
Tackling the Cytotoxic Effect of a Marine Polycyclic Quinone-Type Metabolite: Halenaquinone Induces Molt 4 Cells Apoptosis via Oxidative Stress Combined with the Inhibition of HDAC and Topoisomerase Activities

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
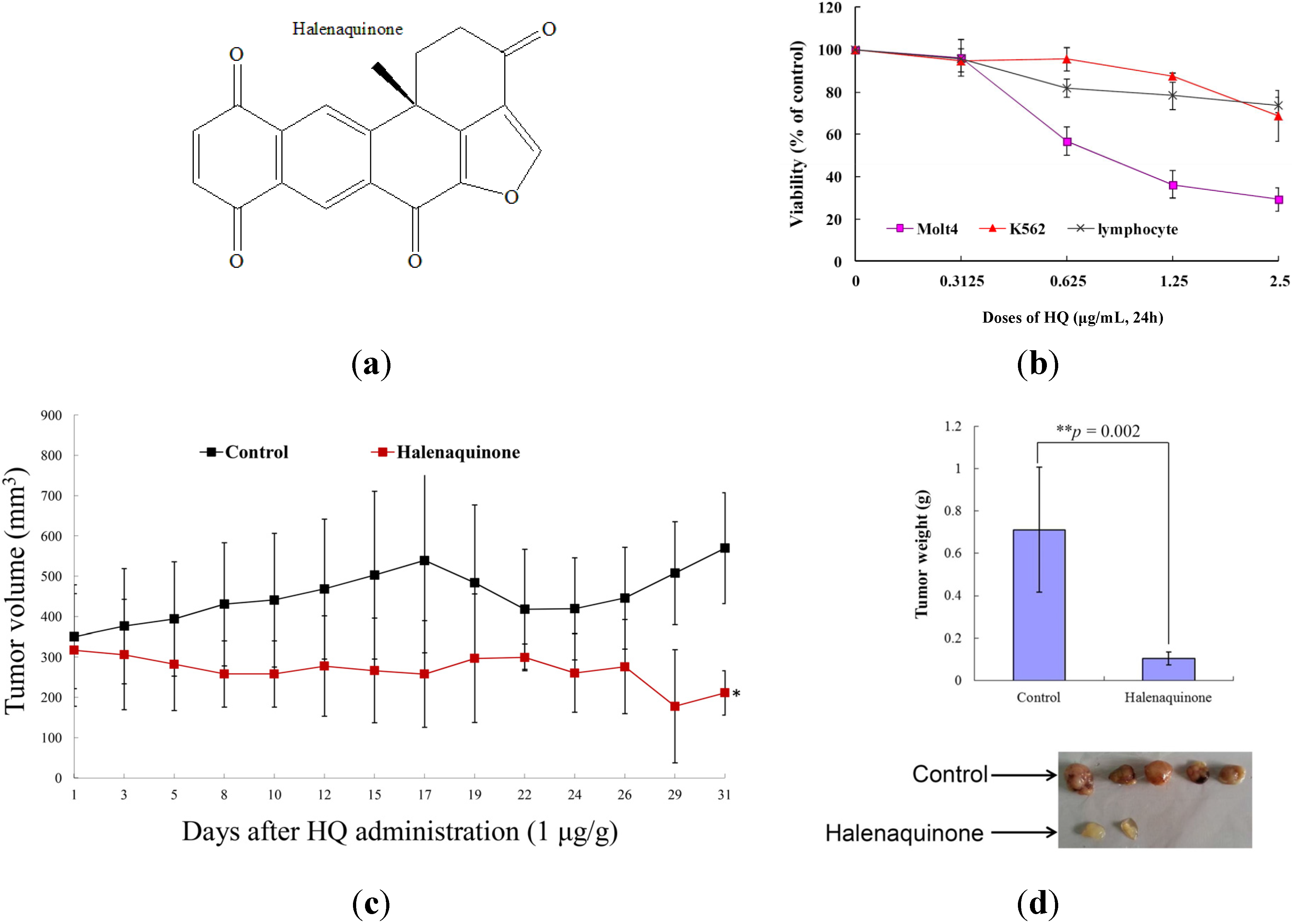
2.1. Effect of HQ on Cellular and Tumor Growth in Vitro Assay and in Vivo Animal Model
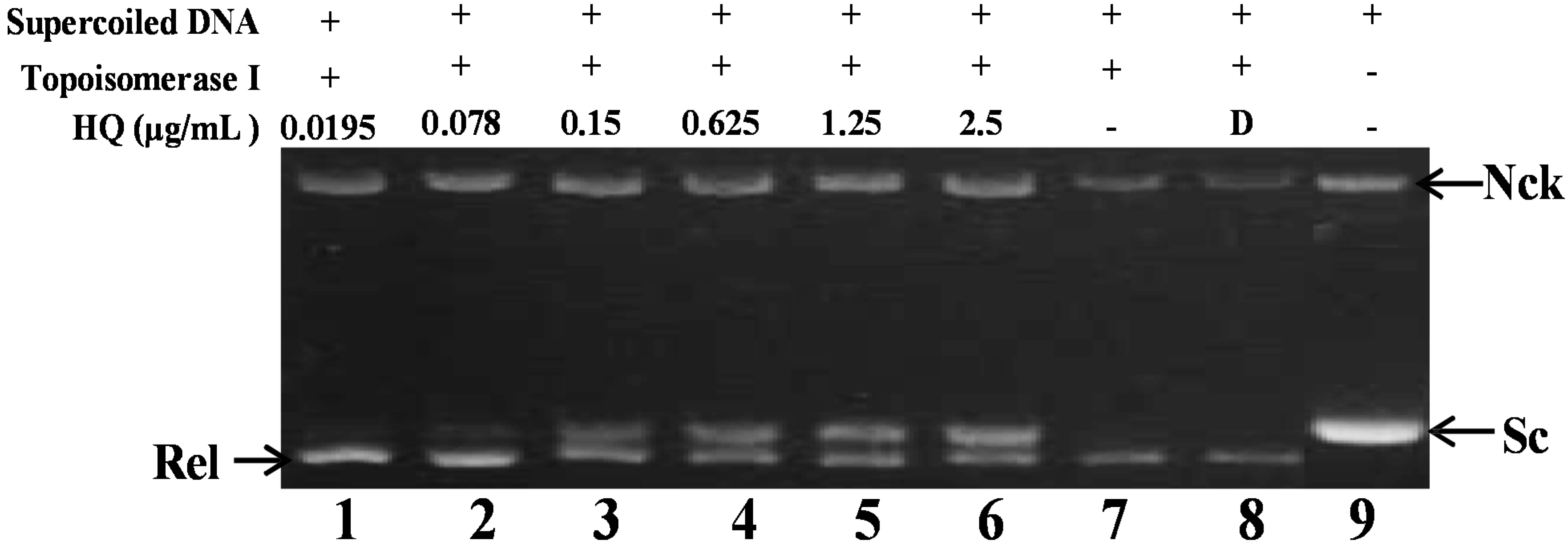
2.2. Effect of HQ on Topoisomerase I and II Activity



2.3. Effect of HQ on Histone Deacetylase Activity

2.4. Effect of HQ on Apoptosis Induction Involved Mitochondrial Dysfunction in Molt 4 Cells



2.5. HQ Induced Apoptosis Is Mediated by Excessive ROS Generation


3. Discussion
4. Experimental Section
4.1. Bioassay Materials
4.2. Preparation of Halenaquinone Stock Solution
4.3. MTT Proliferation Assay
4.4. Annexin V/PI Apoptotic Assay
4.5. Determination of ROS Generation and MMP Disruption
4.6. Assay of Topoisomerase I and II Catalytic Inhibitors and Poisons
4.7. Assays of Caspases 8 and 9 Activities
4.8. Assay of Histone Deacetylase Activity
4.9. Human Leukemia Molt 4 Cells Xenograft Animal Model
4.10. Generation of Rat Lymphocytes
4.11. Statistics
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lakshmaiah, K.C.; Jacob, L.A.; Aparna, S.; Lokanatha, D.; Saldanha, S.C. Epigenetic therapy of cancer with histone deacetylase inhibitors. J. Cancer Res. Ther. 2014, 10, 469–478. [Google Scholar] [PubMed]
- Spange, S.; Wagner, T.; Heinzel, T.; Kramer, O.H. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int. J. Biochem. Cell Biol. 2009, 41, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Florean, C.; Schnekenburger, M.; Grandjenette, C.; Dicato, M.; Diederich, M. Epigenomics of leukemia: From mechanisms to therapeutic applications. Epigenomics 2011, 3, 581–609. [Google Scholar] [CrossRef] [PubMed]
- Schnekenburger, M.; Diederich, M. Epigenetics offer new horizons for colorectal cancer prevention. Curr. Colorectal Cancer Rep. 2012, 8, 66–81. [Google Scholar] [CrossRef] [PubMed]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Kavanaugh, S.M.; White, L.A.; Kolesar, J.M. Vorinostat: A novel therapy for the treatment of cutaneous T-cell lymphoma. Am. J. Health Syst. Pharm. 2010, 67, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Zain, J.; Kaminetzky, D.; O’Connor, O.A. Emerging role of epigenetic therapies in cutaneous T-cell lymphomas. Expert Rev. Hematol. 2010, 3, 187–203. [Google Scholar] [CrossRef] [PubMed]
- Ververis, K.; Hiong, A.; Karagiannis, T.C.; Licciardi, P.V. Histone deacetylase inhibitors (HDACIs): Multitargeted anticancer agents. Biologics 2013, 7, 47–60. [Google Scholar] [PubMed]
- Calderon-Montano, J.M.; Burgos-Moron, E.; Orta, M.L.; Lopez-Lazaro, M. Effect of DNA repair deficiencies on the cytotoxicity of drugs used in cancer therapy—A review. Curr. Med. Chem. 2014, 21, 3419–3454. [Google Scholar] [CrossRef] [PubMed]
- Beretta, G.L.; Gatti, L.; Perego, P.; Zaffaroni, N. Camptothecin resistance in cancer: Insights into the molecular mechanisms of a DNA-damaging drug. Curr. Med. Chem. 2013, 20, 1541–1565. [Google Scholar] [CrossRef] [PubMed]
- Khadka, D.B.; Cho, W.J. Topoisomerase inhibitors as anticancer agents: A patent update. Expert Opin. Ther. Pat. 2013, 23, 1033–1056. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Pendleton, M.; Lindsey, R.H., Jr.; Felix, C.A.; Grimwade, D.; Osheroff, N. Topoisomerase II and leukemia. Ann. N. Y. Acad. Sci. 2014, 1310, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Kakuda, T.; Qi, J.; Hirata, M.; Shintani, T.; Yoshioka, Y.; Okamoto, T.; Oba, Y.; Nakamura, H.; Ojika, M. Novel relationship between the antifungal activity and cytotoxicity of marine-derived metabolite xestoquinone and its family. Biosci. Biotechnol. Biochem. 2005, 69, 1749–1752. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.H.; Slate, D.L.; Moretti, R.; Alvi, K.A.; Crews, P. Marine sponge polyketide inhibitors of protein tyrosine kinase. Biochem. Biophys. Res. Commun. 1992, 184, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Foster, C.; Brisson, M.; Lazo, J.S.; Kingston, D.G. Halenaquinone and xestoquinone derivatives, inhibitors of Cdc25B phosphatase from a Xestospongia sp. Bioorg. Med. Chem. 2005, 13, 999–1003. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, H.; Matsunaga, K.; Saito, M.; Hagiya, S.; Furukawa, K.; Nakamura, H.; Ohizumi, Y. Halenaquinone, a novel phosphatidylinositol 3-kinase inhibitor from a marine sponge, induces apoptosis in PC12 cells. Eur. J. Pharmacol. 2001, 413, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Bourguet-Kondracki, M.L.; Longeon, A.; Dubois, J.; Valentin, A.; Copp, B.R. Chemical and biological explorations of the electrophilic reactivity of the bioactive marine natural product halenaquinone with biomimetic nucleophiles. Bioorg. Med. Chem. Lett. 2011, 21, 1261–1264. [Google Scholar] [CrossRef] [PubMed]
- Takaku, M.; Kainuma, T.; Ishida-Takaku, T.; Ishigami, S.; Suzuki, H.; Tashiro, S. Halenaquinone, a chemical compound that specifically inhibits the secondary DNA binding of RAD. Genes Cells 2011, 16, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, S.; Takeuchi, T.; Kawabata, T.; Kato, H.; Yamakuma, M.; Matsuo, K.; El-Desoky, A.H.; Losung, F.; Mangindaan, R.E.; de Voogd, N.J.; et al. Halenaquinone inhibits RANKL-induced osteoclastogenesis. Bioorg. Med. Chem. Lett. 2014, 24, 5315–5317. [Google Scholar] [CrossRef] [PubMed]
- Bae M-A, T.T.; Tomoko, T.; Kiyosi, K.; Takumi, H.; Masami, I.; Hideyuki, S.; Juni’chi, K. Inhibition of mammalian topoisomease I by xestoquinone and halenaquinone. Biosci. Biotech. Biochem. 1993, 57, 330–331. [Google Scholar] [CrossRef]
- Su, J.H.; Chen, Y.C.; El-Shazly, M.; Du, Y.C.; Su, C.W.; Tsao, C.-W.; Liu, L.-L.; Chou, Y.; Chang, W.-B.; Su, Y.-D.; et al. Towards the small and the beautiful: A small dibromotyrosine derivative from Pseudoceratina sp. sponge exhibits potent apoptotic effect through targeting IKK/NFkappaB signaling pathway. Mar. Drugs 2013, 11, 3168–3185. [Google Scholar] [CrossRef] [PubMed]
- Concepcion, G.P.; Foderaro, T.A.; Eldredge, G.S.; Lobkovsky, E.; Clardy, J.; Barrows, L.R.; Ireland, C.M. Topoisomerase II-mediated DNA cleavage by adocia- and xestoquinones from the Philippine sponge Xestospongia sp. J. Med. Chem. 1995, 38, 4503–4507. [Google Scholar] [CrossRef] [PubMed]
- Shih, H.C.; El-Shazly, M.; Juan, Y.S.; Chang, C.Y.; Su, J.H.; Chen, Y.C.; Shih, S.P.; Chen, H.M.; Wu, Y.C.; Lu, M.C. Cracking the cytotoxicity code: Apoptotic induction of 10-acetylirciformonin B is mediated through ROS generation and mitochondrial dysfunction. Mar. Drugs 2014, 12, 3072–3090. [Google Scholar] [CrossRef] [PubMed]
- Feoktistova, M.; Geserick, P.; Kellert, B.; Dimitrova, D.P.; Langlais, C.; Hupe, M.; Cain, K.; MacFarlane, M.; Häcker, G.; Leverkus, M. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell 2011, 43, 449–463. [Google Scholar] [CrossRef] [PubMed]
- Laurent, D.; Jullian, V.; Parenty, A.; Knibiehler, M.; Dorin, D.; Schmitt, S.; Lozach, O.; Lebouvier, N.; Frostin, M.; Alby, F.; et al. Antimalarial potential of xestoquinone, a protein kinase inhibitor isolated from a Vanuatu marine sponge Xestospongia sp. Bioorg. Med. Chem. 2006, 14, 4477–4482. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Chen, H.; Yin, M.; Ye, X.; Chen, G.; Zhou, X.; Yin, L.; Zhang, C.; Ding, B. MiR-376a and histone deacetylation 9 form a regulatory circuitry in hepatocellular carcinoma. Cell. Physiol. Biochem. 2015, 35, 729–739. [Google Scholar] [PubMed]
- Sheng, J.; Luo, W.; Yu, F.; Gao, N.; Hu, B. MicroRNA-376a sensitizes cells following DNA damage by downregulating MEPE expression. Cancer Biother. Radiopharm. 2013, 28, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Shi, Y.; Guo, X.H.; Ouyang, Y.B.; Wang, S.S.; Liu, D.J.; Wang, A.N.; Li, N.; Chen, D.X. Phosphorylated AKT inhibits the apoptosis induced by DRAM-mediated mitophagy in hepatocellular carcinoma by preventing the translocation of DRAM to mitochondria. Cell Death Dis. 2014, 5, e1078. [Google Scholar] [CrossRef] [PubMed]
- Yadav, N.; Chandra, D. Mitochondrial and postmitochondrial survival signaling in cancer. Mitochondrion 2014, 16, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Oronsky, B.; Oronsky, N.; Knox, S.; Fanger, G.; Scicinski, J. Episensitization: Therapeutic tumor resensitization by epigenetic agents: A review and reassessment. Anticancer Agents Med. Chem. 2014, 14, 1121–1127. [Google Scholar] [CrossRef] [PubMed]
- Matei, D.; Fang, F.; Shen, C.; Schilder, J.; Arnold, A.; Zeng, Y.; Berry, W.A.; Huang, T.; Nephew, K.P. Epigenetic resensitization to platinum in ovarian cancer. Cancer Res. 2012, 72, 2197–2205. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, J.; Granados-Castro, L.F.; Zazueta, C.; Anderica-Romero, A.C.; Chirino, Y.I.; Pedraza-Chaverrí, J. Mitochondria as a target in the therapeutic properties of curcumin. Arch. Pharm. 2014, 347, 873–884. [Google Scholar] [CrossRef]
- Zhang, D.; Li, J.; Wang, F.; Hu, J.; Wang, S.; Sun, Y. 2-Deoxy-d-glucose targeting of glucose metabolism in cancer cells as a potential therapy. Cancer Lett. 2014, 355, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Urra, F.A.; Cordova-Delgado, M.; Pessoa-Mahana, H.; Ramirez-Rodriguez, O.; Weiss-Lopez, B.; Ferreira, J.; Araya-Maturana, R. Mitochondria: A promising target for anticancer alkaloids. Curr. Top. Med. Chem. 2013, 13, 2171–2183. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.M.; Hsu, P.C.; Chen, M.Y.; Li, W.S.; More, S.V.; Lu, K.T.; Wang, Y.C. The novel indole compound SK228 induces apoptosis and FAK/Paxillin disruption in tumor cell lines and inhibits growth of tumor graft in the nude mouse. Int. J. Cancer 2012, 131, 722–732. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Sussman, M.A. Mitochondrial integrity: Preservation through Akt/Pim-1 kinase signaling in the cardiomyocyte. Expert Rev. Cardiovasc. Ther. 2009, 7, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.C.; Lu, M.C.; Chen, H.L.; Tseng, H.I.; Ke, Y.Y.; Wu, Y.C.; Yang, P.Y. Cytotoxicity of calotropin is through caspase activation and downregulation of anti-apoptotic proteins in K562 cells. Cell. Biol. Int. 2009, 33, 1230–1236. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.C.; Chang, F.R.; Wu, T.Y.; Hsu, Y.M.; El-Shazly, M.; Chen, C.F.; Sung, P.J.; Lin, Y.Y.; Lin, Y.H.; Wu, Y.C.; et al. Antileukemia component, dehydroeburicoic acid from Antrodia camphorata induces DNA damage and apoptosis in vitro and in vivo models. Phytomedicine 2012, 19, 788–796. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.-C.; Huang, S.-L.; Chang, F.-R.; Chen, Y.-H.; Chang, T.-T.; Hung, C.-S.; Wang, C.-L.; Chu, Y.-H.; Pan, S.-H.; Wu, Y.-C. Immunostimulatory effect of Antrodia camphorata extract on functional maturation of dendritic cells. Food Chem. 2009, 113, 1049–1057. [Google Scholar] [CrossRef]
- Hewitt, C.W.; Sawyer, S.; Beck, P.A.; Martin, D.C. A simple method for the isolation of platelet-free lymphocyte suspensions from rat whole blood. J. Immunol. Methods 1980, 36, 227–234. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shih, S.-P.; Lee, M.-G.; El-Shazly, M.; Juan, Y.-S.; Wen, Z.-H.; Du, Y.-C.; Su, J.-H.; Sung, P.-J.; Chen, Y.-C.; Yang, J.-C.; et al. Tackling the Cytotoxic Effect of a Marine Polycyclic Quinone-Type Metabolite: Halenaquinone Induces Molt 4 Cells Apoptosis via Oxidative Stress Combined with the Inhibition of HDAC and Topoisomerase Activities. Mar. Drugs 2015, 13, 3132-3153. https://doi.org/10.3390/md13053132
Shih S-P, Lee M-G, El-Shazly M, Juan Y-S, Wen Z-H, Du Y-C, Su J-H, Sung P-J, Chen Y-C, Yang J-C, et al. Tackling the Cytotoxic Effect of a Marine Polycyclic Quinone-Type Metabolite: Halenaquinone Induces Molt 4 Cells Apoptosis via Oxidative Stress Combined with the Inhibition of HDAC and Topoisomerase Activities. Marine Drugs. 2015; 13(5):3132-3153. https://doi.org/10.3390/md13053132
Chicago/Turabian StyleShih, Shou-Ping, Man-Gang Lee, Mohamed El-Shazly, Yung-Shun Juan, Zhi-Hong Wen, Ying-Chi Du, Jui-Hsin Su, Ping-Jyun Sung, Yu-Cheng Chen, Juan-Cheng Yang, and et al. 2015. "Tackling the Cytotoxic Effect of a Marine Polycyclic Quinone-Type Metabolite: Halenaquinone Induces Molt 4 Cells Apoptosis via Oxidative Stress Combined with the Inhibition of HDAC and Topoisomerase Activities" Marine Drugs 13, no. 5: 3132-3153. https://doi.org/10.3390/md13053132
APA StyleShih, S. -P., Lee, M. -G., El-Shazly, M., Juan, Y. -S., Wen, Z. -H., Du, Y. -C., Su, J. -H., Sung, P. -J., Chen, Y. -C., Yang, J. -C., Wu, Y. -C., & Lu, M. -C. (2015). Tackling the Cytotoxic Effect of a Marine Polycyclic Quinone-Type Metabolite: Halenaquinone Induces Molt 4 Cells Apoptosis via Oxidative Stress Combined with the Inhibition of HDAC and Topoisomerase Activities. Marine Drugs, 13(5), 3132-3153. https://doi.org/10.3390/md13053132