
Stock 0.5 mol·L
−1 Fe
III solution was prepared by dissolving 404 mg ferric nitrate nonahydrate in 2 mL 0.1 mol·L
−1 nitric acid, stock 0.5 mol·L
−1 Fe
II solution was prepared by dissolving 279.4 mg ferrous sulphate heptahydrate in 2 mL 0.1 mol·L
−1 sulphuric acid and stock 0.5 mmol·L
−1 MbFe
III solution was prepared by dissolving 8.9 mg myoglobin from equine heart in 1 mL Milli-Q water. Due to the poor solubility of myoglobin, the stock solution was filtered and its concentration was checked by absorbance measurement at 505 nm, with ε = 97 × 10
3 L·mol
−1·cm
−1 [
40]. Dilute iron solutions were then prepared from the stock solution. For stability tests, seven Fe
II solutions (0.5–25 mmol·L
−1) were prepared in sulfuric acid, seven Fe
III solutions (0.05–25 mmol·L
−1) were prepared in nitric acid, and seven MbFe solutions (0.5–500 μmol·L
−1) were prepared in milliQ water. For antioxidant tests, only 1 mmol·L
−1 Fe
II and 25 μmol·L
−1 MbFe
III were used.
4.4. Stability Study and Lipid Peroxidation Inhibition
UV-VIS Spectroscopy. All experiments were performed in glassware protected from light, covered with Teflon stoppers, and kept under magnetic stirring at a constant temperature of 37 °C. Spectra were recorded on a Specord S-600 diode-array spectrophotometer (optical path length = 1 cm), equipped with an eight-cell rail, a magnetic stirring device, and a thermostatic bath (Analytik Jena AG, Jena, Germany). Each treatment was run in triplicate.
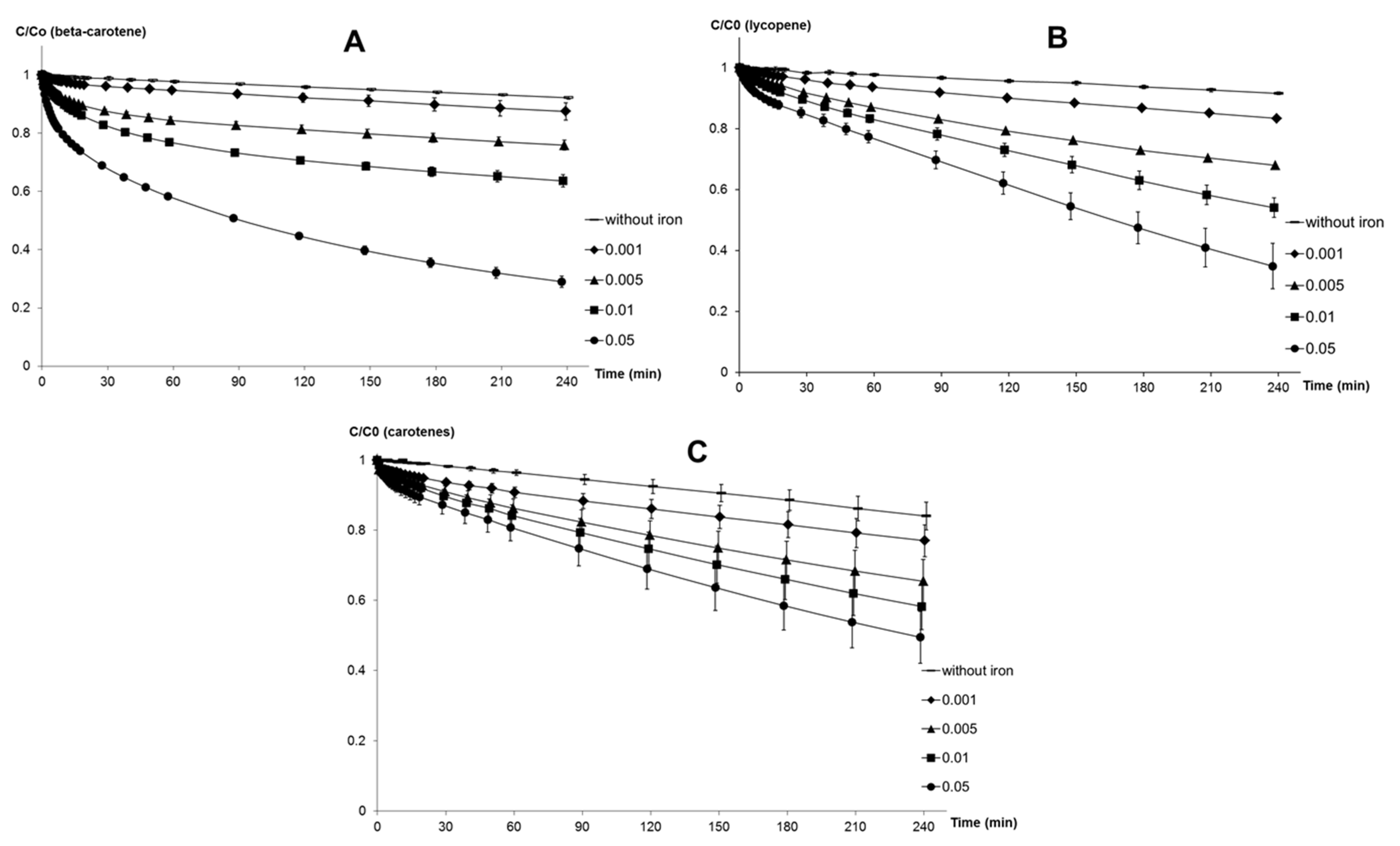
Kinetics of carotenoid oxidation in micelles. Stability measurements were performed at pH 4 and 37 °C. 1.96 mL of the micelle solution containing 4 mmol·L−1 Brij®35 and 10 μmol·L−1 carotenoids alone or in combination were transferred to a macro quartz spectrophotometer cell. At time zero, oxidation was initiated by addition of 40 μL iron solution to the reaction medium. Oxidation was initiated by iron concentrations in the range of 1–1000 μmol·L−1 FeII and FeIII or 0.01–10 μmol·L−1 MbFeIII. Residual carotenoid concentrations were measured directly by UV-VIS spectrometry (normalized to a baseline of acetate buffer alone).
Spectra were recorded at regular intervals for 4 h, from 300 to 1000 nm, and kinetic curves were plotted by extracting the absorbance at the wavelength maximum of each respective carotenoid sample. Results were expressed as a % relative to the initial carotenoid content:
Antioxidant activity in the micelle solution. 1.96 mL of the micelle solution, containing 4 mmol·L−1 Brij®35, 0.7 mmol·L−1 linoleic acid, and increasing concentrations of carotenoids and/or α-tocopherol, rutin or chlorogenic acid (plus the control experiment without antioxidants) was transferred to a macro quartz spectrophotometer cell. At time zero, oxidation was initiated by addition of 40 μL iron solution to the reaction medium. Inhibition of linoleic acid oxidation by the carotenoids was evaluated at pH 4 with initiation by FeII (20 μmol·L−1) or metmyoglobin (0.5 μmol·L−1). At pH 5.8, inhibition of linoleic acid oxidation was evaluated only in the metmyoglobin model.
Lipid peroxidation was followed by measuring the concentration of conjugated dienes (CD: mainly hydroperoxides + minor amounts of alcohols) for about 90 min. Their absorbance was directly recorded by UV-VIS spectroscopy at 234 nm (baseline recorded on acetate buffer or phosphate buffer) [
12,
26]. The residual carotenoid concentration was simultaneously measured at the λ
max value in the visible range.
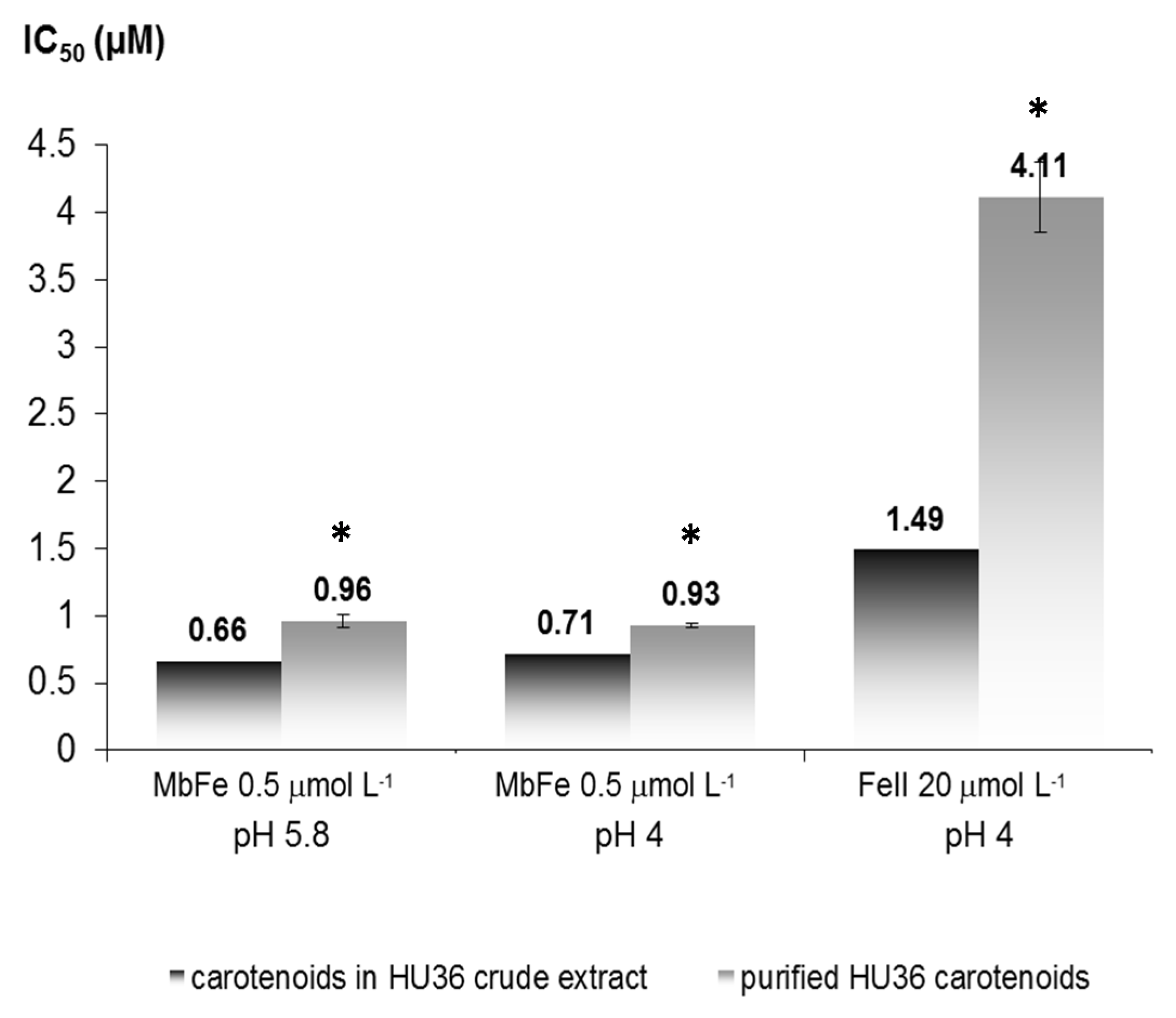
Kinetic curves of CD accumulation were plotted without antioxidant (control) and with increasing concentrations of antioxidant. Inhibition of linoleic acid peroxidation was measured by the delay in hydroperoxide accumulation: the T/T0 ratio was calculated for each antioxidant concentration, with T0 = time required to produce a fixed CD concentration (corresponding to a 0.5 increase in A (234 nm) after iron addition) in the control experiment (no antioxidant), and T = time required to produce the same CD concentration in the presence of the antioxidant. The T/T0 vs. initial antioxidant concentration was plotted for each antioxidant in the three models: MbFeIII at pH 4 and 5.8 and FeII at pH 4. The IC50 value was defined as the antioxidant concentration giving T/T0 = 0.5.
Reduction of Ferrylmyoglobin by the Carotenoids. The experimental procedure was adapted from previous studies [
26]. Ferrylmyoglobin (MbFe
IV=O) was first formed in a spectrophotometer cell (2 mL) by adding small volumes (60 μL) of a concentrated aqueous solution of H
2O
2 (2 mmol·L
−1, final concentration in the cell = 60 μmol·L
−1) to a 60 μmol·L
−1 MbFe
III solution in a pH 7 phosphate buffer containing 4 mmol·L
−1 Brij
®35. Spectral changes featuring the conversion of MbFe
III (specific peak at 505 nm) into MbFe
IV=O (specific peak at 590 nm) were recorded in the visible range until stability was achieved (2–3 min). Then, small volumes (about 50 μL) of a concentrated solution of carotenoid in MeOH/THF (1/1, v/v) were added (final concentrations of the carotenoids in the cell = 0 or 25 or 100 μmol·L
−1) and the reduction of MbFe
IV=O back to MbFe
III was monitored at 590 nm.
Statistical analyses. Each treatment was run in triplicate. The experimental results were expressed as means ± standard deviation. Differences between means were assessed either with the student t-test (when two means were compared) or using ANOVA followed by the post hoc analysis using Tukey-Kramer. A P ≤ 0.05 was considered statistically significant. All statistical analyses were performed using Statview software.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



