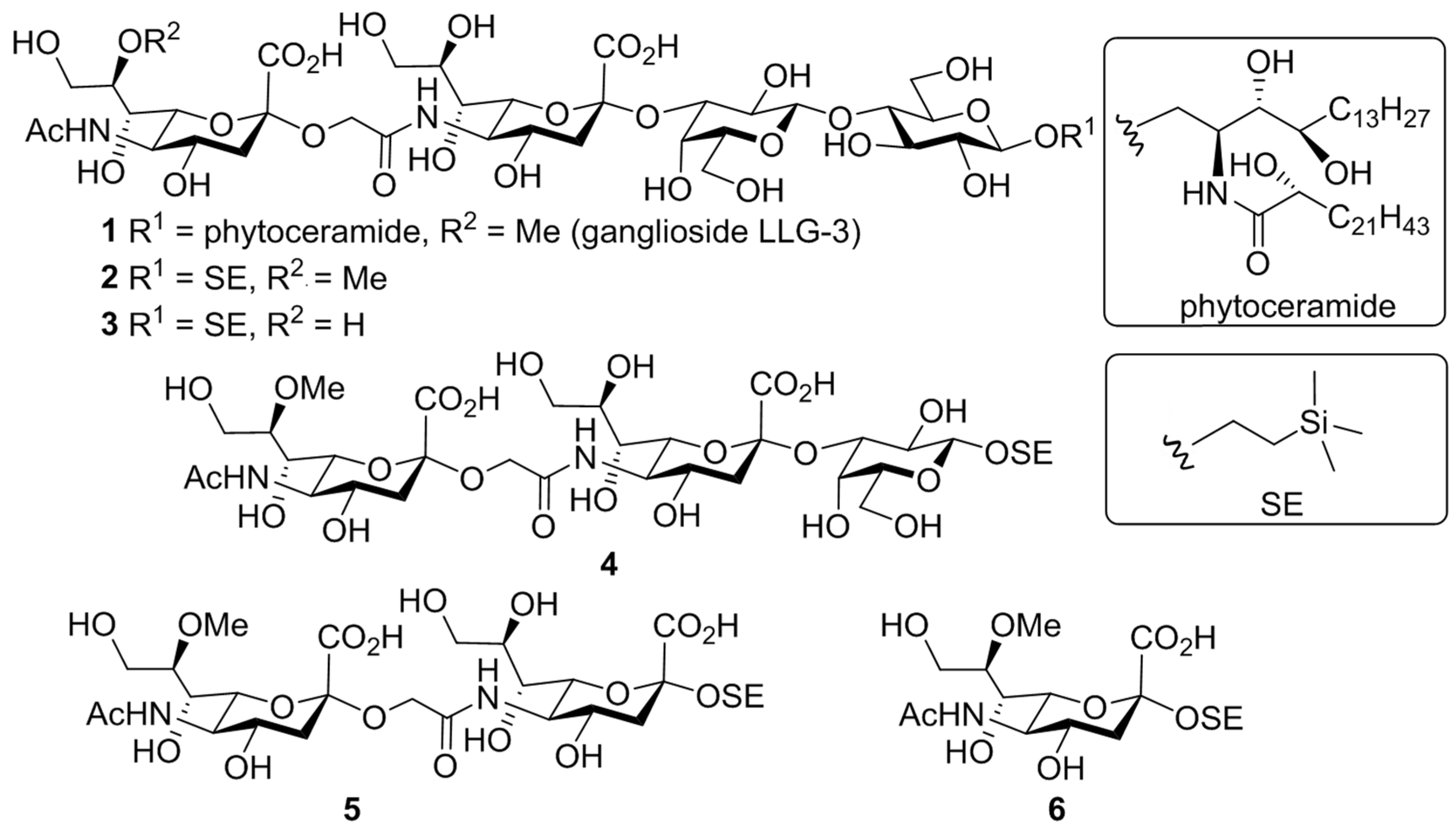
3.1.2. Experimental Procedures
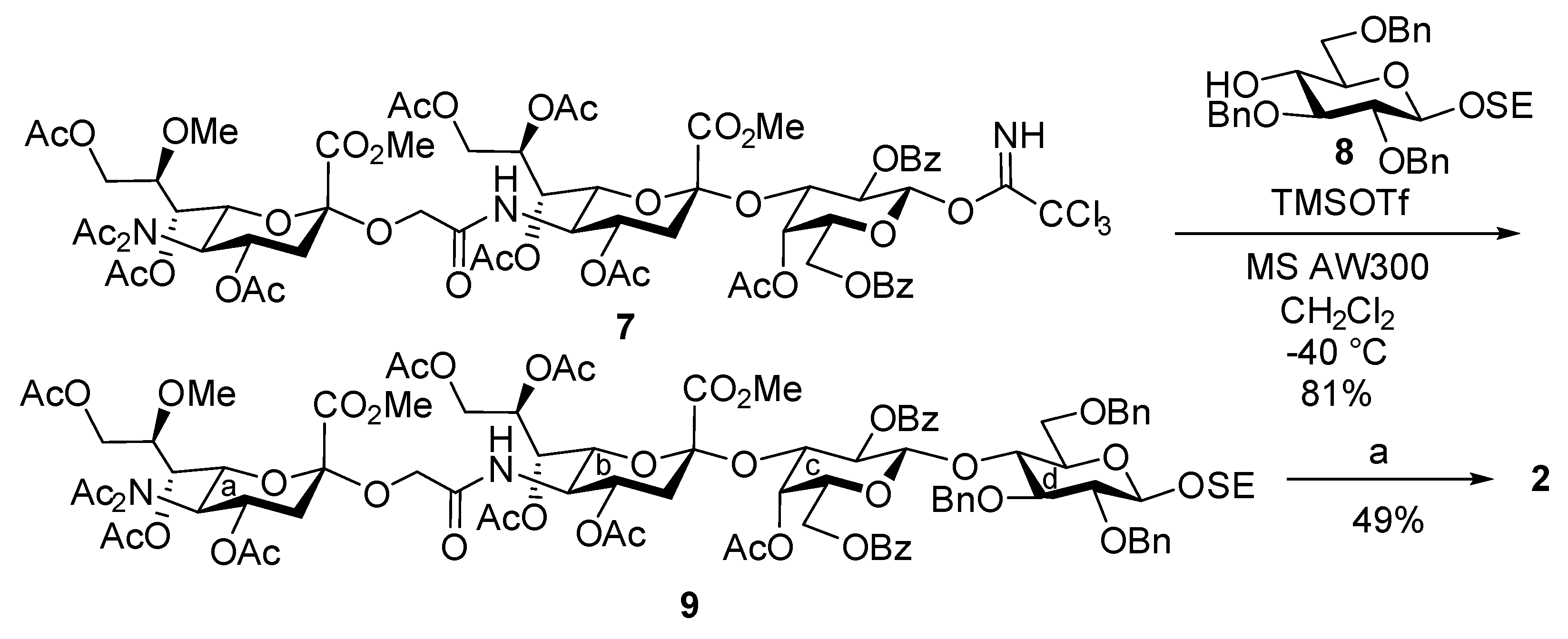
2-(Trimethylsilyl)ethyl [methyl 4,7,8,9-tetra-O-acetyl-3,5-dideoxy-5-(methyl 4,7,9-tri-O-acetyl-5-diacetamido-3,5-dideoxy-8-O-methyl-d-glycero-α-d-galacto-2-nonulopyranosylonate)oxyacetamido-d-glycero-α-d-galacto-2-nonulopyranosylonate]-(2→3)-(4-O-acetyl-2,6-di-O-benzoyl-β-d-galactopyranosyl)-(1→4)-2,3,6-tri-O-benzyl-β-d-glucopyranoside (9). To a mixture of 7 (41.2 mg, 26.6 μmol) and 8 (29.2 mg, 53.2 μmol) in CH2Cl2 (1.0 mL) were added 4 Å molecular sieves (AW300, 60 mg) at room temperature. After stirring for 30 min, the mixture was cooled to –40 °C. TMSOTf (0.1 µL, 5.32 µmol) was then added to the mixture at −40 °C. After stirring for 5 min at −40 °C as the reaction was monitored by TLC (20:1 CHCl3–MeOH), the reaction was quenched by the addition of triethylamine. The solution was diluted with CHCl3 and filtered through Celite. The filtrate was then washed with satd. aq. NaHCO3 and brine. The organic layer was subsequently dried over Na2SO4, and concentrated. The residue was purified by silica gel column chromatography (50:1 to 40:1 toluene–MeOH) to give 9 (41.8 mg, 81%): [α]D +10.0° (c 0.9, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.25–7.17 (m, 25H, 5Ar), 6.03 (d, 1H, JNH,5 = 10.3 Hz, NHb), 5.69 (m, 1H, H-8b), 5.58 (td, 1H, J3eq,4 = 5.2 Hz, J3ax,4 = J4,5 = 10.0 Hz, H-4a), 5.31 (dd, 1H, J1,2 = 8.0 Hz, J2,3 = 10.3 Hz, H-2c), 5.20 (d, 1H, H-1c), 5.11 (dd, 1H, J6,7 = 2.8 Hz, J7,8 = 9.8 Hz, H-7b), 5.07 (d, 1H, J3,4 = 3.5 Hz, H-4c), 4.98 (dd, 1H, J6,7 = 1.7 Hz, J5,6 = 10.3 Hz, H-6a), 4.93 (dd, 1H, J7,8 = 8.0 Hz, H-7a), 5.02 and 4.89 (2d, 2H, PhCH2), 4.87 and 4.66 (2d, 2H, PhCH2), 4.83 (m, 1H, H-4b), 4.76 (dd, 1H, H-3c), 4.39–4.21 (m, 6H, PhCH2, H-9ab, H-6b, H-4d, H-5a), 4.12–3.85 (m, 11H, OCH2, H-9aa, H-9ba, OCH2CH2Si, H-1d, H-3d, H-5b, CO2Me), 3.76 (s, 3H, CO2Me), 3.70–3.59 (m, 2H, H-2d, H-6ac), 3.54 (m, 1H, OCH2CH2Si), 3.47–3.33 (m, 6H, H-6bd, H-5d, H-6bc, OMe), 2.88 (dd, 1H, J3eq,4 = 5.1 Hz, Jgem = 13.2 Hz, H-3eqa), 2.59 (dd, 1H, J3eq,4 = 3.6 Hz, Jgem = 12.1 Hz, H-3eqb), 2.38–1.96 (m, 30H, 10Ac), 1.93 (dd, 1H, J3ax,4 = 10.3 Hz, H-3axa), 1.71 (t, 1H, J3ax,4 = 12.1 Hz, H-3axb), 1.02–0.98 (m, 2H, OCH2CH2Si), 0.00 (s, 9H, SiMe3); 13C NMR (150 MHz, CDCl3) δ 176.5, 175.8, 173.0, 172.9, 172.6, 172.5, 172.4, 172.3, 172.2, 172.1, 171.1, 170.3, 169.8, 167.6, 167.3, 141.4, 140.9, 140.8, 135.4, 135.2, 132.6, 132.3, 132.0, 131.9, 131.9, 130.8, 130.5, 130.4, 130.4, 130.4, 130.4, 130.4, 130.3, 130.3, 130.2, 130.2, 129.7, 129.5, 129.4, 129.4, 129.4, 129.3, 129.3, 129.3, 129.2, 105.1, 103.0, 100.9, 99.1, 85.2, 84.2, 76.3, 74.6, 74.3, 74.0, 72.5, 71.8, 71.5, 71.3, 70.2, 70.2, 69.0, 68.8, 67.7, 67.1, 67.1, 66.8, 66.7, 66.3, 63.2, 61.4, 61.2, 60.9, 57.8, 57.8, 56.3, 52.9, 52.8, 47.8, 37.8, 37.2, 29.4, 27.7, 25.8, 21.1, 20.6, 20.5, 20.5, 20.5, 20.4, 20.1, 18.2, 0.0, 0.0, 0.0; HRMS (ESI) m/z: found [M + Na]+ 1961.6972, C95H118N2O39Si calcd for [M + Na]+ 1961.6973.
2-(Trimethylsilyl)ethyl [5-(5-acetamido-3,5-dideoxy-8-O-methyl-d-glycero-α-d-galacto-2-nonulopyranosylonic acid)oxyacetamido-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonic acid]-(2→3)-(β-d-galactopyranosyl)-(1→4)-β-d-glucopyranoside (2). To a solution of 9 (39.6 mg, 21.0 µmol) in EtOAc (1.0 mL) was added Pd(OH)2/C (20%, 40.0 mg) at room temperature. After stirring for 2 h at room temperature under a hydrogen atmosphere as the reaction was monitored by TLC (20:1 CHCl3–MeOH), the mixture was filtered through Celite. The filtrate was concentrated, and the crude residue was exposed to high vacuum. The resulting residue was then dissolved in pyridine (1.0 mL), and lithium chloride (5.2 mg, 126 µmol) was added at room temperature. After stirring for 18 h under reflux as the reaction was monitored by TLC (5:2:0.2 CHCl3–MeOH–H2O), the reaction mixture was co-evaporated with toluene. The crude residue was purified by gel filtration column chromatography (Sephadex LH-20) using MeOH as eluent. The purified product was exposed to high vacuum, and then dissolved in 0.1 M aq NaOH (2.1 mL). After stirring for 42 h at room temperature and another 10 h at 40 °C as the reaction was monitored by TLC (4:6:1 CHCl3–MeOH–H2O), the reaction mixture was neutralized with Dowex (H+) resin. The resin was filtered through cotton wool and the filtrate was then evaporated. The residue was purified by silica gel column chromatography (12:8:1 to 4:6:1 CHCl3–MeOH–H2O), followed by gel filtration column chromatography (Sephadex LH-20) using MeOH–H2O (5:1) as eluent to give 2 (10.7 mg, 49%): [α]D –41.9° (c 0.2, MeOH); 1H NMR (500 MHz, CD3OD) δ 4.45 (d, 1H, J1,2 = 7.6 Hz, H-1), 4.33 (d, 1H, J1,2 = 8.2 Hz, H-1), 4.34 and 4.27 (2d, 2H, OCH2), 4.04–3.21 (m, 31H, H-4a, H-5a, H-6a, H-7a, H-8a, H-9aa, H-9ba, H-4b, H-5b, H-6b, H-7b, H-8b, H-9ab, H-9bb, H-2c, H-3c, H-4c, H-5c, H-6ac, H-6bc, H-2d, H-3d, H-4d, H-5d, H-6ad, H-6bd, OCH2CH2Si, OMe), 2.78 (dd, 1H, J3eq,4 = 3.4 Hz, Jgem = 12.6 Hz, H-3eq), 2.49 (dd, 1H, J3eq,4 = 4.6 Hz, Jgem = 12.6 Hz, H-3eq), 2.00 (s, 3H, Ac), 1.77–1.69 (m, 2H, H-3axa, H-3axb), 1.07–0.92 (m, 2H, OCH2CH2Si), 0.00 (s, 9H, SiMe3); 13C NMR (125 MHz, CD3OD) δ 175.5, 174.8, 174.8, 174.5, 104.5, 103.3, 101.5, 101.1, 81.4, 80.3, 77.2, 76.7, 76.2, 74.4, 74.3, 74.0, 73.0, 71.1, 70.8, 69.3, 69.0, 69.0, 69.0, 68.8, 68.6, 63.8, 63.7, 62.5, 61.6, 61.4, 59.1, 53.7, 53.5, 41.4, 40.3, 22.9, 19.0, −1.4, −1.4, −1.4; HRMS (ESI) m/z: found [M − 2H + Na]− 1075.3631, C40H70N2O28Si calcd for [M − 2H + Na]− 1075.3631.
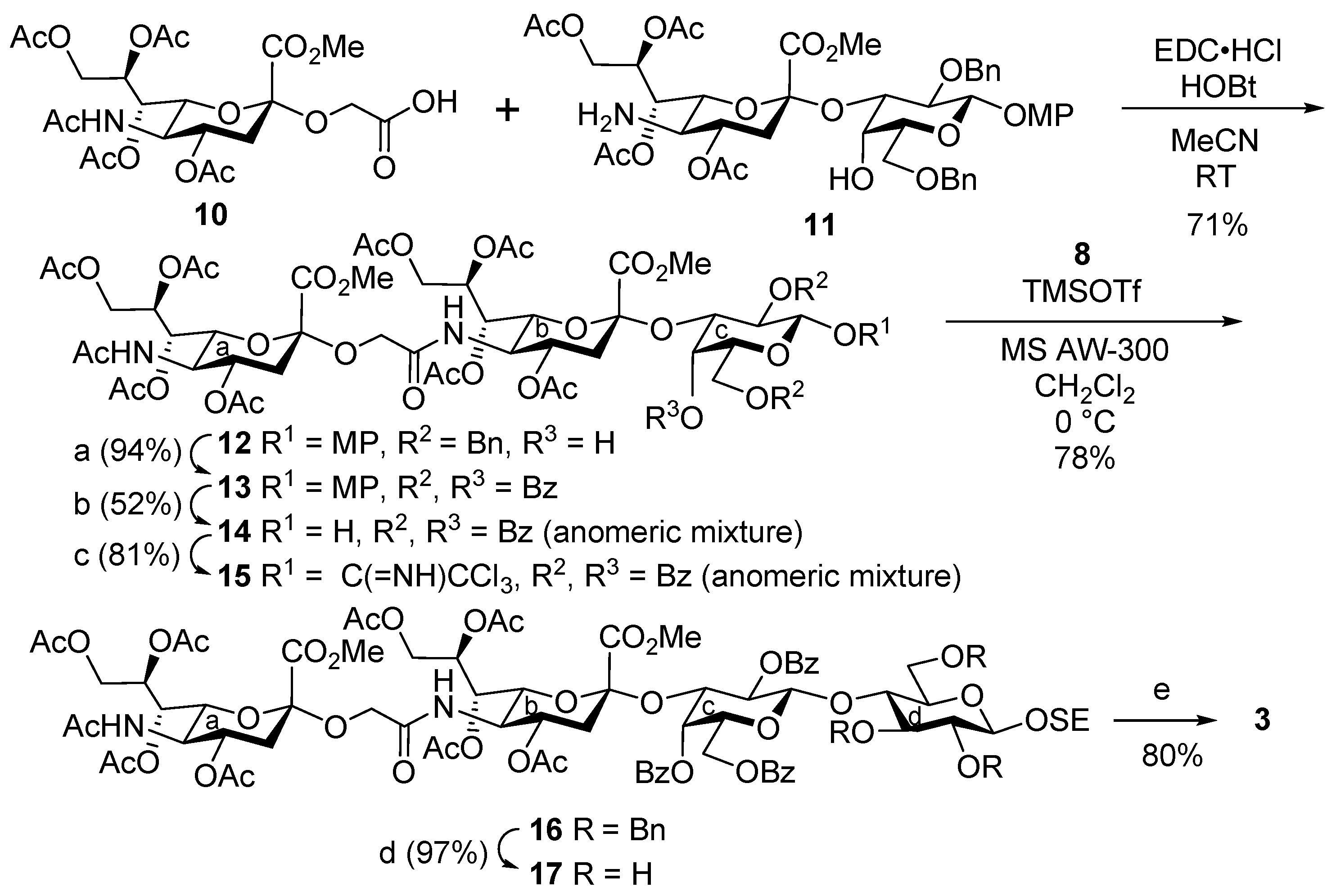
4-Methoxyphenyl [methyl 4,7,8,9-tetra-O-acetyl-3,5-dideoxy-5-(methyl 5-acetamido-4,7,8,9-tetra-O-acetyl-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonate)oxyacetamido-d-glycero-α-d-galacto-2-nonulopyranosylonate]-(2→3)-2,6-di-O-benzyl-β-d-galactopyranoside (12). To a solution of 10 (414 mg, 735 μmol) and 11 (788 mg, 878 μmol) in acetonitrile (15.1 mL) were added EDC·HCl (260 mg, 1.36 mmol) and HOBt (50.8 mg, 377 μmol) at room temperature. After stirring for 7 h at room temperature as the reaction was monitored by TLC (15:1 CHCl3–MeOH), the mixture was diluted with EtOAc. The solution was then washed with 2 M HCl, satd. aq. NaHCO3 and brine. The organic layer was subsequently dried over Na2SO4, and concentrated. The resulting residue was purified by silica gel column chromatography (55:1 CHCl3–MeOH) to give 12 (762 mg, 71%): [α]D +3.5° (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.42–6.78 (m, 14H, 3Ar), 6.10 (d, 1H, JNH,5 = 10.0 Hz, NHb), 5.43 (td, 1H, J8,9a = 3.0 Hz, J7,8 = J8,9b = 10.5 Hz, H-8a), 5.33–5.32 (m, 2H, H-7b, H-8b), 5.23 (dd, 1H, J6,7 = 2.3 Hz, H-7a), 5.04 (d, 1H, JNH,5 = 10.0 Hz, NHa), 4.95–4.87 (m, 4H, H-4a, H-4b, H-1c, PhCH2), 4.82 (d, 1H, Jgem = 11.5 Hz, PhCH2), 4.57 (2d, 2H, Jgem = 11.5 Hz, PhCH2), 4.34 (dd, 1H, Jgem = 12.5 Hz, H-9aa), 4.29–4.26 (m, 2H, H-9ab, H-3c), 4.18–4.06 (m, 6H, H-5a, H-6a, H-5b, H-6b, H-9bb, OCH2), 3.95 (dd, 1H, H-9ba), 3.86–3.76 (m, 15H, H-2c, H-4c, H-5c, H-6ac, H-6bc, OCH2, 3OMe), 2.72 (d, 1H, JOH,4 = 3.0 Hz, OHc), 2.67 (dd, 1H, J3eq,4 = 5.0 Hz, Jgem = 13.0 Hz, H-3eqa), 2.62 (dd, 1H, J3eq,4 = 4.5 Hz, Jgem = 12.5 Hz, H-3eqb), 2.14–1.89 (m, 29H, H-3axa, H-3axb, 9Ac); 13C NMR (125 MHz, CDCl3) δ 170.9, 170.6, 170.5, 170.3, 170.2, 170.0, 170.0, 169.8, 168.6, 167.6, 155.2, 151.7, 138.9, 138.2, 128.3, 128.2, 127.9, 127.6, 127.6, 127.4, 118.6, 114.5, 102.8, 98.5, 98.1, 75.7, 75.0, 73.6, 73.1, 73.0, 72.9, 69.4, 68.8, 68.5, 68.2, 67.5, 67.2, 63.8, 62.6, 62.2, 55.6, 53.2, 53.1, 49.3, 48.6, 37.5, 37.0, 23.2, 21.2, 21.0, 20.8, 20.8, 20.7, 20.7; HRMS (ESI) m/z: found [M + Na]+ 1451.4899, C67H84N2O32 calcd for [M + Na]+ 1451.4899.
4-Methoxyphenyl [methyl 4,7,8,9-tetra-O-acetyl-3,5-dideoxy-5-(methyl 5-acetamido-4,7,8,9-tetra-O-acetyl-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonate)oxyacetamido-d-glycero-α-d-galacto-2-nonulopyranosylonate]-(2→3)-2,4,6-tri-O-benzoyl-β-d-galactopyranoside (13). To a solution of 12 (650 mg, 455 µmol) in EtOAc (9.1 mL) was added Pd(OH)2/C (20%, 650 mg) at room temperature. After stirring for 30 min at room temperature under a hydrogen atmosphere as the reaction was monitored by TLC (15:1 CHCl3–MeOH), the mixture was filtered through Celite. The filtrate was concentrated, and the crude residue was exposed to high vacuum. Then, it was dissolved in pyridine (2.3 mL). Benzoic anhydride (618 mg, 2.73 mmol) and DMAP (5.6 mg, 45.5 µmol) were added to the mixture at 0 °C. After stirring for 11 h at room temperature as the reaction was monitored by TLC (15:1 CHCl3–MeOH), the reaction was quenched by the addition of MeOH at 0 °C. The mixture was co-evaporated with toluene and the residue was then diluted with CHCl3, and washed with 2 M HCl, H2O and satd. aq. NaHCO3. The organic layer was subsequently dried over Na2SO4, and concentrated. The resulting residue was purified by silica gel column chromatography (50:1 CHCl3–MeOH) to give 13 (666 mg, 94%): [α]D +48.0° (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.18–6.68 (m, 19H, 4Ar), 5.94 (d, 1H, JNH,5 = 10.4 Hz, NHb), 5.69 (dd, 1H, J1,2 = 8.0 Hz, J2,3 = 10.1 Hz, H-2c), 5.63 (td, 1H, J8,9a = 2.5 Hz, J8,9b = 6.4 Hz, J7,8 = 9.6 Hz, H-8b), 5.40 (d, 1H, J3,4 = 3.3 Hz, H-4c), 5.34–5.26 (m, 3H, H-7a, H-8a, H-1c), 5.14–5.10 (m, 2H, NHa, H-7b), 4.97 (dd, 1H, H-3c), 4.90 (td, 1H, J3eq,4 = 4.7 Hz, J3ax,4 = J4,5 = 10.3 Hz, H-4a), 4.84 (td, 1H, J3eq,4 = 4.5 Hz, J3ax,4 = J4,5 = 10.6 Hz, H-4b), 4.49 (dd, 1H, J5,6a = 7.6 Hz, Jgem = 11.4 Hz, H-6ac), 4.43 (dd, 1H, J5,6b = 5.6 Hz, H-6bc), 4.31–4.23 (m, 3H, H-5c, H-9aa, H-9ab), 4.13 (dd, 1H, J6,7 = 2.0 Hz, J5,6 = 10.8 Hz, H-6a), 4.08–4.02 (m, 3H, H-5a, H-9ba, OCH2), 3.93–3.86 (m, 5H, H-5b, H-9bb, OMe), 3.82 (s, 3H, OMe), 3.75–3.69 (m, 5H, H-6b, OCH2, OMe), 2.64 (dd, 1H, Jgem = 12.9 Hz, H-3eqa), 2.55 (dd, 1H, Jgem = 12.7 Hz, H-3eqb), 2.24–1.88 (m, 25H, 8Ac, H-3axa), 1.68 (t, 1H, H-3axb), 1.44 (s, 3H, Ac); 13C NMR (125 MHz, CDCl3) δ 170.9, 170.6, 170.6, 170.3, 170.2, 170.1, 170.0, 169.8, 168.5, 168.3, 167.6, 165.9, 165.4, 155.5, 151.5, 133.4, 133.2, 133.1, 130.2, 130.1, 129.8, 129.3, 128.5, 128.5, 128.4, 119.0, 114.4, 101.2, 98.4, 96.9, 77.6, 72.9, 71.9, 71.3, 71.0, 68.9, 68.8, 68.5, 68.4, 67.5, 67.1, 66.7, 63.8, 68.5, 68.4, 67.5, 67.1, 66.7, 63.8, 62.7, 62.5, 62.2, 55.6, 53.3, 53.2, 49.3, 48.2, 37.5, 37.4, 23.2, 21.5, 21.0, 20.8, 20.7, 20.6, 20.3; HRMS (ESI) m/z: found [M + Na]+ 1583.4748, C74H84N2O35 calcd for [M + Na]+ 1583.4747.
[Methyl 4,7,8,9-tetra-O-acetyl-3,5-dideoxy-5-(methyl 5-acetamido-4,7,8,9-tetra-O-acetyl-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonate)oxyacetamido-d-glycero-α-d-galacto-2-nonulopyranosylonate]-(2→3)-2,4,6-tri-O-benzoyl-d-galactopyranose (14). To a suspension of 13 (200 mg, 128 µmol) in acetonitrile/toluene/H2O (6:5:3, 2.6 mL) was added CAN (702 mg, 1.28 mmol) at 0 °C. After stirring for 5 min at 0 °C as the reaction was monitored by TLC (30:1 CHCl3–MeOH), the mixture was diluted with EtOAc, and then washed with H2O, satd. aq. NaHCO3 and brine. The organic layer was subsequently dried over Na2SO4, and concentrated. The residue was roughly purified by silica gel column chromatography (45:1 CHCl3–MeOH). The crude product was exposed to high vacuum, and then dissolved in pyridine (1.0 mL). Benzoic anhydride (66.3 mg, 293 µmol) and DMAP (1.2 mg, 9.8 µmol) were added to the solution at 0 °C. After stirring for 5.5 h at room temperature as the reaction was monitored by TLC (30:1 CHCl3–MeOH, developed twice), the reaction was quenched by the addition of MeOH at 0 °C. The mixture was co-evaporated with toluene, and the residue was then diluted with CHCl3, and washed with 2 M HCl, H2O and satd. aq. NaHCO3. The organic layer was subsequently dried over Na2SO4, and concentrated. The resulting residue was purified by silica gel column chromatography (50:1 CHCl3–MeOH). The obtained product was exposed to high vacuum, and then dissolved in DMF (890 µL). Hydrazine acetate (1.2 mg, 9.8 µmol) was added to the solution at 0 °C. After stirring for 9 h at room temperature as the reaction was monitored by TLC (30:1 CHCl3–MeOH, developed twice), the reaction mixture was diluted with EtOAc, and washed with H2O and brine. The organic layer was subsequently dried over Na2SO4, and concentrated. The resulting residue was purified by silica gel column chromatography (40:1 CHCl3–MeOH) to give 14 (96.2 mg, 52%, anomeric mixture): 1H NMR (500 MHz, CDCl3) δ 8.23–7.39 (m, 30H, 6Ph), 6.03 (d, 2H, JNH,5 = 10.3 Hz, NHb, NHb), 5.68–5.63 (m, 2H, H-8b, H-8b), 5.60–5.58 (m, 2H, H-2c, H-2c), 5.50 (d, 1H, J3,4 = 2.8 Hz, H-4c), 5.46 (d, 1H, J3,4 = 3.0 Hz, H-4c), 5.34–5.19 (m, 9H, H-8a, H-8a, H-7a, H-7a, NHa, NHa, H-1c, H-7b, H-7b), 5.14–5.08 (m, 2H, H-1c, H-3c), 5.03 (dd, 1H, J2,3 = 10.3 Hz, H-3c), 4.93–4.86 (m, 4H, H-4a, H-4a, H-4b, H-4b), 4.61 (m, 1H, H-5c), 4.53–4.45 (m, 3H, H-6ac, H-6ac, H-5c), 4.36–4.25 (m, 6H, H-6bc, H-9ab, H-9ab, H-6c, H-9aa, H-9aa), 4.15–3.96 (m, 12H, H-9ba, 2OCH2, H-5a, H-5a, H-5b, H-5b, H-9ba, H-6a, H-6a, H-9bb, H-9bb), 3.89, 3.84 and 3.83 (3s, 12H, 4OMe), 3.79–3.74 (m, 4H, 2OCH2, H-6b, H-6b), 2.67–2.63 (m, 2H, H-3eqa, H-3eqa), 2.55–2.51 (m, 2H, H-3eqb, H-3eqb), 2.22–1.88 (m, 50H, 16Ac, H-3axa, H-3axa), 1.70–1.66 (m, 7H, 2Ac, H-3axb), 1.65 (t, 1H, J3ax,4 = Jgem = 12.4 Hz, H-3axb); 13C NMR (125 MHz, CDCl3) δ 171.4, 171.0, 170.9, 170.7, 170.6, 170.3, 170.3, 170.2, 170.2, 170.0, 169.9, 168.7, 168.7, 168.3, 168.2, 167.6, 167.3, 165.9, 165.9, 165.9, 165.7, 133.5, 133.4, 133.3, 133.1, 133.1, 130.3, 130.2, 130.0, 129.9, 129.8, 129.8, 129.5, 129.3, 128.6, 128.6, 128.5, 128.3, 128.3, 98.4, 97.3, 97.0, 96.1, 92.0, 77.6, 73.5, 72.9, 72.5, 72.3, 71.2, 70.8, 69.9, 69.5, 68.8, 68.6, 68.5, 68.1, 67.7, 67.1, 67.0, 66.9, 63.7, 62.7, 62.6, 62.3, 62.2, 53.3, 53.3, 53.2, 49.3, 48.4, 37.7, 37.6, 37.4, 31.9, 29.7, 29.4, 23.2, 22.7, 21.5, 21.4, 21.0, 20.8, 20.7, 20.6; HRMS (ESI) m/z: found [M + Na]+ 1477.4329, C67H78N2O34 calcd for [M + Na]+ 1477.4328.
[Methyl 4,7,8,9-tetra-O-acetyl-3,5-dideoxy-5-(methyl 5-acetamido-4,7,8,9-tetra-O-acetyl-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonate)oxyacetamido-d-glycero-α-d-galacto-2-nonulopyranosylonate]-(2→3)-2,4,6-tri-O-benzoyl-d-galactopyranosyl trichloroacetimidate (15). To a solution of 14 (96.2 mg, 66.1 µmol) in CH2Cl2 (1.3 mL) were added trichloroacetonitrile (132 µL, 1.32 mmol) and DBU (2.0 µL, 13.2 µmol) at 0 °C. After stirring for 1.5 h at 0 °C as the reaction was monitored by TLC (30:1 CHCl3–MeOH, developed twice), the reaction mixture was evaporated. The resulting residue was purified by silica gel column chromatography (50:1 CHCl3–MeOH) to give 15 (85.8 mg, 81%, α:β = 1:1.4): 1H NMR (500 MHz, CDCl3); α isomer: δ 8.62 (s, 1H, C=NH), 8.19–7.36 (m, 15H, 3Ph), 6.87 (d, 1H, J1,2 = 3.8 Hz, H-1c), 6.10 (d, 1H, JNH,5 = 10.4 Hz, NHb), 5.72 (d, 1H, J3,4 = 3.0 Hz, H-4c), 5.67–5.62 (m, 2H, H-8b, H-2c), 5.52 (dd, 1H, J2,3 = 10.6 Hz, H-3c), 5.35 (dd, 1H, J6,7 = 2.2 Hz, H-7b), 5.32–5.25 (m, 3H, H-7a, H-8a, NHa), 4.92–4.85 (m, 2H, H-4a, H-4b), 4.52 (dd, 1H, J5,6a = 6.4 Hz, Jgem = 11.2 Hz, H-6ac), 4.35 (m, 1H, H-6bc), 4.26–4.21 (m, 2H, H-5c, H-9ab), 4.19–4.06 (m, 6H, H-5b, H-9bb, H-9aa, OCH2, H-5a, H-9ba), 3.96 (dd, 1H, J6,7 = 2.2 Hz, J5,6 = 10.7 Hz, H-6a), 3.86 (s, 3H, OMe), 3.83–3.80 (m, 5H, OCH2, H-6b, OMe), 2.68 (dd, 1H, J3eq,4 = 4.7 Hz, Jgem = 12.3 Hz, H-3eqa), 2.55 (dd, 1H, J3eq,4 = 4.6 Hz, Jgem = 12.4 Hz, H-3eqb), 2.21–1.88 (m, 28H, 9Ac, H-3axa), 1.68 (t, 1H, J3ax,4 = 12.4 Hz, H-3axb); β isomer: δ 8.70 (s, 1H, C=NH), 8.19–7.36 (m, 15H, 3Ph), 6.28 (d, 1H, J1,2 = 8.2 Hz, H-1c), 5.96 (d, 1H, JNH,5 = 10.3 Hz, NHb), 5.72 (m, 1H, H-2c), 5.64 (m, 1H, H-8b), 5.48 (d, 1H, J3,4 = 3.0 Hz, H-4c), 5.32–5.25 (m, 3H, H-8a, H-7a, NHa), 5.13 (dd, 1H, J6,7 = 2.6 Hz, J7,8 = 9.3 Hz, H-7b), 5.05 (dd, 1H, J2,3 = 10.0 Hz, H-3c), 4.93–4.82 (m, 2H, H-4a, H-4b), 4.56 (dd, 1H, J5,6a = 6.0 Hz, Jgem = 10.8 Hz, H-6ac), 4.42 (m, 1H, H-5c), 4.37–4.33 (m, 2H, H-6bc, H-9ab), 4.27 (m, 1H, H-9aa), 4.09–4.03 (m, 4H, H-6a, H-9ba, H-5a, OCH2), 3.94–3.88 (m, 2H, H-9bb, H-5b), 3.87 and 3.82 (2s, 6H, 2OMe), 3.74–3.70 (m, 2H, OCH2, H-6b), 2.64 (dd, 1H, J3eq,4 = 4.6 Hz, Jgem = 12.7 Hz, H-3eqa), 2.52 (dd, 1H, J3eq,4 = 4.4 Hz, Jgem = 12.3 Hz, H-3eqb), 2.21–1.88 (m, 28H, 9Ac, H-3axa), 1.61 (t, 1H, J3ax,4 = 12.3 Hz, H-3axb); 13C NMR (125 MHz, CDCl3); anomeric mixture (α:β = 1:1.4) δ 170.8, 170.8, 170.6, 170.6, 170.5, 170.5, 170.3, 170.2, 170.1, 169.9, 169.8, 169.7, 169.6, 168.6, 168.5, 168.2, 167.6, 167.5, 165.8, 165.8, 165.8, 165.7, 165.5, 165.0, 161.1, 160.8, 133.6, 133.3, 133.1, 133.0, 132.9, 130.1, 130.0, 129.9, 129.9, 129.8, 129.7, 129.3, 129.3, 128.6, 128.5, 128.3, 128.2, 128.1, 98.4, 96.9, 96.6, 96.5, 94.2, 90.9, 90.4, 77.2, 72.9, 72.8, 72.0, 71.9, 71.2, 70.1, 69.9, 69.3, 68.8, 68.7, 68.5, 68.1, 67.6, 67.4, 67.3, 67.1, 66.7, 63.7, 63.7, 62.6, 62.5, 62.1, 61.9, 53.2, 53.2, 49.2, 48.7, 48.1, 38.5, 37.4, 37.4, 37.3, 31.9, 29.6, 29.3, 23.1, 22.6, 21.4, 21.2, 21.0, 20.9, 20.8, 20.7, 20.7, 20.6, 20.6, 20.2; HRMS (ESI) m/z: found [M + Na]+ 1620.3426, C69H78Cl3N3O34 calcd for [M + Na]+ 1620.3425.
2-(Trimethylsilyl)ethyl [methyl 4,7,8,9-tetra-O-acetyl-3,5-dideoxy-5-(methyl 5-acetamido-4,7,8,9-tetra-O-acetyl-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonate)oxyacetamido-d-glycero-α-d-galacto-2-nonulopyranosylonate]-(2→3)-(2,4,6-tri-O-benzoyl-β-d-galactopyranosyl)-(1→4)-2,3,6-tri-O-benzyl-β-d-glucopyranoside (16). To a mixture of 15 (63.3 mg, 39.0 µmol) and 8 (43.6 mg, 79.0 µmol) in CH2Cl2 (1.6 mL) was added 4 Å molecular sieves (AW300, 150 mg) at room temperature. After stirring for 1 h, the mixture was cooled to 0 °C, and TMSOTf (0.7 µL, 4.0 µmol) was added at 0 °C. After stirring for 2 h at 0 °C as the reaction was monitored by TLC (30:1 CHCl3–MeOH), the reaction was quenched by the addition of satd. aq. NaHCO3. The solution was diluted with CHCl3 and filtered through Celite. The filtrate was then washed with satd. aq. NaHCO3 and brine. The organic layer was subsequently dried over Na2SO4, and concentrated. The resulting residue was purified by silica gel column chromatography (60:1 CHCl3–MeOH) to give 16 (55.8 mg, 78%): [α]D +2.4° (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.25–7.07 (m, 30H, 6Ph), 5.92 (d, 1H, JNH,5 = 10.4 Hz, NHb), 5.72 (td, 1H, J8,9a = 2.4 Hz, J7,8 = J8,9b = 9.7 Hz, H-8b), 5.48 (dd, 1H, J1,2 = 8.0 Hz, J2,3 = 9.9 Hz, H-2c), 5.34–5.27 (m, 3H, H-7a, PhCH2, H-8a), 5.23 (d, 1H, H-1c), 5.15–5.06 (m, 3H, NHa, H-4c, H-7b), 4.93–4.79 (m, 5H, H-4a, H-3c, 2PhCH2, H-4b), 4.66 (d, 1H, Jgem = 11.2 Hz, PhCH2), 4.42 (2d, 2H, Jgem = 12.0 Hz, PhCH2), 4.33–4.24 (m, 3H, H-6ac, H-1d, H-6a), 4.15–3.84 (m, 17H, H-9aa, H-5a, H-9ba, H-6ad, OCH2CH2Si, H-6bc, H-5c, H-5b, OCH2, H-9ab, H-9bb, 2OMe), 3.74–3.61 (m, 4H, H-4d, H-6b, H-6bd, OCH2), 3.56–3.51 (m, 2H, OCH2CH2Si, H-3d), 3.38–3.33 (m, 2H, H-2d, H-5d), 2.65 (dd, 1H, J3eq,4 = 4.7 Hz, Jgem = 12.9 Hz, H-3eqa), 2.50 (dd, 1H, J3eq,4 = 4.6 Hz, Jgem = 12.8 Hz, H-3eqb), 2.19–1.89 (m, 25H, 8Ac, H-3axa), 1.64 (t, 1H, J3ax,4 = 12.8 Hz, H-3axb), 1.46 (s, 3H, Ac), 1.04–0.99 (m, 2H, OCH2CH2Si), 0.01 (s, 9H, SiMe3); 13C NMR (125 MHz, CDCl3) δ 170.8, 170.6, 170.3, 170.2, 170.2, 170.0, 169.8, 168.5, 168.2, 167.6, 165.7, 165.5, 165.0, 139.0, 138.7, 138.6, 133.2, 132.9, 130.3, 130.0, 129.9, 129.9, 129.7, 129.4, 128.6, 128.4, 128.2, 128.2, 128.1, 128.0, 127.9, 127.4, 127.2, 127.2, 127.1, 126.9, 102.8, 100.7, 98.4, 96.9, 82.9, 81.9, 77.6, 74.9, 74.7, 74.4, 72.8, 72.0, 71.8, 71.7, 70.8, 69.1, 68.9, 68.7, 68.4, 68.2, 67.3, 67.1, 66.7, 63.7, 62.8, 62.1, 61.7, 53.2, 53.2, 49.3, 48.0, 37.5, 35.4, 29.7, 23.2, 21.4, 21.0, 20.8, 20.7, 20.7, 20.6, 20.4, 18.5, −1.5; HRMS (ESI) m/z: found [M + Na]+ 2009.6970, C99H118N2O39Si calcd for [M + Na]+ 2009.6973.
2-(Trimethylsilyl)ethyl [methyl 4,7,8,9-tetra-O-acetyl-3,5-dideoxy-5-(methyl 5-acetamido-4,7,8,9-tetra-O-acetyl-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonate)oxyacetamido-d-glycero-α-d-galacto-2-nonulopyranosylonate]-(2→3)-(2,4,6-tri-O-benzoyl-β-d-galactopyranosyl)-(1→4)-β-d-glucopyranoside (17). To a solution of 16 (48.8 mg, 24.0 µmol) in EtOAc (490 µL) was added Pd(OH)2/C (20%, 48.8 mg) at room temperature. After stirring for 2 h at room temperature under a hydrogen atmosphere as the reaction was monitored by TLC (30:1 CHCl3–MeOH, developed twice), the mixture was filtered through Celite. The filtrate was concentrated and the residue was purified by silica gel column chromatography (50:1 to 40:1 CHCl3–MeOH) to give 17 (39.8 mg, 97%): [α]D +155.0° (c 0.1, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.34–7.42 (m, 15H, 3Ph), 5.97 (d, 1H, JNH,5 = 10.4 Hz, NHb), 5.87 (td, 1H, J8,9a = 2.3 Hz, J7,8 = J8,9b = 9.6 Hz, H-8b), 5.55 (dd, 1H, J1,2 = 8.0 Hz, J2,3 = 10.0 Hz, H-2c), 5.36–5.28 (m, 4H, H-7a, H-8a, H-4c, NHa), 5.07 (dd, 1H, J6,7 = 2.6 Hz, H-7b), 5.02 (d, 1H, H-1c), 4.96–4.89 (m, 2H, H-3c, H-4a), 4.82 (td, 1H, J3eq,4 = 4.4 Hz, J3ax,4 = J4,5 = 11.7 Hz, H-4b), 4.56–4.51 (m, 2H, OCH2, H-9ab), 4.38–4.25 (m, 5H, OCH2, H-9aa, H-1d, H-9ba, H-6ac), 4.15 (dd, 1H, J6,7 = 1.7 Hz, J5,6 = 10.7 Hz, H-6a), 4.11–4.04 (m, 3H, H-5c, H-5a, H-6bc), 3.98–3.76 (m, 10H, OCH2CH2Si, H-6ad, H-5b, 2OMe, H-9bb), 3.73–3.64 (m, 4H, H-6bd, H-4d, H-5d, H-6b), 3.57–3.50 (m, 2H, H-3d, OCH2CH2Si), 3.43 (t, 1H, J1,2 = J2,3 = 8.5 Hz, H-2d), 3.26 (d, 1H, JOH,2 = 9.4 Hz, OH-2d), 2.98 (s, 1H, OH-4d), 2.65 (dd, 1H, J3eq,4 = 4.6 Hz, Jgem = 12.8 Hz, H-3eqa), 2.56–2.53 (m, 2H, H-3eqb, OH-6d), 2.25–1.90 (m, 25H, 8Ac, H-3axa), 1.63 (t, 1H, Jgem = 12.5 Hz, H-3axb), 1.09–0.94 (m, 2H, OCH2CH2Si), 0.02 (s, 9H, SiMe3); 13C NMR (125 MHz, CDCl3) δ 172.3, 170.9, 170.8, 170.6, 170.2, 170.2, 169.9, 169.9, 168.5, 168.2, 167.5, 165.9, 165.7, 165.1, 133.5, 133.3, 133.2, 130.5, 130.0, 129.9, 129.4, 129.1, 129.0, 128.6, 128.5, 128.2, 101.8, 101.6, 98.3, 96.8, 79.8, 77.6, 77.3, 77.0, 76.7, 74.5, 74.3, 73.8, 72.8, 71.7, 71.5, 71.2, 70.6, 68.8, 68.7, 68.4, 68.2, 67.3, 67.3, 67.0, 66.5, 63.9, 63.6, 62.3, 62.1, 59.9, 53.3, 53.2, 49.2, 47.9, 37.4, 37.3, 31.9, 30.0, 29.6, 29.3, 27.0, 23.1, 22.6, 21.4, 21.0, 20.8, 20.8, 20.7, 20.6, 20.5, 18.1, 14.1, −1.5; HRMS (ESI) m/z: found [M + Na]+ 1739.5563, C78H100N2O39Si calcd for [M + Na]+ 1739.5565.
2-(Trimethylsilyl)ethyl [5-(5-acetamido-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonic acid)oxyacetamido-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonic acid]-(2→3)-(β-d-galactopyranosyl)-(1→4)-β-d-glucopyranoside (3). To a solution of 17 (39.8 mg, 23.0 µmol) in pyridine (2.3 mL) was added lithium chloride (27.3 mg, 345 µmol) at room temperature. After stirring for 18 h under reflux as the reaction was monitored by TLC (10:1 CHCl3–MeOH, 4:1:0.1 CHCl3–MeOH–H2O), the reaction mixture was co-evaporated with toluene. The resulting residue was purified by gel filtration column chromatography (Sephadex LH-20) using MeOH as eluent. The product obtained was exposed to high vacuum and then dissolved in 0.1 M aq NaOH (250 µL). After stirring for 1 week at room temperature as the reaction was monitored by TLC (5:4:1 CHCl3–MeOH–H2O), the reaction mixture was neutralized with Dowex (H+) resin. The resin was filtered through cotton wool, and the filtrate was concentrated. The residue was purified by silica gel column chromatography (6:4:1 CHCl3–MeOH–H2O) followed by gel filtration column chromatography (Sephadex LH-20) using MeOH as eluent to give 3 (17.2 mg, 80%): [α]D –0.5° (c 0.1, MeOH); 1H NMR (500 MHz, CD3OD) δ 4.50 (d, 1H, J1,2 = 7.8 Hz, H-1c), 4.39 (d, 1H, J1,2 = 7.9 Hz, H-1d), 4.32 (d, 1H, Jgem = 15.6 Hz, OCH2), 4.13 (d, 1H, OCH2), 4.09 (dd, 1H, J3,4 = 3.0 Hz, J2,3 = 9.8 Hz, H-3c), 4.01 (td, 1H, OCH2CH2Si), 3.96–3.48 (m, 25H, H-4a, H-5a, H-6a, H-7a, H-8a, H-9aa, H-9ba, H-4b, H-5b, H-6b, H-7b, H-8b, H-9ab, H-9bb, H-2c, H-4c, H-5c, H-6ac, H-6bc, H-2d, H-4d, H-5d, H-6ad, H-6bd, OCH2CH2Si), 3.27 (t, 1H, J2,3 = J3,4 = 7.9 Hz, H-3d), 2.81 (m, 2H, H-3eqa, H-3eqb), 2.04 (s, 3H, Ac), 1.79 (m, 2H, H-3axa, H-3axb), 1.08 (td, 1H, OCH2CH2Si), 0.98 (td, 1H, OCH2CH2Si), 0.03 (s, 9H, SiMe3); 13C NMR (125 MHz, CD3OD) δ 175.9, 175.0, 174.4, 173.8, 104.3, 103.2, 101.5, 101.0, 80.1, 77.1, 76.6, 76.1, 76.0, 74.3, 74.2, 74.0, 72.7, 69.7, 69.3, 68.7, 64.2, 64.0, 53.4, 49.8, 49.7, 49.6, 49.5, 49.5, 49.1, 41.2, 22.8, 18.9, −1.5; HRMS (ESI) m/z: found [M − 2H + Na]− 1061.3476, C39H66N2O28Si calcd for [M − 2H + Na]− 1061.3475.
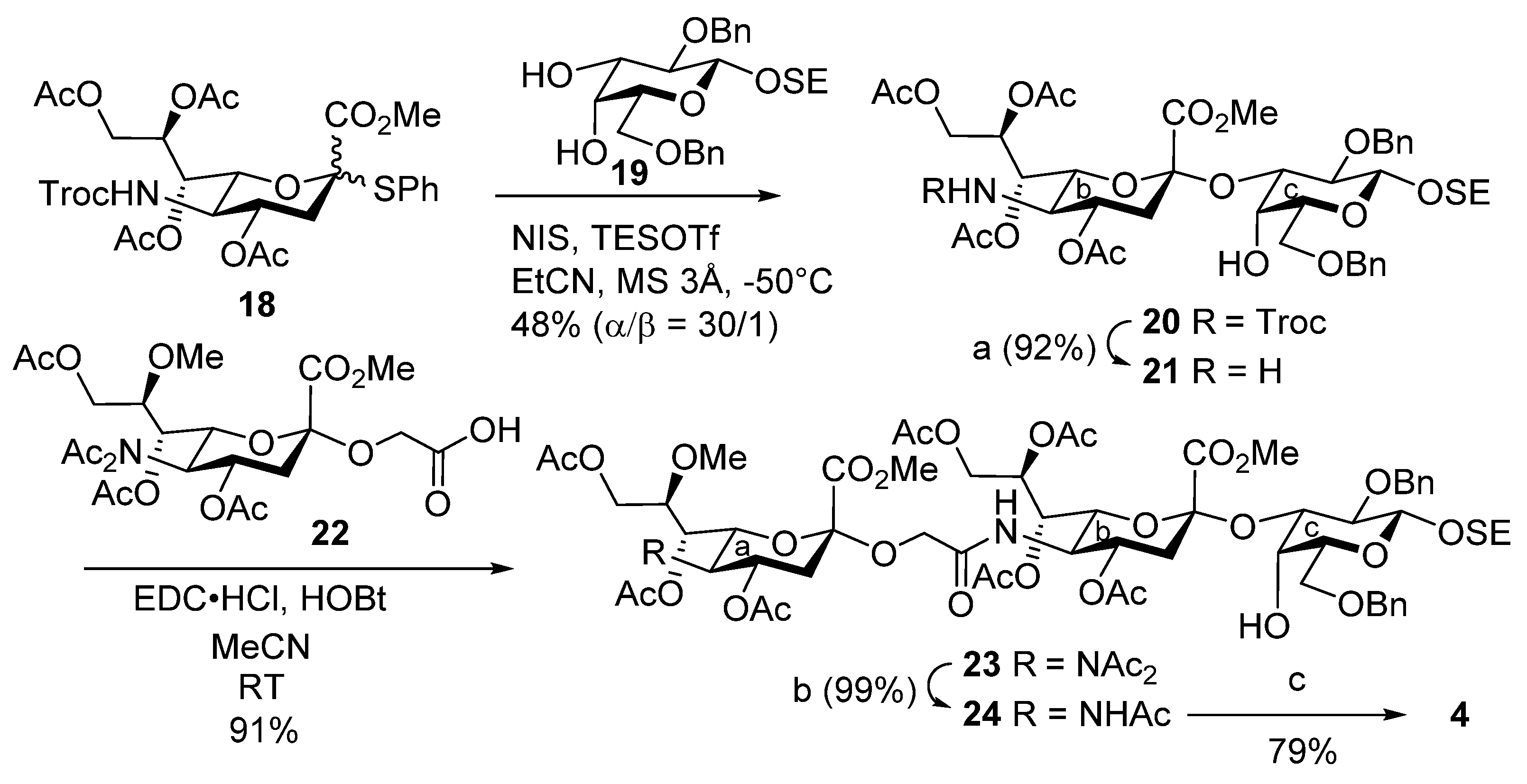
2-(Trimethylsilyl)ethyl [methyl 4,7,8,9-tetra-O-acetyl-3,5-dideoxy-5-(2,2,2-trichloroethoxycarbamoyl)-d-glycero-α-d-galacto-2-nonulopyranosylonate]-(2→3)-2,6-di-O-benzyl-β-d-galactopyranoside (20). To a mixture of 18 (100 mg, 218 µmol) and 19 (157 mg, 218 µmol) in propionitrile (2.2 mL) were added 3 Å molecular sieves (325 mg) and NIS (75.4 mg, 336 µmol) at room temperature. After stirring for 1 h, the mixture was cooled to −50 °C. TESOTf (7.5 µL, 3.3 µmol) was then added to the mixture at −50 °C. After stirring for 3.5 h at −50 °C as the reaction was monitored by TLC (1:1 EtOAc–n-hexane), the solution was diluted with CHCl3 and filtered through Celite. The filtrate was then washed with satd. aq. Na2S2O3, satd. aq. NaHCO3 and brine. The organic layer was subsequently dried over Na2SO4, and concentrated. The residue was purified by silica gel column chromatography (1:7 EtOAc–toluene) to give 20 (107 mg, 46%) and its β-isomer (4 mg, 2%): α-isomer: [α]D –8.0° (c 0.1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.42–7.23 (m, 10H, 2Ph), 5.40 (m, 1H, H-8b), 5.36 (dd, 1H, J6,7 = 2.0 Hz, J7,8 = 8.6 Hz, H-7b), 4.95 (td, 1H, J3eq,4 = 4.8 Hz, J3ax,4 = J4,5 = 11.5 Hz, H-4b), 4.87 (d, 1H, Jgem = 12.4 Hz, OCH2), 4.84 (d, 1H, Jgem = 11.7 Hz, OCH2), 4.79 (d, 1H, JNH,5 = 10.3 Hz, NHb), 4.71 (d, 1H, OCH2), 4.57 (s, 2H, OCH2), 4.47 (d, 1H, OCH2), 4.43 (d, 1H, J1,2 = 7.6 Hz, H-1c), 4.26 (dd, 1H, J8,9a = 2.8 Hz, Jgem = 12.7 Hz, H-9ab), 4.13–4.08 (m, 2H, H-3c, H-6b), 4.04–3.99 (m, 2H, OCH2CH2Si, H-9bb), 3.81–3.58 (m, 9H, H-5b, H-4c, H-5c, H-6ac, H-6bc, OCH2CH2Si, OMe), 3.51 (dd, 1H, J2,3 = 9.7 Hz, H-2c), 2.72 (m, 2H, H-3eqb, OH-4c), 2.09–1.92 (m, 13H, 4Ac, H-3axb), 1.02 (t, 2H, OCH2CH2Si), 0.00 (s, 9H, SiMe3); 13C NMR (100 MHz, CDCl3) δ 170.6, 170.3, 169.9, 168.5, 154.1, 139.1, 138.2, 128.4, 128.1, 127.8, 127.6, 127.6, 103.2, 97.7, 95.3, 77.6, 75.7, 74.5, 73.5, 72.6, 72.2, 69.3, 68.5, 68.3, 68.2, 67.4, 67.3, 62.2, 53.1, 51.5, 37.0, 21.1, 20.8, 20.6, 18.5, −1.4; MALDI m/z: found [M + Na]+ 1088.32, C46H62Cl3NO19Si calcd for [M + Na]+ 1088.16.
2-(Trimethylsilyl)ethyl (methyl 4,7,8,9-tetra-O-acetyl-5-amino-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonate)-(2→3)-2,6-di-O-benzyl-β-d-galactopyranoside (21). To a solution of 20 (20.0 mg, 18.7 µmol) in acetonitrile/AcOH (4:1, 1.3 mL) was added zinc powder (100 mg) at room temperature. After stirring for 40 min at room temperature as the reaction was monitored by TLC (15:1 CHCl3–MeOH), the mixture was filtered through Celite. The filtrate was concentrated, and the resulting residue was purified by silica gel column chromatography (60:1 CHCl3–MeOH) to give 21 (15.2 mg, 92%): [α]D –11.5° (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.39–7.26 (m, 10H, 2Ph), 5.45 (s, 2H, H-7b, H-8b), 4.82 (d, 1H, Jgem = 11.5 Hz, PhCH2), 4.70 (d, 1H, PhCH2), 4.64–4.58 (m, 3H, H-4b, PhCH2), 4.42 (d, 1H, J1,2 = 7.8 Hz, H-1c), 4.29 (m, 1H, H-9ab), 4.12 (m, 1H, H-9bb), 4.08–4.00 (m, 2H, H-3c, OCH2CH2Si), 3.83–3.72 (m, 7H, H-4c, OMe, H-6ac, H-6b, H-6bc), 3.63–3.58 (m, 2H, H-5c, OCH2CH2Si), 3.51 (dd, 1H, J2,3 = 9.5 Hz, H-2c), 2.66 (dd, 1H, J3eq,4 = 4.6 Hz, Jgem = 12.9 Hz, H-3eqb), 2.59–2.53 (m, 2H, OH-4c, H-5b), 2.11, 2.04 and 1.93 (3s, 12H, 4Ac), 1.80 (t, 1H, J3ax,4 = 12.9 Hz, H-3axb), 1.05–1.01 (m, 2H, OCH2CH2Si), 0.00 (s, 9H, SiMe3); 13C NMR (125 MHz, CDCl3) δ 170.6, 170.6, 170.2, 169.9, 168.5, 139.1, 138.2, 128.3, 128.1, 127.9, 127.6, 127.6, 127.4, 103.2, 98.0, 77.6, 77.6, 75.6, 75.1, 74.9, 73.5, 72.8, 71.9, 69.3, 68.4, 68.1, 67.9, 67.3, 62.1, 52.9, 51.0, 36.3, 29.7, 21.1, 21.0, 20.7, 20.5, 18.5, −1.4; HRMS (ESI) m/z: found [M + Na]+ 914.3604, C43H61NO17Si calcd for [M + Na]+ 914.3601.
2-(Trimethylsilyl)ethyl [methyl 4,7,8,9-tetra-O-acetyl-3,5-dideoxy-5-(methyl 4,7,9-tri-O-acetyl-5-diacetamido-3,5-dideoxy-8-O-methyl-d-glycero-α-d-galacto-2-nonulopyranosylonate)oxyacetamido-d-glycero-α-d-galacto-2-nonulopyranosylonate]-(2→3)-2,6-di-O-benzyl-β-d-galactopyranoside (23). To a mixture of 22 (86.2 mg, 153 μmol) and 21 (209 mg, 230 μmol) in acetonitrile (3.1 mL) were added EDC·HCl (52.8 mg, 275 µmol) and HOBt (10.3 mg, 76 μmol) at room temperature. After stirring for 5 h at room temperature as the reaction was monitored by TLC (15:1 CHCl3–MeOH), the mixture was diluted with EtOAc. The solution was then washed with 2 M HCl, satd. aq. NaHCO3 and brine. The organic layer was subsequently dried over Na2SO4, and concentrated. The resulting residue was purified by silica gel column chromatography (55:1 CHCl3–MeOH) to give 23 (199 mg, 91%): [α]D −15.0° (c 0.1, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.42–7.24 (m, 10H, 2Ph), 6.21 (d, 1H, JNH,5 = 10.2 Hz, NHb), 5.61 (td, 1H, J3eq,4 = 5.5 Hz, J3ax,4 = J4,5 = 9.5 Hz, H-4a), 5.44 (td, 1H, J8,9a = 2.6 Hz, J7,8 = J8,9b = 8.3 Hz, H-8b), 5.25 (dd, 1H, J6,7 = 2.1 Hz, H-7b), 4.99 (dd, 1H, J6,7 = 1.4 Hz, J5,6 = 10.3 Hz, H-6a), 4.94 (dd, 1H, J7,8 = 8.3 Hz, H-7a), 4.90–4.84 (m, 2H, H-4b, PhCH2), 4.72 (d, 1H, Jgem = 11.8 Hz, PhCH2), 4.59 (2d, 2H, Jgem = 13.2 Hz, PhCH2), 4.45 (d, 1H, J1,2 = 7.7 Hz, H-1c), 4.32–4.25 (m, 3H, H-9aa, H-9ab, H-5a), 4.22–3.93 (m, 11H, OCH2, H-3c, H-5b, H-6b, OCH2, H-9bb, H-9ba, OCH2CH2Si, CO2Me), 3.81–3.59 (m, 9H, CO2Me, H-4c, H-6ac, H-6bc, H-5c, H-8a, OCH2CH2Si), 3.52 (dd, 1H, J2,3 = 9.5 Hz, H-2c), 3.45 (s, 3H, OMe), 2,89 (dd, 1H, Jgem = 13.2 Hz, H-3eqa), 2.62–2.57 (m, 2H, OH-4c, H-3eqb), 2.38–1.93 (m, 29H, 9Ac, H-3axb, H-3axa), 1.04–1.01 (m, 2H, OCH2CH2Si), 0.01 (s, 9H, SiMe3); 13C NMR (125 MHz, CDCl3) δ 174.2, 173.6, 170.8, 170.3, 169.9, 169.8, 169.0, 168.7, 167.6, 139.2, 138.2, 128.3, 128.1, 127.7, 127.6, 127.5, 127.3, 103.1, 98.7, 97.9, 77.7, 77.6, 77.2, 76.6, 75.7, 74.8, 73.5, 72.8, 72.6, 70.4, 69.3, 68.6, 68.5, 68.2, 68.0, 67.3, 67.0, 63.5, 62.4, 61.5, 58.1, 56.6, 53.2, 53.0, 48.5, 38.0, 37.0, 29.6, 28.0, 26.1, 22.6, 21.1, 20.9, 20.8, 20.7, 20.7, 20.7, 18.4, 14.1, −1.5; HRMS (ESI) m/z: found [M + Na]+ 1459.5345, C66H92N2O31Si calcd for [M + Na]+ 1459.5346.
2-(Trimethylsilyl)ethyl [methyl 4,7,8,9-tetra-O-acetyl-3,5-dideoxy-5-(methyl 5-acetamido-4,7,9-tri-O-acetyl-3,5-dideoxy-8-O-methyl-d-glycero-α-d-galacto-2-nonulopyranosylonate)oxyacetamido-d-glycero-α-d-galacto-2-nonulopyranosylonate]-(2→3)-2,6-di-O-benzyl-β-d-galactopyranoside (24). To a solution of 23 (49.5 mg, 34.0 µmol) in THF (1.4 mL) was added hydrazine acetate (9.5 mg, 100 µmol) at 0 °C. After stirring for 5 h at room temperature as the reaction was monitored by TLC (15:1 CHCl3–MeOH), the mixture was diluted with EtOAc. The solution was then washed with 2 M HCl, H2O, satd. aq. NaHCO3 and brine. The organic layer was subsequently dried over Na2SO4, and concentrated. The resulting residue was purified by silica gel column chromatography (50:1 CHCl3–MeOH) to give 24 (46.9 mg, 99%): [α]D −65.0° (c 0.1, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.42–7.24 (m, 10H, 2Ph), 6.23 (d, 1H, JNH,5 = 10.0 Hz, NHb), 5.44 (td, 1H, J8,9a = 2.7 Hz, J7,8 = J8,9b = 8.0 Hz, H-8b), 5.28 (d, 1H, JNH,5 = 10.0 Hz, NHa), 5.23 (dd, 1H, J6,7 = 2.1 Hz, H-7b), 5.13 (dd, 1H, J6,7 = 1.8 Hz, J7,8 = 9.3 Hz, H-7a), 5.01 (td, 1H, J3eq,4 = 4.8 Hz, J3ax,4 = J4,5 = 11.5 Hz, H-4a), 4.92–4.84 (m, 2H, H-4b, PhCH2), 4.73 (d, 1H, Jgem = 11.8 Hz, PhCH2), 4.59 (2d, 2H, Jgem = 13.5 Hz, PhCH2), 4.44 (d, 1H, J1,2 = 7.7 Hz, H-1c), 4.31 (dd, 1H, Jgem = 12.4 Hz, H-9ab), 4.25–3.90 (m, 12H, OCH2, H-9aa, H-3c, H-5b, H-5a, H-9ba, OCH2CH2Si, H-9bb, OCH2, CO2Me), 3.81–3.59 (m, 9H, CO2Me, H-4c, H-6ac, H-6bc, H-8a, H-5c, OCH2CH2Si), 3.54–3.49 (m, 4H, H-2c, OMe), 2.74 (dd, 1H, Jgem = 12.9 Hz, H-3eqa), 2.64 (br s, 1H, OH-4c), 2.59 (dd, 1H, J3eq,4 = 4.6 Hz, Jgem = 13.0 Hz, H-3eqb), 2.13–1.88 (m, 26H, 8Ac, H-3axb, H-3axa), 1.04–1.01 (m, 2H, OCH2CH2Si), 0.01 (s, 9H, SiMe3); 13C NMR (125 MHz, CDCl3) δ 170.8, 170.8, 170.5, 170.3, 170.2, 170.0, 169.8, 168.8, 168.7, 167.8, 139.1, 138.2, 128.3, 128.1, 127.7, 127.6, 127.5, 127.3, 103.1, 98.6, 98.0, 77.7, 77.6, 77.2, 75.9, 75.7, 74.8, 73.5, 72.8, 72.6, 72.6, 69.3, 69.1, 68.6, 68.4, 68.2, 68.0, 67.4, 67.3, 63.5, 62.5, 61.7, 58.4, 53.1, 53.0, 49.2, 48.7, 37.5, 36.9, 31.9, 29.6, 23.2, 22.6, 21.1, 20.8, 20.7, 20.7, 20.6, 18.4, 14.1, −1.5; HRMS (ESI) m/z: found [M + Na]+ 1417.5242, C64H90N2O30Si calcd for [M + Na]+ 1417.5240.
2-(Trimethylsilyl)ethyl [5-(5-acetamido-3,5-dideoxy-8-O-methyl-d-glycero-α-d-galacto-2-nonulopyranosylonic acid)oxyacetamido-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonic acid]-(2→3)-β-d-galactopyranoside (4). To a solution of 24 (46.9 mg, 33.0 µmol) in EtOAc (1.3 mL) was added Pd(OH)2/C (20%, 46.9 mg) at room temperature. After stirring for 1 h at room temperature under a hydrogen atmosphere as the reaction was monitored by TLC (15:1 CHCl3–MeOH), the mixture was filtered through Celite. The filtrate was concentrated and the crude residue obtained was exposed to high vacuum overnight. The resulting residue was then dissolved in pyridine (3.3 mL), and lithium chloride (27.3 mg, 495 µmol) was added at room temperature. After stirring for 42 h under reflux as the reaction was monitored by TLC (4:1:0.1 CHCl3–MeOH–AcOH), the reaction mixture was co-evaporated with toluene. The obtained residue was purified by gel filtration column chromatography (Sephadex LH-20) using MeOH as eluent. The product was exposed to high vacuum and then dissolved in 0.1 M aq NaOH (5.0 mL). After stirring for 2 h at room temperature as the reaction was monitored by TLC (6:4:1 CHCl3–MeOH–H2O), the reaction mixture was neutralized with Dowex (H+) resin. The resin was filtered through cotton wool, and the filtrate was then evaporated. The residue was purified by silica gel column chromatography (6:4:1 CHCl3–MeOH–H2O), followed by gel filtration column chromatography (Sephadex LH-20) using MeOH as eluent to give 4 (23.3 mg, 79%): [α]D +75.0° (c 0.1, MeOH); 1H NMR (500 MHz, CD3OD) δ 4.52–3.31 (m, 28H, H-4a, H-5a, H-6a, H-7a, H-8a, H-9aa, H-9ba, H-4b, H-5b, H-6b, H-7b, H-8b, H-9ab, H-9bb, H-1c, H-2c, H-3c, H-4c, H-5c, H-6ac, H-6bc, OCH2, OCH2CH2Si, OMe), 2.88 (br d, 1H, H-3eqa), 2.86 (br d, 1H, H-3eqb), 2.02 (s, 3H, Ac), 1.76–1.74 (m, 2H, H-3axa, H-3axb), 1.09–0.94 (m, 2H, OCH2CH2Si), 0.07 (s, 9H, SiMe3); 13C NMR (125 MHz, CD3OD) δ 175.5, 175.3, 175.3, 175.2, 104.2, 81.4, 79.4, 78.0, 76.4, 74.7, 74.4, 73.1, 70.8, 69.3, 68.9, 68.0, 63.4, 62.6, 61.7, 59.1, 54.0, 53.7, 49.8, 49.7, 49.6, 49.5, 42.0, 30.7, 22.8, 19.1, −1.6; HRMS (ESI) m/z: found [M − 2H + Na]− 913.3095, C34H58N2O23Si calcd for [M − 2H + Na]− 913.3097.
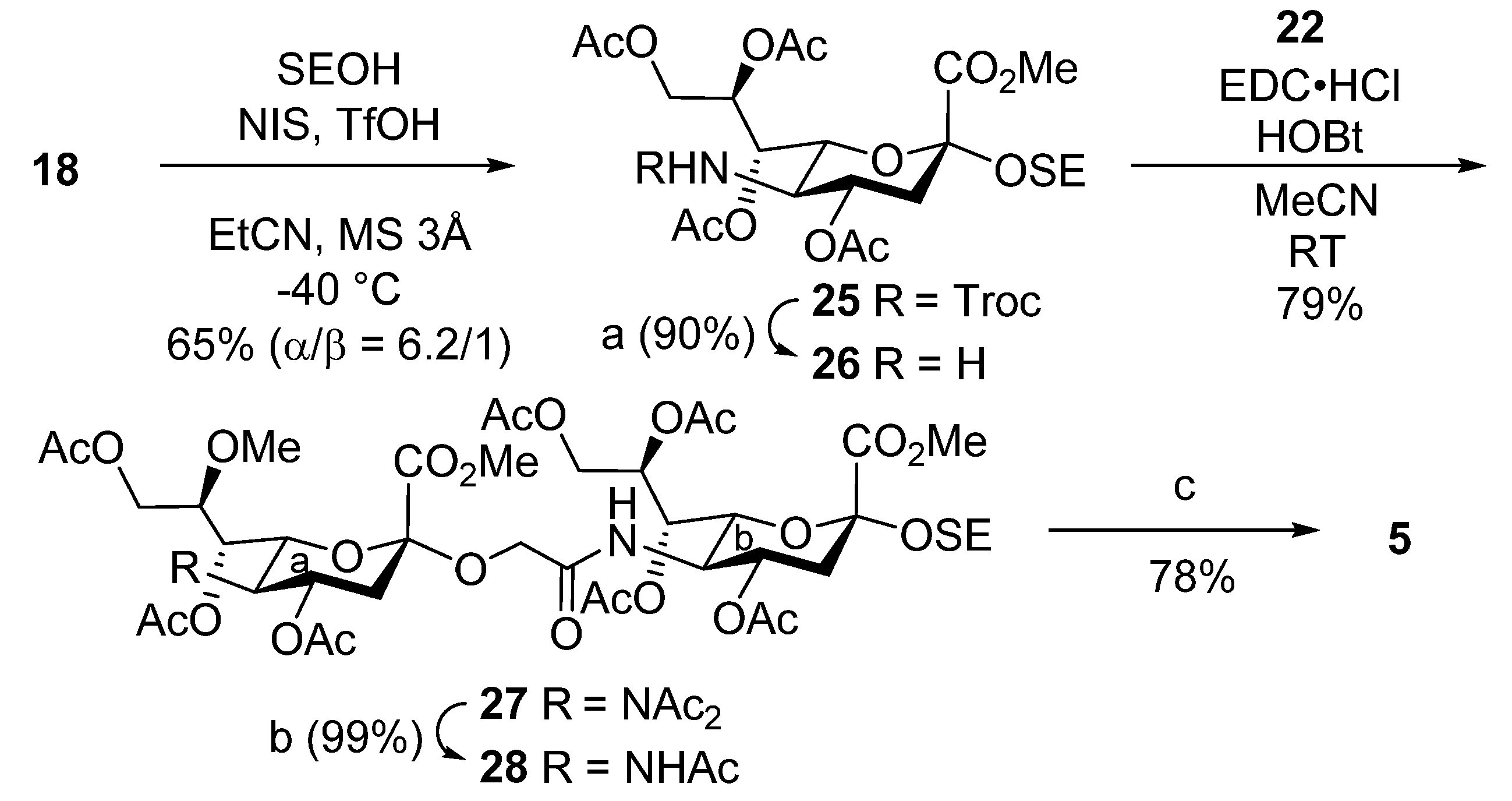
Methyl [2-(trimethylsilyl)ethyl 4,7,8,9-tetra-O-acetyl-3,5-dideoxy-5-(2,2,2-trichloroethoxycarbamoyl)-d-glycero-α-d-galacto-2-nonulopyranosid]onate (25). To a mixture of 18 (1.0 g, 1.39 mmol) and 2-(trimethylsilyl)ethanol (1.0 mL, 6.97 mmol) in propionitrile (14 mL) were added 3 Å molecular sieves (1.4 g) and NIS (470 mg, 2.08 mmol) at room temperature. After stirring for 1 h, the mixture was cooled to −40 °C. TfOH (18.3 µL, 210 µmol) was then added to the mixture at −40 °C. After stirring for 5 days at −40 °C as the reaction was monitored by TLC (1:5 EtOAc–toluene), the reaction was quenched by the addition of triethylamine. The solution was diluted with CHCl3 and filtered through Celite. The filtrate was then washed with satd. aq. Na2S2O3 and brine. The organic layer was subsequently dried over Na2SO4, and concentrated. The residue was purified by silica gel column chromatography (1:5 EtOAc–toluene) to give 25 (654 mg, 65%, α:β = 6.2:1): [α]D –5.0° (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 5.39 (s, 2H, H-7, H-8), 4.96–4.84 (m, 3H, H-4, OCH2, NH), 4.48 (d, 1H, Jgem = 12.1 Hz, OCH2), 4.29 (dd, 1H, J8,9a = 1.8 Hz, Jgem = 11.7 Hz, H-9a), 4.19–4.12 (m, 2H, H-6, H-9b), 3.89 (m, 1H, OCH2CH2Si), 3.80 (s, 3H, OMe), 3.64 (q, 1H, J4,5 = J5,6 = J5,NH = 10.3 Hz, H-5), 3.34 (m, 1H, OCH2CH2Si), 2.64 (dd, 1H, J3eq,4 = 4.7 Hz, Jgem = 12.6 Hz, H-3eq), 2.14–2.01 (m, 12H, 4Ac), 1.87 (t, 1H, J3ax,4 = 12.6 Hz, H-3ax), 0.91–0.85 (m, 2H, OCH2CH2Si), 0.03 (s, 9H, SiMe3); 13C NMR (125 MHz, CDCl3) δ 170.6, 170.4, 170.1, 169.7, 168.3, 154.1, 98.4, 95.4, 74.5, 71.8, 68.6, 68.3, 67.5, 62.6, 62.2, 52.6, 51.8, 38.4, 21.0, 20.8, 20.8, 20.7, 18.0, −1.4; HRMS (ESI) m/z: found [M + Na]+ 746.1180, C26H40Cl3NO14Si calcd for [M + Na]+ 746.1176.
Methyl [2-(trimethylsilyl)ethyl 4,7,8,9-tetra-O-acetyl-5-amino-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosid]onate (26). To a solution of 25 (40.0 mg, 55.0 µmol) in acetonitrile/AcOH (4:1, 1.8 mL) was added zinc powder (200 mg) at room temperature. After stirring for 5 h at room temperature as the reaction was monitored by TLC (15:1 CHCl3–MeOH), the mixture was filtered through Celite. The filtrate was concentrated and the residue was purified by silica gel column chromatography (60:1 CHCl3–MeOH) to give 26 (27.0 mg, 90%): [α]D –8.5° (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 5.39 (s, 2H, H-7, H-8), 4.96–4.84 (m, 3H, H-4, OCH2, NH), 4.48 (d, 1H, Jgem = 12.1 Hz, OCH2), 4.29 (dd, 1H, J8,9a = 1.8 Hz, Jgem = 11.7 Hz, H-9a), 4.19–4.12 (m, 2H, H-6, H-9b), 3.90–3.87 (m, 1H, OCH2CH2Si), 3.80 (s, 3H, OMe), 3.64 (q, 1H, J4,5 = J5,6 = J5,NH = 10.3 Hz, H-5), 3.35–3.32 (m, 1H, OCH2CH2Si), 2.64 (dd, 1H, J3eq,4 = 4.7 Hz, Jgem = 12.6 Hz, H-3eq), 2.14–2.01 (m, 12H, 4Ac), 1.87 (t, 1H, J3ax,4 = 12.6 Hz, H-3ax), 0.91–0.85 (m, 2H, OCH2CH2Si), 0.03 (s, 9H, SiMe3); 13C NMR (125 MHz, CDCl3) δ 170.8, 170.7, 170.3, 169.9, 168.2, 98.3, 74.6, 71.9, 68.0, 67.9, 62.3, 62.0, 52.4, 51.2, 37.8, 21.0, 20.8, 20.7, 17.9, –1.4; HRMS (ESI) m/z: found [M + Na]+ 572.2137, C23H39NO12Si calcd for [M + Na]+ 572.2134.
2-(Trimethylsilyl)ethyl [methyl 4,7,8,9-tetra-O-acetyl-3,5-dideoxy-5-(methyl 4,7,9-tri-O-acetyl-5-diacetamido-3,5-dideoxy-8-O-methyl-d-glycero-α-d-galacto-2-nonulopyranosylonate)oxyacetamido-d-glycero-α-d-galacto-2-nonulopyranosid]onate (27). To a solution of 22 (50.0 mg, 76.0 μmol) and 26 (76.0 mg, 143 μmol) in acetonitrile (3.1 mL) were added EDC·HCl (26.0 mg, 137 μmol) and HOBt (5.0 mg, 38.0 μmol) at room temperature. After stirring for 5 h at room temperature as the reaction was monitored by TLC (15:1 CHCl3–MeOH), the mixture was diluted with EtOAc. The solution was then washed with 2 M HCl, satd. aq. NaHCO3 and brine. The organic layer was subsequently dried over Na2SO4, and concentrated. The resulting residue was purified by silica gel column chromatography (80:1 CHCl3–MeOH) to give 27 (66.0 mg, 79%): [α]D –2.0° (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 6.20 (d, 1H, JNH,5 = 9.8 Hz, NHb), 5.62 (td, 1H, J3eq,4 = 5.6 Hz, J3ax,4 = J4,5 = 9.9 Hz, H-4a), 5.42 (td, 1H, J8,9a = 2.8 Hz, J7,8 = J8,9b = 8.4 Hz, H-8b), 5.26 (dd, 1H, J6,7 = 1.5 Hz, H-7b), 5.00 (dd, 1H, J6,7 = 1.5 Hz, J5,6 = 10.3 Hz, H-6a), 4.94 (dd, 1H, J7,8 = 8.4 Hz, H-7a), 4.86 (td, 1H, J3eq,4 = 5.6 Hz, J3ax,4 = J4,5 = 7.8 Hz, H-4b), 4.32–3.99 (m, 9H, H-9ab, H-9aa, OCH2, H-5a, H-5b, H-9ba, H-9bb, H-6b), 3.94–3.88 (m, 4H, CO2Me, OCH2CH2Si), 3.82 (s, 3H, CO2Me), 3.55 (td, 1H, J8,9a = 2.9 Hz, J8,9b = 4.2 Hz, H-8a), 3.46 (s, 3H, OMe), 3.32 (m, 1H, OCH2CH2Si), 2.90 (dd, 1H, Jgem = 13.2 Hz, H-3eqa), 2.63 (dd, 1H, Jgem = 12.6 Hz, H-3eqb), 2.38 and 2.31 (2s, 6H, 2Ac), 2.16–1.91 (m, 23H, 7Ac, H-3axa, H-3axb), 0.93–0.85 (m, 2H, OCH2CH2Si), 0.04 (s, 9H, SiMe3); 13C NMR (125 MHz, CDCl3) δ 174.2, 173.6, 170.8, 170.5, 170.4, 170.1, 169.9, 169.8, 169.7, 168.9, 168.5, 167.5, 98.6, 98.5, 77.2, 76.5, 72.4, 70.4, 68.7, 68.3, 68.0, 67.4, 67.0, 63.5, 62.6, 62.4, 61.4, 58.1, 56.6, 53.2, 52.9, 48.7, 38.3, 38.0, 29.6, 28.0, 26.1, 21.0, 20.9, 20.8, 20.8, 20.8, 20.7, 20.7, 17.9, −1.3, −1.7; HRMS (ESI) m/z: found [M + Na]+ 1117.3883, C46H70N2O26Si calcd for [M + Na]+ 1117.3884.
Methyl [2-(trimethylsilyl)ethyl 4,7,8,9-tetra-O-acetyl-3,5-dideoxy-5-(methyl 5-acetamido-4,7,9-tri-O-acetyl-3,5-dideoxy-8-O-methyl-d-glycero-α-d-galacto-2-nonulopyranosylonate)oxyacetamido-d-glycero-α-d-galacto-2-nonulopyranosid]onate (28). To a solution of 27 (65.6 mg, 59.0 µmol) in THF (2.4 mL) was added hydrazine acetate (16.6 mg, 180 µmol) at 0 °C. After stirring for 3.5 h at room temperature as the reaction was monitored by TLC (15:1 CHCl3–MeOH), the mixture was diluted with EtOAc. The solution was then washed with 2 M HCl, H2O, satd. aq. NaHCO3 and brine. The organic layer was subsequently dried over Na2SO4, and concentrated. The resulting residue was purified by silica gel column chromatography (50:1 CHCl3–MeOH) to give 28 (61.3 mg, 99%): [α]D −14.0° (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 6.23 (d, 1H, JNH,5 = 9.7 Hz, NHb), 5.42 (td, 1H, J8,9a = 2.6 Hz, J7,8 = J8,9b = 6.1 Hz, H-8b), 5.27–5.23 (m, 2H, NHa, H-7b), 5.13 (dd, 1H, J6,7 = 1.8 Hz, J7,8 = 9.3 Hz, H-7a), 5.01 (td, 1H, J3eq,4 = 4.6 Hz, J3ax,4 = J4,5 = 11.6 Hz, H-4a), 4.88 (td, 1H, J3eq,4 = 4.6 Hz, J3ax,4 = J4,5 = 10.0 Hz, H-4b), 4.31 (dd, 1H, Jgem = 12.4 Hz, H-9ab), 4.28–4.01 (m, 8H, H-9aa, OCH2, H-5b, H-6a, H-9bb, H-5a, H-9ba, H-6b), 3.94–3.88 (m, 5H, OCH2, CO2Me, OCH2CH2Si), 3.82 (s, 3H, CO2Me), 3.69 (td, 1H, J8,9a = 3.2 Hz, J8,9b = 9.3 Hz, H-8a), 3.49 (s, 3H, OMe), 3.32 (td, 1H, OCH2CH2Si), 2.74 (dd, 1H, Jgem = 12.9 Hz, H-3eqa), 2.63 (dd, 1H, Jgem = 12.8 Hz, H-3eqb), 2.15–1.89 (m, 26H, 8Ac, H-3axa, H-3axb), 0.96–0.84 (m, 2H, OCH2CH2Si), 0.03 (s, 9H, SiMe3); 13C NMR (125 MHz, CDCl3) δ 170.8, 170.8, 170.5, 170.4, 170.2, 169.8, 168.7, 168.6, 167.8, 98.6, 98.6, 77.2, 75.9, 72.6, 72.5, 69.1, 68.6, 68.5, 68.0, 67.8, 63.5, 62.6, 62.5, 61.7, 58.4, 53.1, 52.6, 49.2, 48.9, 38.3, 37.5, 29.6, 23.2, 21.0, 20.8, 20.8, 20.7, 17.9, −1.5; HRMS (ESI) m/z: found [M + Na]+ 1075.3772, C44H68N2O25Si calcd for [M + Na]+ 1075.3773.
3,5-Dideoxy-[2-(trimethylsilyl)ethyl 5-acetamido-3,5-dideoxy-8-O-methyl-d-glycero-α-d-galacto-2-nonulopyranosylonic acid]oxyacetamido-d-glycero-α-d-galacto-2-nonulopyranosidonic acid (5). To a solution of 28 (61.3 mg, 58.0 µmol) in pyridine (5.8 mL) was added lithium chloride (36.9 mg, 870 µmol) at room temperature. After stirring for 22 h under reflux as the reaction was monitored by TLC (6:4:0.1 CHCl3–MeOH–AcOH), the reaction mixture was co-evaporated with toluene. The crude residue was purified by gel filtration column chromatography (Sephadex LH-20) using MeOH as eluent. The product was exposed to high vacuum and then dissolved in 0.1 M aq NaOH (5.0 mL). After stirring for 2 h at room temperature as the reaction was monitored by TLC (5:4:1 CHCl3–MeOH–H2O), the reaction mixture was neutralized with Dowex (H+) resin. The resin was filtered through cotton wool and the filtrate was then evaporated. The residue was purified by silica gel column chromatography (6:4:1 CHCl3–MeOH–H2O) followed by gel filtration column chromatography (Sephadex LH-20) using MeOH as eluent to give 5 (33.0 mg, 78%): [α]D –11.5° (c 0.1, MeOH); 1H NMR (500 MHz, CD3OD) δ 4.31–3.35 (m, 21H, H-4a, H-5a, H-6a, H-7a, H-8a, H-9aa, H-9ba, H-4b, H-5b, H-6b, H-7b, H-8b, H-9ab, H-9bb, OCH2, OCH2CH2Si, OMe), 2.82 (dd, 1H, J3eq,4 = 4.2 Hz, Jgem = 12.5 Hz, H-3eqa), 2.62 (br d, 1H, H-3eqb), 1.99 (s, 3H, Ac), 1.72 (t, 1H, J3ax,4 = Jgem = 11.7 Hz, H-3axb), 1.59 (t, 1H, J3ax,4 = 12.5 Hz, H-3axa), 0.92–0.85 (m, 2H, OCH2CH2Si), 0.01 (s, 9H, SiMe3); 13C NMR (125 MHz, CD3OD) δ 175.0, 174.9, 174.2, 173.3, 101.6, 101.3, 81.4, 79.5, 79.4, 74.6, 74.0, 73.1, 70.0, 69.2, 69.0, 64.2, 62.5, 61.5, 58.9, 54.1, 54.0, 49.9, 49.7, 49.6, 49.3, 42.7, 41.3, 30.7, 22.7, −1.5; HRMS (ESI) m/z: found [M − 2H + Na]− 751.2572, C28H50N2O18Si calcd for [M − 2H + Na]− 751.2575.
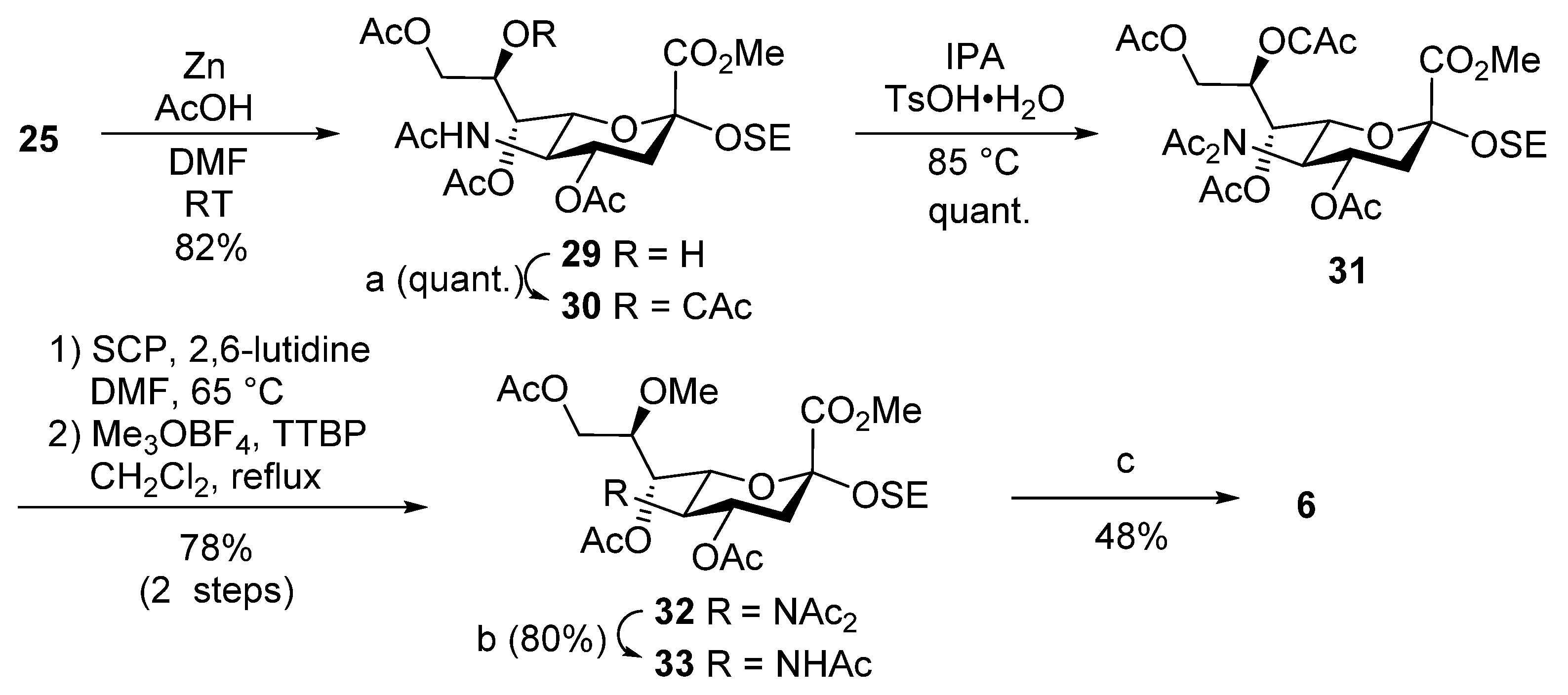
Methyl [2-(trimethylsilyl)ethyl 5-acetamido-4,7,9-tri-O-acetyl-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosid]onate (29). To a solution of 25 (100 mg, 138 µmol) in DMF/AcOH (4:1, 4.6 mL) was added zinc powder (500 mg) at room temperature. After stirring for 12 h at room temperature as the reaction was monitored by TLC (15:1 CHCl3–MeOH), the mixture was filtered through Celite. The filtrate was concentrated and the residue was purified by silica gel column chromatography (60:1 CHCl3–MeOH) to give 29 (62.5 mg, 82%): [α]D –12.5° (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 5.46 (d, 1H, JNH,5 = 10.2 Hz, NH), 5.13 (dd, 1H, J6,7 = 2.2 Hz, J7,8 = 8.7 Hz, H-7), 4.84 (td, 1H, J3eq,4 = 5.6 Hz, J3ax,4 = J4,5 = 7.3 Hz, H-4), 4.18–4.13 (m, 3H, H-9a, H-5, H-8), 4.09–4.05 (m, 2H, H-9b, OH), 3.91–3.86 (m, 5H, H-6, OMe, OCH2CH2Si), 3.44 (m, 1H, OCH2CH2Si), 2.70 (dd, 1H, Jgem = 13.0 Hz, H-3eq), 2.12–1.93 (m, 10H, 3Ac, H-3ax), 1.88 (s, 3H, Ac), 0.92–0.88 (m, 2H, OCH2CH2Si), 0.00 (s, 9H, SiMe3); 13C NMR (125 MHz, CDCl3) δ 170.9, 170.9, 170.3, 170.3, 169.8, 98.6, 77.2, 72.6, 69.5, 68.9, 68.3, 64.7, 62.2, 53.4, 49.0, 37.4, 23.0, 20.9, 20.8, 17.8, −1.4; HRMS (ESI) m/z: found [M + Na]+ 572.2137, C23H39NO12Si calcd for [M + Na]+ 572.2134.
Methyl [2-(trimethylsilyl)ethyl 5-acetamido-4,7,9-tri-O-acetyl-8-O-chloroacetyl-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosid]onate (30). To a solution of 29 (72.4 mg, 130 µmol) in THF (1.3 mL) were added chloroacetic anhydride (33.8 mg, 197 µmol) and DMAP (1.6 mg, 13.0 µmol) at 0 °C. After stirring for 12 h at room temperature as the reaction was monitored by TLC (20:1 CHCl3–MeOH), the reaction was quenched by the addition of MeOH at 0 °C. The residue was then diluted with EtOAc, which was subsequently washed with 2 M HCl, H2O, satd. aq. NaHCO3 and brine. The organic layer was dried over Na2SO4, and concentrated. The resulting residue was purified by silica gel column chromatography (60:1 CHCl3–MeOH) to give 30 (70.8 mg, quant.): [α]D –10.0° (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 5.48 (m, 1H, H-8), 5.34 (dd, 1H, J6,7 = 1.6 Hz, J7,8 = 9.0 Hz, H-7), 5.26 (d, 1H, JNH,5 = 9.6 Hz, NH), 4.81 (td, 1H, J3eq,4 = 5.2 Hz, J3ax,4 = J4,5 = 7.8 Hz, H-4), 4.34–4.30 (m, 2H, H-9a, OCH2), 4.17 (d, 1H, Jgem = 15.0 Hz, OCH2), 4,12–4.07 (m, 3H, H-5, H-6, H-9b), 3.86 (m, 1H, OCH2CH2Si), 3.80 (s, 3H, OMe), 3,26 (m, 1H, OCH2CH2Si), 2.58 (dd, 1H, Jgem = 12.9 Hz, H-3eq), 2.15, 2.04 and 2.03 (3s, 9H, 3Ac), 1.95 (t, 1H, H-3ax), 1.88 (s, 3H, Ac), 0.94–0.82 (m, 2H, OCH2CH2Si), 0.03 (s, 9H, SiMe3); 13C NMR (125 MHz, CDCl3) δ 172.5, 172.0, 171.7, 170.2, 167.8, 100.0, 78.7, 78.7, 78.5, 78.2, 73.7, 71.3, 70.5, 68.4, 64.2, 63.6, 54.1, 50.8, 42.7, 39.7, 31.1, 24.6, 22.3, 22.3, 22.1, 19.4, −1.4; HRMS (ESI) m/z: found [M + Na]+ 648.1851, C25H40ClNO13Si calcd for [M + Na]+ 648.1850.
Methyl [2-(trimethylsilyl)ethyl 4,7,9-tri-O-acetyl-8-O-chloroacetyl-5-diacetamido-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosid]onate (31). To a solution of 30 (71.2 mg, 113 µmol) in isopropenyl acetate (4.5 mL) was added p-toluenesulfonic acid monohydrate (2.0 mg, 11.3 µmol) at room temperature. After stirring for 7 h at 85 °C as the reaction was monitored by TLC (20:1 CHCl3–MeOH), the reaction was quenched by the addition of triethylamine. The residue was then diluted with EtOAc, and washed with H2O and brine. The organic layer was subsequently dried over Na2SO4, and concentrated. The resulting residue was purified by silica gel column chromatography (100:1 CHCl3–MeOH) to give 31 (75.5 mg, quant.): [α]D +9.0° (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 5.48–5.43 (m, 2H, H-8, H-4), 5.16 (dd, 1H, J6,7 = 1.9 Hz, J7,8 = 8.7 Hz, H-7), 4.99 (dd, 1H, J5,6 = 10.1 Hz, H-6), 4.35 (d, 1H, Jgem = 15.1 Hz, OCH2), 4.31 (dd, 1H, J8,9a = 2.6 Hz, Jgem = 12.7 Hz, H-9a), 4.21 (t, 1H, J4,5 = 10.1 Hz, H-5), 4.16–4.12 (m, 2H, OCH2, H-9b), 3.89 (m, 1H, OCH2CH2Si), 3.83 (s, 3H, OMe), 3.44 (m, 1H, OCH2CH2Si), 2.76 (dd, 1H, J3eq,4 = 5.2 Hz, Jgem = 12.9 Hz, H-3eq), 2.38–1.98 (m, 15H, 5Ac), 1.84 (t, 1H, J3ax,4 = 12.9 Hz, H-3ax), 0.90–0.85 (m, 2H, OCH2CH2Si), 0.03 (s, 9H, SiMe3); 13C NMR (125 MHz, CDCl3) δ 174.4, 173.5, 170.5, 169.6, 167.9, 166.4, 98.5, 77.2, 69.8, 69.3, 66.8, 66.7, 62.5, 61.6, 56.9, 52.7, 41.2, 39.3, 27.9, 26.0, 20.9, 20.7, 20.6, 17.9, −1.4; HRMS (ESI) m/z: found [M + Na]+ 690.1956, C27H42ClNO14Si calcd for [M + Na]+ 690.1955.
Methyl [2-(trimethylsilyl)ethyl 4,7,9-tri-O-acetyl-5-diacetamido-3,5-dideoxy-8-O-methyl-d-glycero-α-d-galacto-2-nonulopyranosid]onate (32). To a solution of 31 (79.5 mg, 118 µmol) in DMF (4.5 mL) were added 1-selenocarbamoylpiperidine (45.4 mg, 238 µmol) and 2,6-lutidine (20 µL, 177 µmol) at room temperature. After stirring for 1 h at 65 °C as the reaction was monitored by TLC (1:3 EtOAc–n-hexane), the mixture was diluted with EtOAc. The solution was then washed with 2 M HCl, H2O, satd. aq. NaHCO3 and brine. The organic layer was subsequently dried over Na2SO4, and concentrated. The crude residue was exposed to high vacuum. The residue was then dissolved in CH2Cl2 (4.7 mL). Trimethyloxonium tetrafluoroborate (87.3 mg, 590 µmol) and 2,4,6-tri-tert-butylpyrimidine (161 mg, 650 µmol) were added to the mixture at room temperature. After stirring for 4 h under reflux as the reaction was monitored by TLC (1:3 EtOAc–n-hexane), the reaction was quenched by the addition of iced water. The residue was then diluted with EtOAc, and washed with H2O, satd. aq. NaHCO3 and brine. The organic layer was subsequently dried over Na2SO4, and concentrated. The resulting residue was purified by silica gel column chromatography (1:3 EtOAc–n-hexane) to give 32 (55.6 mg, 78%): [α]D +5.0° (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 5.56 (td, 1H, J3eq,4 = 5.4 Hz, J3ax,4 = J4,5 = 10.4 Hz, H-4), 4.99 (dd, 1H, J6,7 = 1.4 Hz, J7,8 = 7.5 Hz, H-7), 4.94 (dd, 1H, J5,6 = 10.4 Hz, H-6), 4.34 (dd, 1H, J8,9a = 1.4 Hz, Jgem = 10.1 Hz, H-9a), 4.15 (t, 1H, H-5), 4.09 (dd, 1H, J8,9b = 5.0 Hz, H-9b), 3.93 (m, 1H, OCH2CH2Si), 3.87 (s, 3H, CO2Me), 3.76 (m, 1H, OCH2CH2Si), 3.47 (s, 3H, OMe), 2.80 (dd, 1H, Jgem = 13.0 Hz, H-3eq), 2.36–1.99 (m, 15H, 5Ac), 1.85 (near t, 1H, H-3ax), 0.93–0.89 (m, 2H, OCH2CH2Si), 0.03 (s, 9H, SiMe3); 13C NMR (125 MHz, CDCl3) δ 174.4, 173.8, 170.8, 170.1, 169.7, 168.0, 98.7, 69.9, 68.3, 67.1, 62.0, 61.9, 58.1, 57.4, 52.6, 38.7, 28.0, 25.9, 20.9, 20.8, 20.8, 18.0, −1.5; HRMS (ESI) m/z: found [M + Na]+ 628.2398, C26H43NO13Si calcd for [M + Na]+ 628.2396.
Methyl [2-(trimethylsilyl)ethyl 5-acetamido-4,7,9-tri-O-acetyl-3,5-dideoxy-8-O-methyl-d-glycero-α-d-galacto-2-nonulopyranosid]onate (33). To a solution of 32 (46.7 mg, 77.0 µmol) in THF (3.1 mL) was added hydrazine acetate (21.3 mg, 230 µmol) at 0 °C. After stirring for 4 h at room temperature as the reaction was monitored by TLC (20:1 CHCl3–MeOH), the mixture was diluted with EtOAc. The solution was then washed with 2 M HCl, H2O, satd. aq. NaHCO3 and brine. The organic layer was subsequently dried over Na2SO4, and concentrated. The resulting residue was purified by silica gel column chromatography (60:1 CHCl3–MeOH) to give 33 (34.8 mg, 80%): [α]D –12.0° (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 5.15–5.13 (m, 2H, H-7, NH), 4.92 (td, 1H, J3eq,4 = 5.0 Hz, J3ax,4 = J4,5 = 12.5 Hz, H-4), 4.26 (dd, 1H, J8,9a = 3.3 Hz, Jgem = 12.3 Hz, H-9a), 4.11–4.08 (m, 2H, H-9b, H-6), 4.03 (t, 1H, J5,NH = 12.5 Hz, H-5), 3.91 (m, 1H, OCH2CH2Si), 3.83 (s, 3H, CO2Me), 3.77 (m, 1H, H-8), 3.54–3.49 (m, 4H, OMe, OCH2CH2Si), 2.62 (dd, 1H, Jgem = 12.5 Hz, H-3eq), 2.15–1.97 (m, 9H, 3Ac), 1.91 (t, 1H, H-3ax), 1.75 (s, 3H, Ac), 0.95–0.88 (m, 2H, OCH2CH2Si), 0.03 (s, 9H, SiMe3); 13C NMR (125 MHz, CDCl3) δ 170.9, 170.8, 170.1, 170.0, 168.5, 98.7, 77.6, 76.2, 72.1, 69.4, 68.3, 62.0, 61.8, 58.4, 52.5, 49.5, 37.7, 29.7, 23.2, 20.9, 20.9, 20.8, 18.0, −1.6; HRMS (ESI) m/z: found [M + Na]+ 586.2292, C24H41NO12Si calcd for [M + Na]+ 586.2290.
2-(Trimethylsilyl)ethyl 5-acetamido-3,5-dideoxy-8-O-methyl-d-glycero-α-d-galacto-2-nonulopyranosidonic acid (6). To a solution of 33 (34.8 mg, 61.0 µmol) in pyridine (6.2 mL) was added lithium chloride (39.2 mg, 930 µmol) at room temperature. After stirring for 22 h under reflux as the reaction was monitored by TLC (6:4:0.1 CHCl3–MeOH–AcOH), the reaction mixture was co-evaporated with toluene. The crude residue was purified by gel filtration column chromatography (Sephadex LH-20) using MeOH as eluent. The product was exposed to high vacuum, and then dissolved in 0.1 M aq NaOH (5.0 mL). After stirring for 2 h at room temperature as the reaction was monitored by TLC (5:4:1 CHCl3–MeOH–H2O), the reaction mixture was neutralized with Dowex (H+) resin. The resin was filtered through cotton wool and the filtrate was then evaporated. The residue was purified by silica gel column chromatography (6:4:1 CHCl3–MeOH–H2O) followed by gel filtration column chromatography (Sephadex LH-20) using MeOH as eluent to give 6 (12.3 mg, 48%): [α]D −3.0° (c 1.0, MeOH); 1H NMR (500 MHz, D2O) δ 3.98 (dd, 1H, J8,9a = 2.5 Hz, Jgem = 12.0 Hz, H-9a), 3.91–3.83 (m, 3H, OCH2CH2Si, H-7, H-5), 3.74–3.59 (m, 4H, H-4, H-9b, OCH2CH2Si, H-6), 3.48–3.44 (m, 4H, H-8, OMe), 2.65 (dd, 1H, J3eq,4 = 4.5 Hz, Jgem = 12.5 Hz, H-3eq), 2.02 (s, 3H, Ac), 1.69 (t, 1H, J3ax,4 = 12.5 Hz, H-3ax), 0.96–0.92 (m, 2H, OCH2CH2Si), 0.01 (s, 9H, SiMe3); 13C NMR (125 MHz, CD3OD) δ 175.1, 171.3, 100.3, 81.4, 74.5, 69.2, 68.8, 62.7, 61.2, 58.5, 54.1, 49.6, 49.5, 49.3, 49.1, 48.5, 42.2, 22.7, 19.1, −1.3; HRMS (ESI) m/z: found [M − H]− 422.1851, C17H33NO9Si calcd for [M − H]− 422.1852.


 and
and 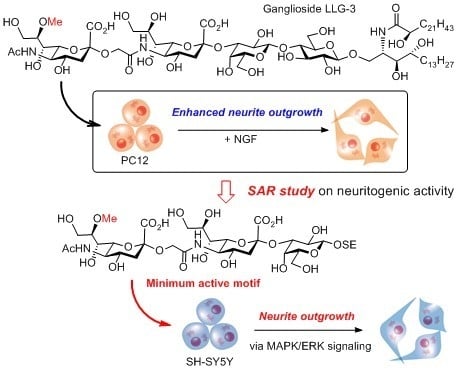
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
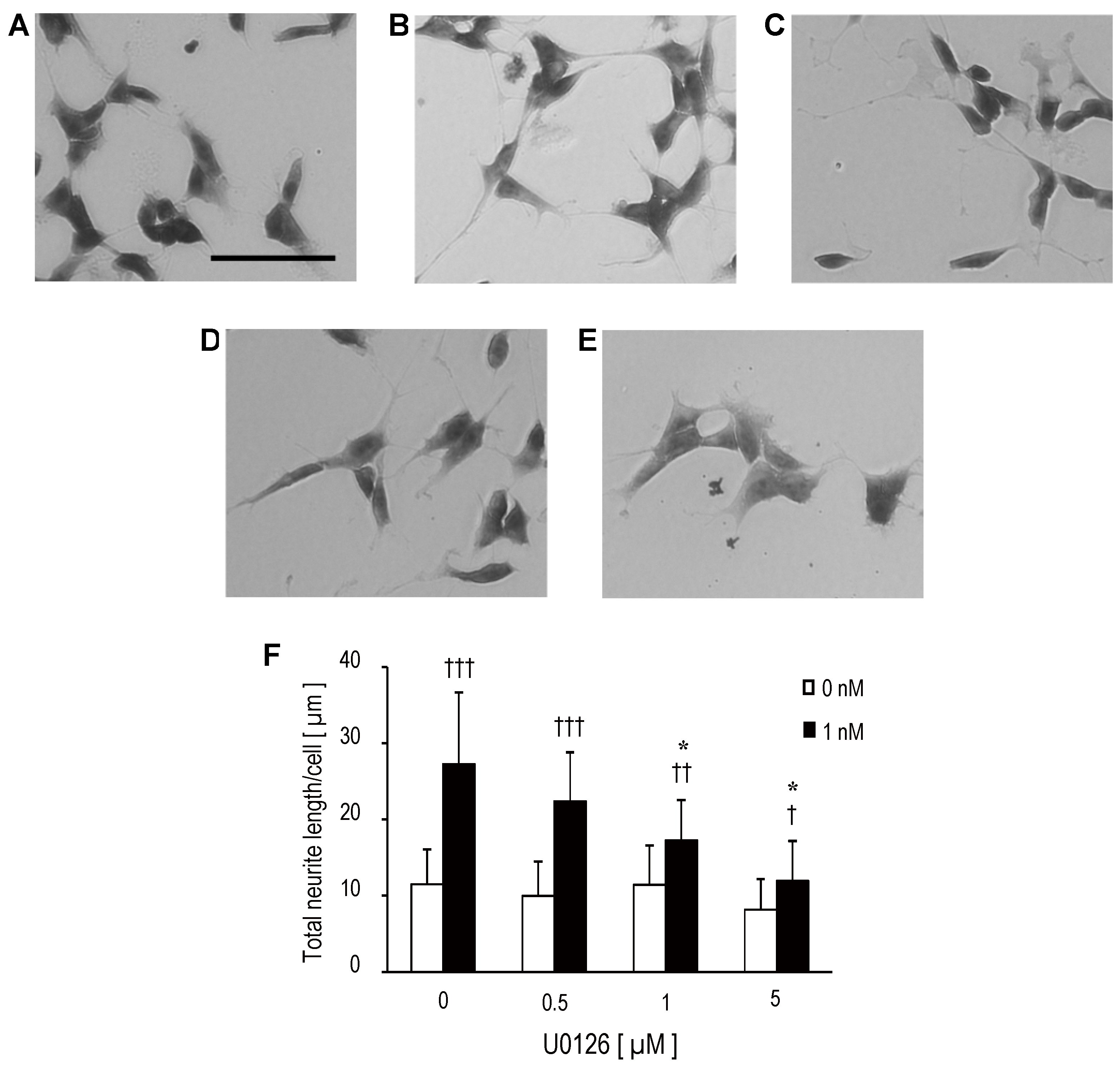{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










