
Antitumor and Antimicrobial Activity of Some Cyclic Tetrapeptides and Tripeptides Derived from Marine Bacteria


Abstract
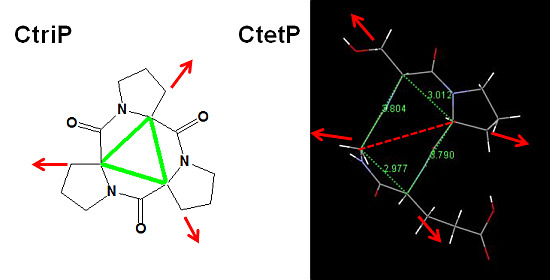
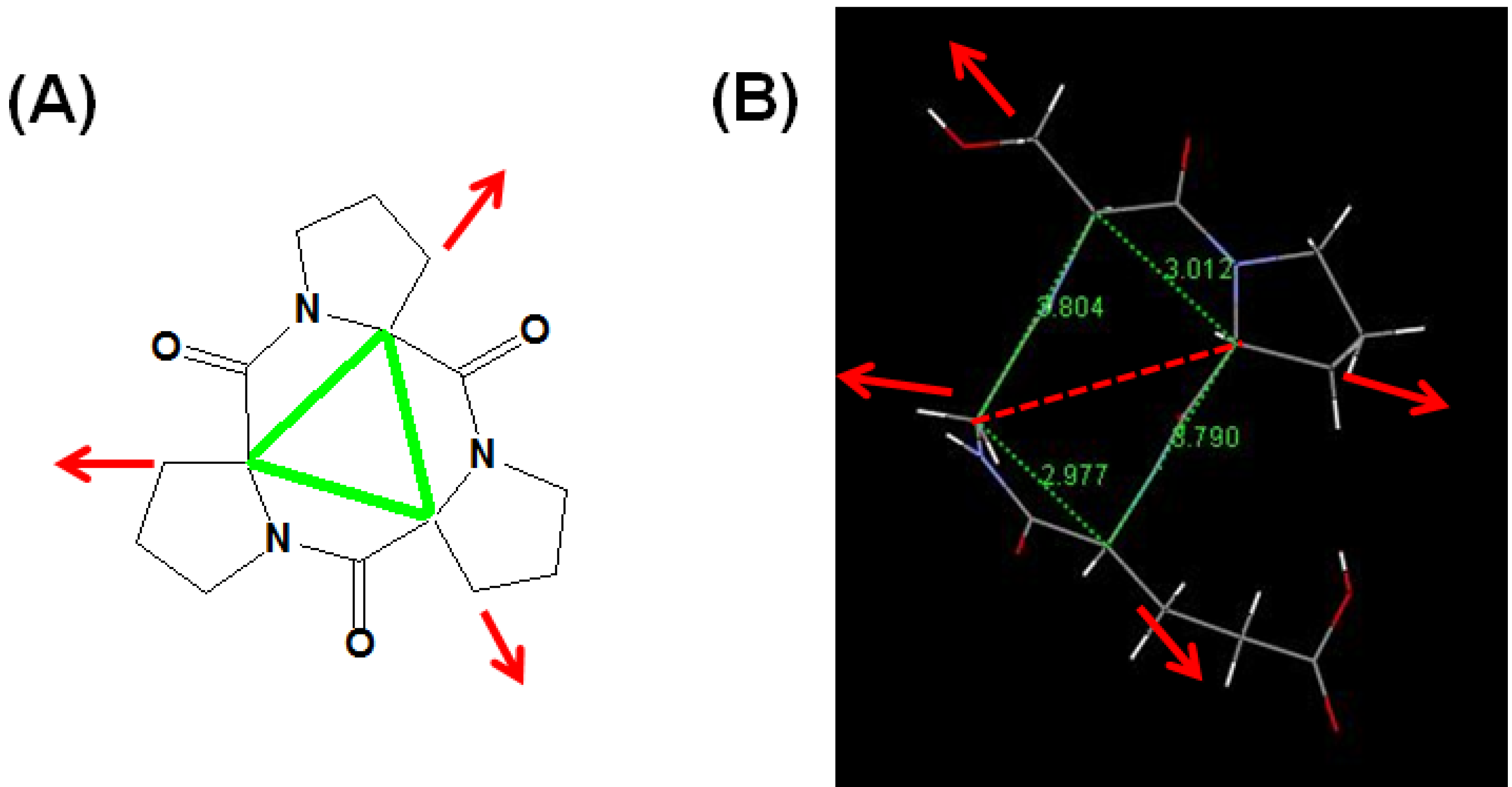
:
1. Introduction
2. Results and Discussion
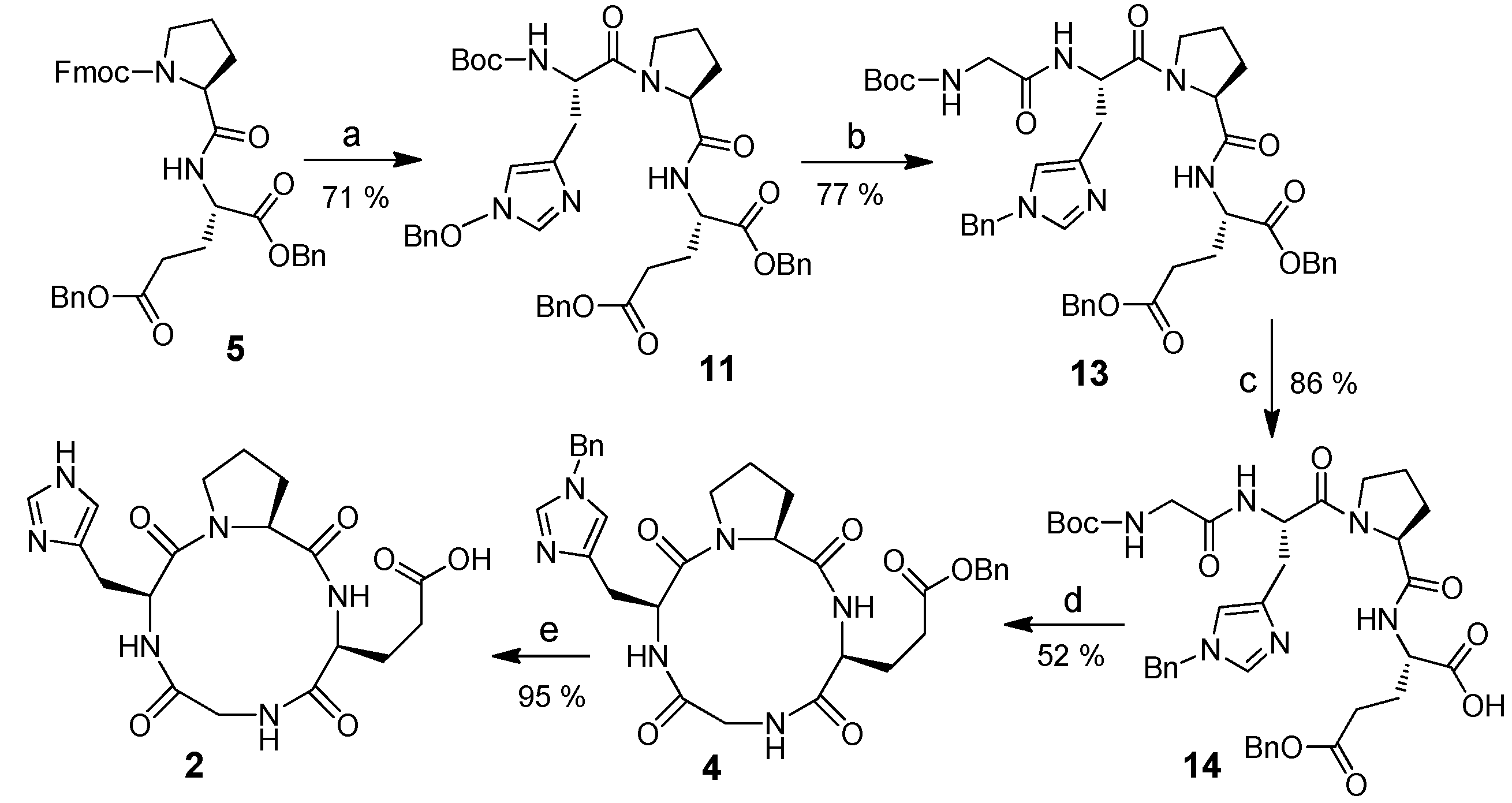
2.1. Cyclic Tetrapeptides Syntheses


2.2. Biological Activities of CtetPs


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MIC (μM) a | Growth Inhibition Assay (μM) b | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| R 1 | R 2 | E. coli | P. aer. | S. aureus | A549 | DBTRG | HepG2 | LNCaP | |
| CtetP 1 | HO– | H | --- | --- | --- | --- | --- | --- | --- |
| CtetP 2 |  | H | --- | --- | --- | --- | --- | --- | --- |
| CtetP 3 | BnO– | Bn | --- | --- | 60 | 108 | 100 | --- | 104 |
| CtetP 4 |  | Bn | --- | --- | 120 | 92 | 80 | --- | 84 |
| Tetracycline | 4.5 | 68 | 1.1 | ||||||
| Taxol | 0.0026 | 0.01 | 0.0065 | 0.0018 | |||||
2.3. Biological Activities of CtriPs
2.4. Structure and Activity Relationship of CtetPs and CtriPs

3. Experimental Section
3.1. General
3.2. Bacterial Strains and Culture Conditions
3.3. Chemicals
3.3.1. Fmoc-l-prolyl-l-glutamyl Dibenzyl Ester (5)
3.3.2. l-Prolyl-l-glutamyl Dibenzyl Ester (6)
3.3.3. Boc-l-seryl(OBn)-l-Prolyl-l-glutamate Dibenzyl Ester (7)
3.3.4. l-Seryl(OBn)-l-Prolyl-l-glutamate Dibenzyl Ester (8)
3.3.5. Boc-glycyl-l-seryl(OBn)-l-Prolyl-l-glutamate Dibenzyl Ester (9)
3.3.6. Boc-glycyl-l-sery(OBn)-l-Prolyl-l-glutamic acid-α-COOH-γ-OBn (10)
3.3.7. Cyclo(glycyl-l-seryl(OBn)-l-Prolyl-l-glutamyl(OBn)) (3)
3.3.8. Cyclo(glycyl-l-seryl-l-Prolyl-l-glutamyl) (1)
3.3.9. Boc-l-histidinyl(Bn)-l-Prolyl-l-glutamyl Dibenzyl Ester (11)
3.3.10. l-Histidinyl(Bn)-l-Prolyl-l-glutamyl Dibenzyl Ester (12)
3.3.11. Boc-glycyl-l-histidinyl(Bn)-l-Prolyl-l-glutamyl Dibenzyl Ester (13)
3.3.12. Boc-glycyl-l-histidinyl(Bn)-l-Prolyl-l-glutamic acid-γ(OBn) (14)
3.3.13. Cyclo(glycyl-l-histidinyl(Bn)-l-Prolyl-l-glutamyl(OBn) (4)
3.3.14. Cyclo(glycyl-l-histidinyl-l-Prolyl-l-glutamyl) (2)
3.4. Minimal Inhibitory Concentrations (MICs) Assay
3.5. Cell Lines and Culture
3.6. MTT Assay for Cancer Cell Lines (Growth Inhibition Assay)
4. Conclusions
Acknowledgments
Author Contributions
Supplementary Information
Conflicts of Interest
References
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Fjell, C.D.; Hiss, J.A.; Hancock, R.E.W.; Schneider, G. Designing antimicrobial peptides: Form follows function. Nat. Rev. Drug Discov. 2012, 11, 37–51. [Google Scholar]
- Harris, F.; Dennison, S.R.; Singh, J.; Phoenix, D.A. On the selectivity and efficacy of defense peptides with respect to cancer cells. Med. Res. Rev. 2013, 33, 190–234. [Google Scholar] [CrossRef] [PubMed]
- Craik, D.J.; Fairlie, D.P.; Liras, S.; Price, D. The future of peptide-based drugs. Chem. Biol. Drug Des. 2013, 81, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.J.; Falla, T.J. Host defense peptides for use as potential therapeutics. Curr. Opin. Investig. Drugs 2009, 10, 164–171. [Google Scholar] [PubMed]
- Nicolas, P. Multifunctional host defense peptides: Intracellular-targeting antimicrobial peptides. FEBS J. 2009, 276, 6483–6496. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, F.C.; Rigden, D.J.; Franco, O.L. Prediction of antimicrobial peptides based on the adaptive neuro-fuzzy inference system application. Biopolymers 2012, 98, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Arbor, S.; Marshall, G.R. A virtual library of constrained cyclic tetrapeptides that mimics all four side-chain orientations for over half the reverse turns in the protein data bank. J. Comput. Aided Mol. Des. 2009, 23, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-C.; Kim, J.-Y.; Lee, J.-K.; Yoo, S.; Kim, H.; Seo, C.H.; Nah, J.-W.; Hahm, K.-S.; Park, Y. Synthetic diastereomeric-antimicrobial peptide: Antibacterial activity against multiple drug resistant clinical isolates. Biopolymers 2010, 96, 130–136. [Google Scholar] [CrossRef]
- Marr, A.K.; Gooderham, W.J.; Hancock, R.E.W. Antibacterial peptides for therapeutic use: Obstacles and realistic outlook. Curr. Opin. Pharmacol. 2006, 6, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Lopez, S.; Kim, H.-S.; Choi, E.C.; Delgado, M.; Granja, J.R.; Khasanov, A.; Kraehenbuehl, K.; Long, G.; Weinberger, D.A.; Wilcoxen, K.M.; et al. Antibacterial agents based on the cyclic d,l-α-peptide architecture. Nature 2001, 412, 452–455. [Google Scholar] [CrossRef] [PubMed]
- Dartois, V.; Sanchez-Quesada, J.; Cabezas, E.; Chi, E.; Dubbelde, C.; Dunn, C.; Granja, J.; Gritzen, C.; Weinberger, D.; Ghadiri, M.R.; et al. Systemic antibacterial activity of novel synthetic cyclic peptides. Antimicrob. Agents Chemother. 2005, 49, 3302–3310. [Google Scholar] [CrossRef] [PubMed]
- Torfoss, V.; Isaksson, J.; Ausbacher, D.; Brandsdal, B.-O.; Flaten, G.E.; Anderssen, T.; Cavalcanti-Jacobsen Cde, A.; Havelkova, M.; Nguyen, L.T.; Vogele, H.J.; et al. Improved anticancer potency by head-to-tail cyclization of short cationic anticancer peptides containing a lipophilic β2,2-amino acid. J. Pept. Sci. 2012, 18, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Lian, W.; Upadhyaya, P.; Rhodes, C.A.; Liu, Y.; Pei, D. Screening bicyclic peptide libraries for protein-protein interaction inhibitors: Discovery of a tumor necrosis factor-α antagonist. J. Am. Chem. Soc. 2013, 135, 11990–11995. [Google Scholar] [CrossRef] [PubMed]
- Lian, W.; Jiang, B.; Qian, Z.; Pei, D. Cell-permeable bicyclic peptide inhibitors against intracellular proteins. J. Am. Chem. Soc. 2014, 136, 9830–9833. [Google Scholar] [CrossRef] [PubMed]
- Polcyn, P.; Jurczak, M.; Rajnisz, A.; Solecka, J.; Urbanczyk-Lipkowska, Z. Design of antimicrobially active small amphiphilic peptide dendrimers. Molecules 2009, 14, 3881–3905. [Google Scholar] [CrossRef] [PubMed]
- Janiszewska, J.; Swieton, J.; Lipkowski, A.W.; Urbanczyk-Lipkowska, Z. Low molecular mass peptide dendrimers that express antimicrobial properties. Bioorganic Med. Chem. 2003, 13, 3711–3713. [Google Scholar] [CrossRef]
- Santos, S.; Torcato, I.; Castanho, M.A. Biomedical applications of dipeptides and tripeptides. Biopolymers 2012, 98, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Tyagi, P.; Tai, D.F.; Lee, G.H.; Peng, S.H. A lead(II) 3D coordination polymer based on a marine cyclic peptide motif. Molecules 2013, 18, 4972–4985. [Google Scholar] [CrossRef] [PubMed]
- Aracil, J.M.; Badre, A.; Fadli, M.; Jeanty, G.; Banaigs, B.; Francisco, C.; Lafargue, F.; Heitz, A.; Aumelas, A. Nouveaux cyclotétrapeptides isolés de l’ascidie cystodytes delle chiajei. Tetrahedron Lett. 1991, 32, 2609–2612. [Google Scholar] [CrossRef]
- Kawagishi, H.; Somoto, A.; Kuranari, J.; Kimura, A.; Chiba, S. A novel cyclotetrapeptide produced by Lactobacillus helveticus as a tyrosinase inhibitor. Tetrahedron Lett. 1993, 34, 3439–3440. [Google Scholar] [CrossRef]
- Kawai, M.; Jasensky, R.D.; Rich, D.H. Conformational analysis by NMR spectrometry of the highly substituted cyclic tetrapeptides, chlamydocin and Ala4-chlamydocin. Evidence for a unique amide bond sequence in dimethyl sulfoxide-d6. J. Am. Chem. Soc. 1983, 105, 4456–4462. [Google Scholar] [CrossRef]
- Kijima, M.; Yoshida, M.; Sugita, K.; Horinouchi, S.; Beppu, T. Trapoxin, an antitumor cyclic tetrapeptide, is an irreversible inhibitor of mammalian histone deacetylase. J. Biol. Chem. 1993, 268, 22429–22435. [Google Scholar] [PubMed]
- Singh, S.B.; Zink, D.L.; Polishook, J.D.; Dombrowski, A.W.; Darkin-Rattray, S.J.; Schamtz, D.M.; Goetz, M.A. Apicidins: Novel cyclic tetrapeptides as coccidiostats and antimalarial agents from Fusarium pallidoroseum. Tetrahedron Lett. 1996, 37, 8077–8080. [Google Scholar] [CrossRef]
- Scott, R.W.; DeGrado, W.F.; Tew, G.N. De novo designed synthetic mimics of antimicrobial peptides. Curr. Opin. Biotech. 2008, 19, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Thaker, H.D.; Som, A.; Ayaz, F.; Lui, D.; Pan, W.; Scott, R.W.; Anguita, J.; Tew, G.N. Synthetic mimics of antimicrobial peptides with immunomodulatory responses. J. Am. Chem. Soc. 2012, 134, 11088–11091. [Google Scholar] [CrossRef] [PubMed]
- Duggan, P.J.; Lewis, R.J.; Lok, Y.P.; Lumsden, N.G.; Tuck, K.L.; Yang, A. Low molecular weight non-peptide mimics of ω-conotoxin GVIA. Bioorganic Med. Chem. Lett. 2009, 19, 2763–2765. [Google Scholar] [CrossRef]
- Ling, L.L.; Schneider, T.; Peoples, A.J.; Spoering, A.L.; Engels, I.; Conlon, B.P.; Mueller, A.; Schaberle, T.F.; Hughes, D.E.; Epstein, S.; et al. A new antibiotic kills pathogens without detectable resistance. Nature 2015, 517, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Somogyi, L.; Haberhauer, G.; Rebek, J. Improved synthesis of functionalized molecular platforms related to marine cyclopeptides. Tetrahedron 2001, 57, 1699–1708. [Google Scholar] [CrossRef]
- Dube, H.; Durola, F.; Ajami, D.; Rebek, J. Molecular switching in nanospaces. J. Chin. Chem. Soc. 2010, 57, 595–603. [Google Scholar] [CrossRef]
- Mitova, M.; Popov, S.; de Rosa, S. Cyclic peptides from a Ruegeria strain of bacteria associated with the sponge Suberites domuncula. J. Nat. Prod. 2004, 67, 1178–1181. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.A.; Kang, C.; Chia, C.S.B. Solid-phase synthesis and NMR structural studies of the marine antibacterial cyclic tetrapeptide: Cyclo[GSPE]. Int. J. Pept. Res. Ther. 2010, 16, 145–152. [Google Scholar] [CrossRef]
- Lim, H.A.; Tan, L.T.; Chia, C.S.B. Exploring solution-phase cyclization and sulfamyl safety-catch resin strategies for the total synthesis of the marine antimicrobial cyclic tetrapeptide cyclo(GSPE). Int. J. Pept. Res. Ther. 2013, 19, 25–31. [Google Scholar] [CrossRef]
- Schwalbe, R.; Steele-Moore, L.; Goodwin, A.C. Antimicrobial Susceptibility Testing Protocols; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Torfoss, V.; Ausbacher, D.; Cavalcanti-Jacobsen, C.dA.; Hansen, T.; Brandsdal, B.-O.; Havelkova, M.; Strom, M.B. Synthesis of anticancer heptapeptides containing a unique lipophilic β2,2-amino acid building block. J. Pept. Sci. 2012, 18, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Held-Kuznetsov, V.; Rotem, S.; Assaraf, Y.G.; Mor, A. Host-defense peptide mimicry for novel antitumor agents. FASEB J. 2009, 23, 4299–4307. [Google Scholar] [CrossRef] [PubMed]
- Steinstraesser, L.; Schubert, C.; Hauk, J.; Becerikli, M.; Stricker, I.; Koeller, M.; Hatt, H.; von Duering, M.; Shai, Y.; Steinau, H.-U.; et al. Oncolytic designer host defense peptide suppresses growth of human liposarcoma. Int. J. Cancer 2011, 128, 2994–3004. [Google Scholar] [CrossRef] [PubMed]
- Tai, D.-F.; Lin, Y.-F. Molecularly imprinted cavities template the macrocyclization of tetrapeptides. Chem. Commun. 2008, 43, 5598–5600. [Google Scholar] [CrossRef]
- Lin, Y.-F.; Lee, S.-M.; Chen, C.-T.; Lu, K.-H.; Tai, D.-F. On-surface cyclization of tetrapeptides using molecularly imprinted polymers as non-covalent auxiliaries. J. Chin. Chem. Soc. 2009, 56, 127–134. [Google Scholar] [CrossRef]
- Schweizer, F. Cationic amphiphilic peptides with cancer-selective toxicity. Eur. J. Pharmacol. 2009, 625, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Yeung, A.T.Y.; Gellatly, S.L.; Hancock, R.E.W. Multifunctional cationic host defence peptides and their clinical applications. Cell. Mol. Life Sci. 2011, 68, 2161–2176. [Google Scholar] [CrossRef] [PubMed]
- Kacprzyk, L.; Rydengard, V.; Morgelin, M.; Davoudi, M.; Pasupuleti, M.; Malmsten, M.; Schmidtchen, A. Antimicrobial activity of histidine-rich peptides is dependent on acidic conditions. Biochim. Biophys. Acta Biomembr. 2007, 1768, 2667–2680. [Google Scholar] [CrossRef]
- Mason, A.J.; Moussaoui, W.; Abdelrahman, T.; Boukhari, A.; Bertani, P.; Marquette, A.; Shooshtarizaheh, P.; Moulay, G.; Boehm, N.; Guerold, B.; et al. Structural determinants of antimicrobial and antiplasmodial activity and selectivity in histidine-rich amphipathic cationic peptides. J. Biol. Chem. 2009, 284, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Stefanucci, A.; Pinnen, F.; Feliciani, F.; Cacciatore, I.; Lucente, G.; Mollica, A. Conformationally Constrained Histidines in the Design of Peptidomimetics: Strategies for the χ-Space Control. Int. J. Mol. Sci. 2011, 12, 2853–2890. [Google Scholar] [CrossRef] [PubMed]
- Bose, A.K.; Strube, R.E. Some derivatives of aspartic and glutamic acids. J. Pharm. Sci. 1963, 9, 847–851. [Google Scholar] [CrossRef]
- Miyazawa, T.; Ogura, M.; Nakajo, S.; Yamada, T. Synthesis of monoesters of N-protected α-aminodicarboxylic acids via the microbial protease-catalyzed regioselective hydrolysis of their diesters. Biotechnol. Tech. 1998, 12, 431–434. [Google Scholar] [CrossRef]
- Cheng, C.-T.; Lo, V.; Chen, J.; Chen, W.-C.; Lin, C.-Y.; Lin, H.-C.; Yang, C.-H.; Sheh, L. Synthesis and DNA nicking studies of a novel cyclic peptide: Cyclo[Lys-Trp-Lys-Ahx-]. Bioorganic Med. Chem. 2001, 9, 1493–1948. [Google Scholar] [CrossRef]
- Makovitzki, A.; Baram, J.; Shai, Y. Antimicrobial lipopolypeptides composed of palmitoyl di- and tricationic peptides: In vitro and in vivo activities, self-assembly to nanostructures, and a plausible mode of action. Biochemistry 2008, 47, 10630–10636. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Lin, S.-H.; Shiuan, D.; Tai, D.-F. Syntheses of Some α-Cyclic Tripeptides as Potential Inhibitors for HMG-CoA Reductase. Amino Acids 2015, 47. [Google Scholar] [CrossRef] [PubMed]
- Druyan, M.E.; Coulter, C.L.; Walter, R.; Kartha, G.; Arnbady, G.K. Structure and conformation of cyclo(tri-l-prolyl) in the crystalline state. J. Am. Chem. Soc. 1976, 98, 5496–5502. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chakraborty, S.; Tai, D.-F.; Lin, Y.-C.; Chiou, T.-W. Antitumor and Antimicrobial Activity of Some Cyclic Tetrapeptides and Tripeptides Derived from Marine Bacteria. Mar. Drugs 2015, 13, 3029-3045. https://doi.org/10.3390/md13053029
Chakraborty S, Tai D-F, Lin Y-C, Chiou T-W. Antitumor and Antimicrobial Activity of Some Cyclic Tetrapeptides and Tripeptides Derived from Marine Bacteria. Marine Drugs. 2015; 13(5):3029-3045. https://doi.org/10.3390/md13053029
Chicago/Turabian StyleChakraborty, Subrata, Dar-Fu Tai, Yi-Chun Lin, and Tzyy-Wen Chiou. 2015. "Antitumor and Antimicrobial Activity of Some Cyclic Tetrapeptides and Tripeptides Derived from Marine Bacteria" Marine Drugs 13, no. 5: 3029-3045. https://doi.org/10.3390/md13053029
APA StyleChakraborty, S., Tai, D. -F., Lin, Y. -C., & Chiou, T. -W. (2015). Antitumor and Antimicrobial Activity of Some Cyclic Tetrapeptides and Tripeptides Derived from Marine Bacteria. Marine Drugs, 13(5), 3029-3045. https://doi.org/10.3390/md13053029