
Crambescin C1 Exerts a Cytoprotective Effect on HepG2 Cells through Metallothionein Induction




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
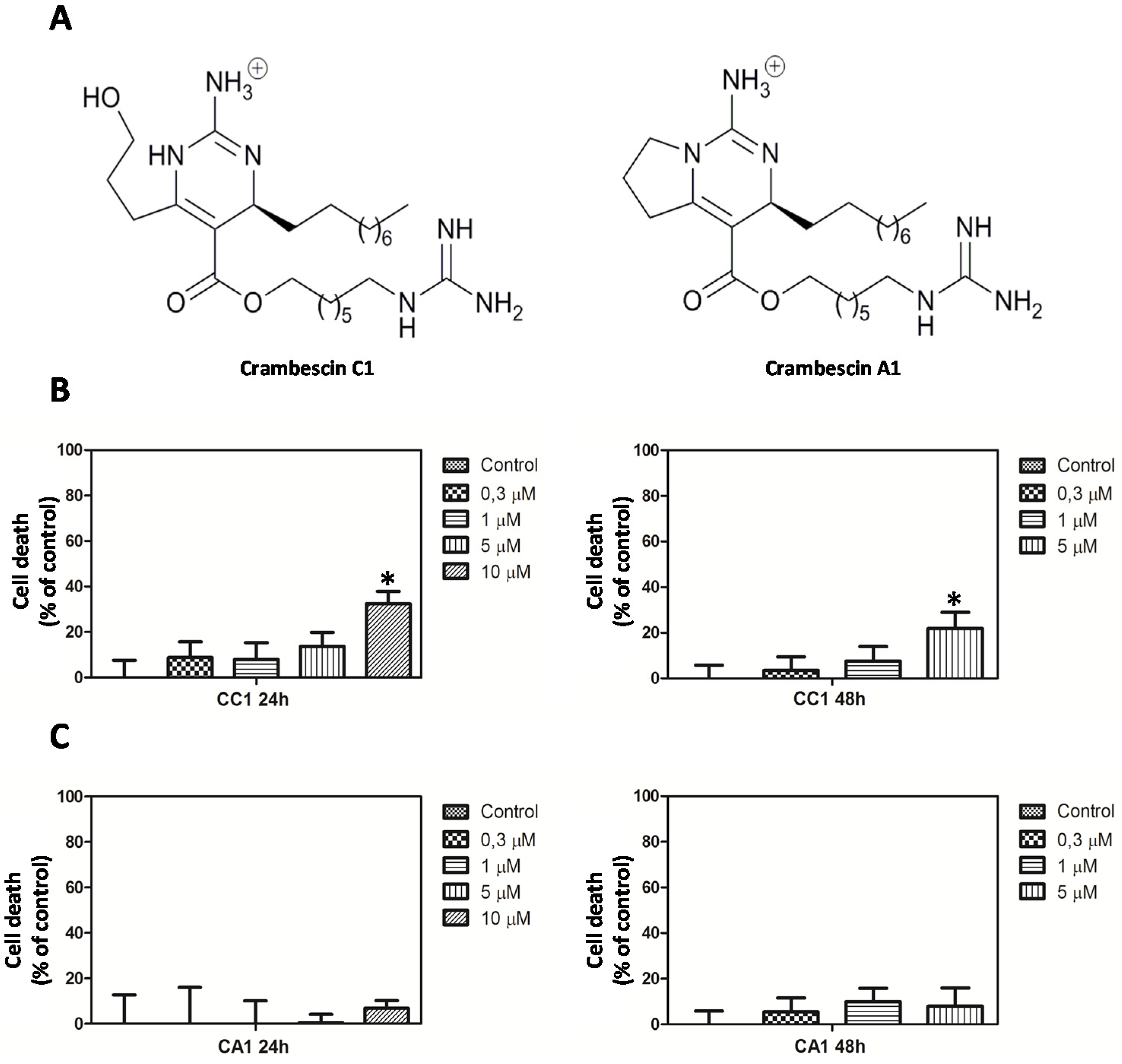
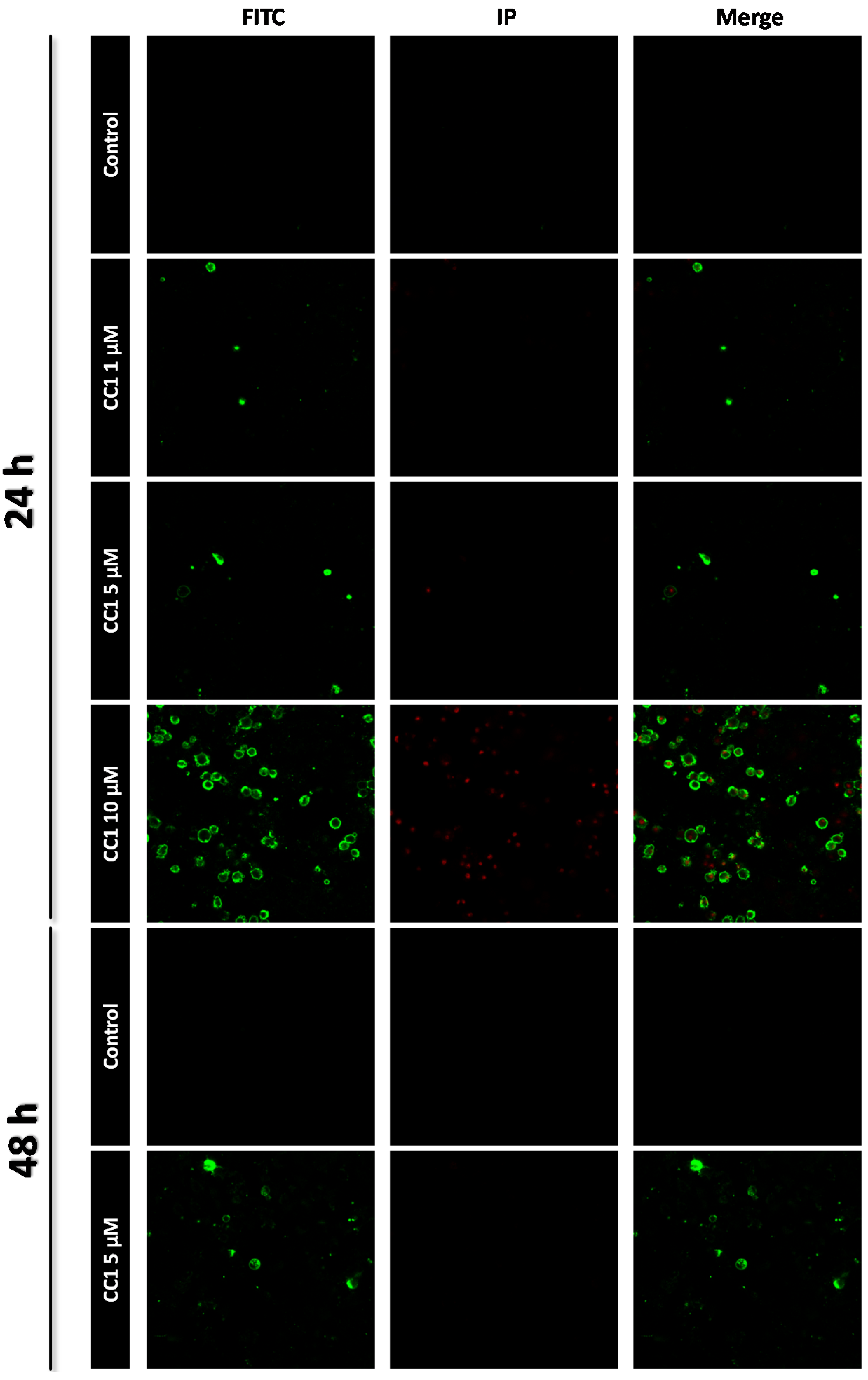
2.1. CC1 Inhibits Cell Proliferation and Induces Cell Death at High Doses


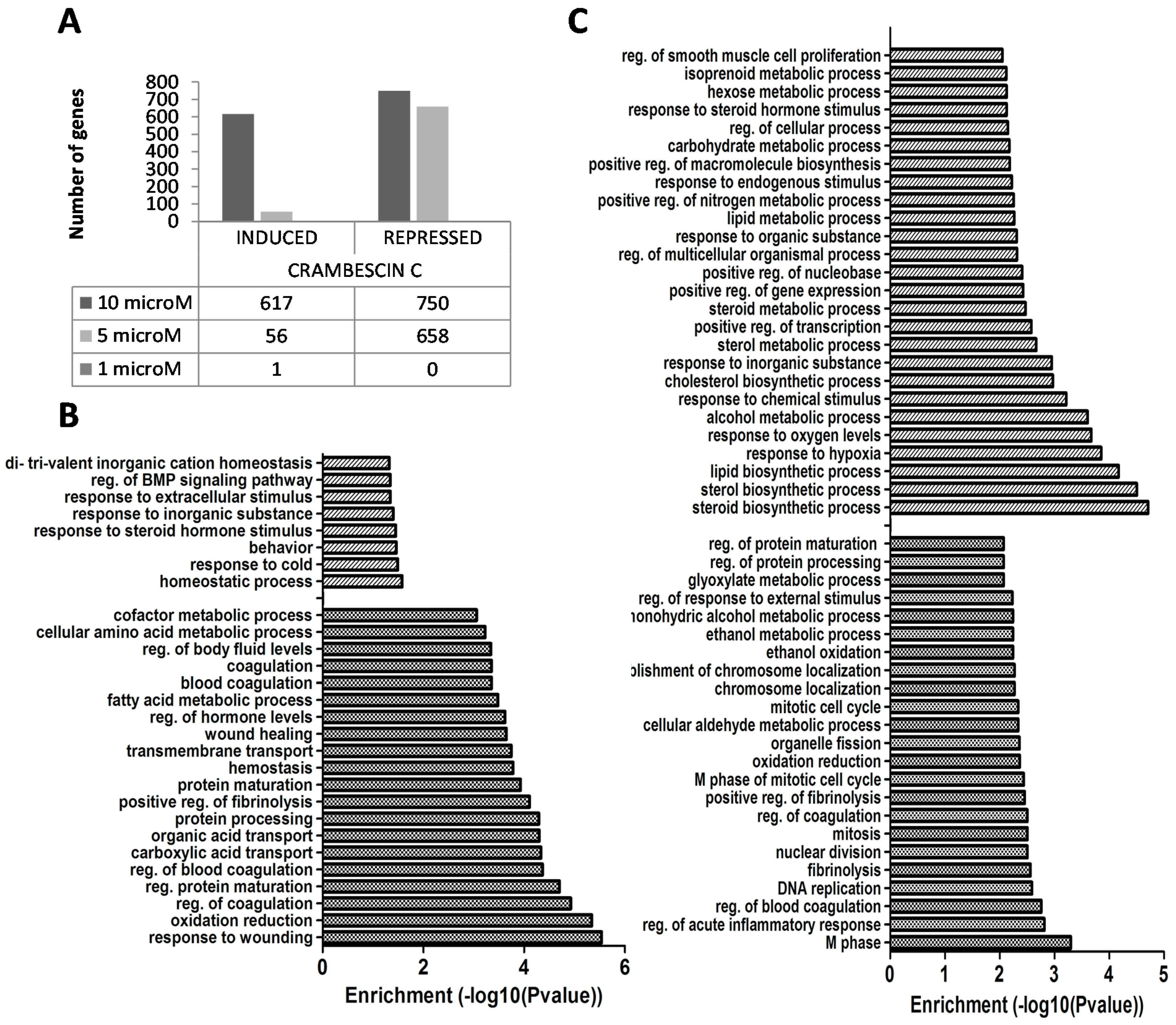
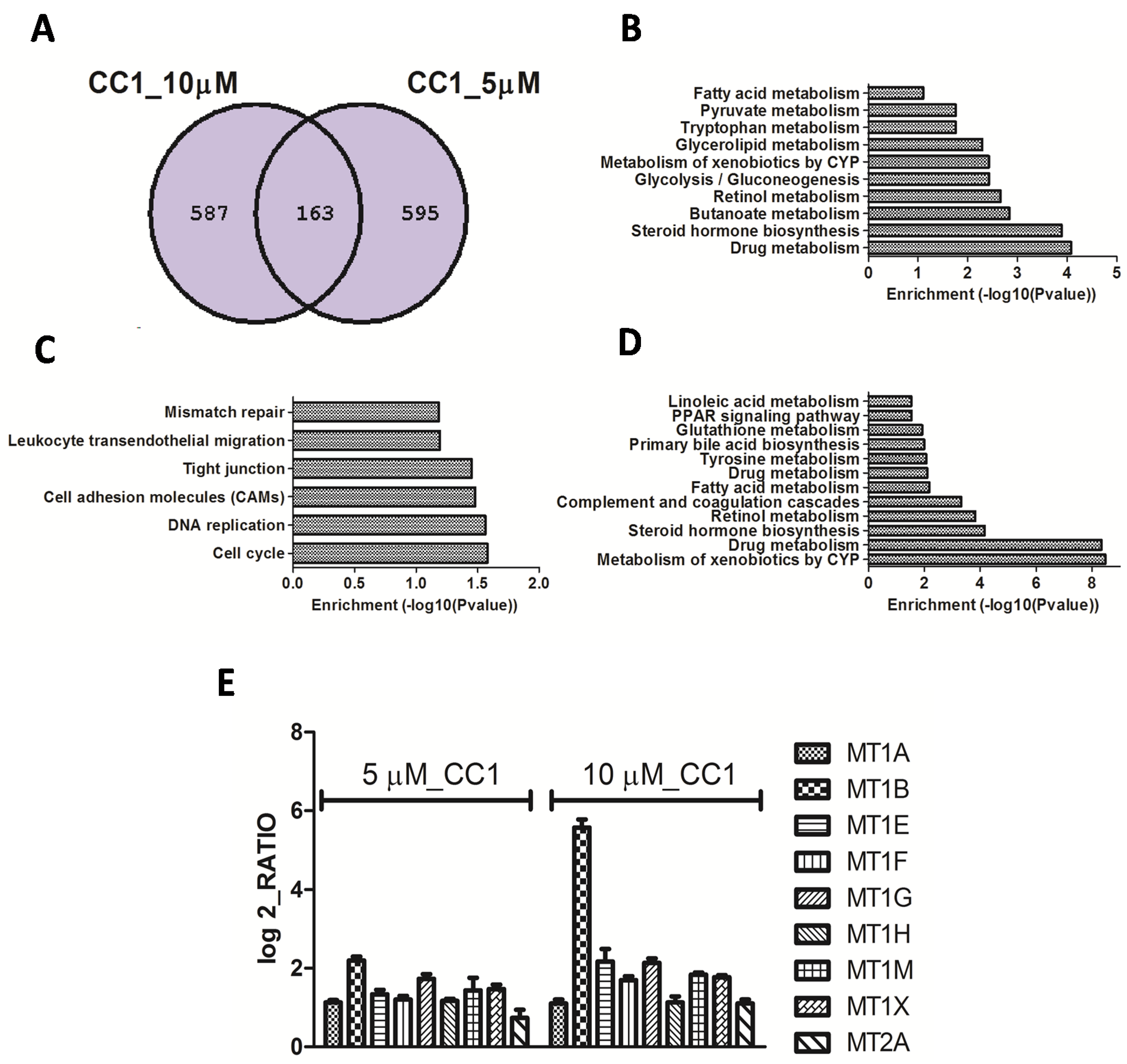
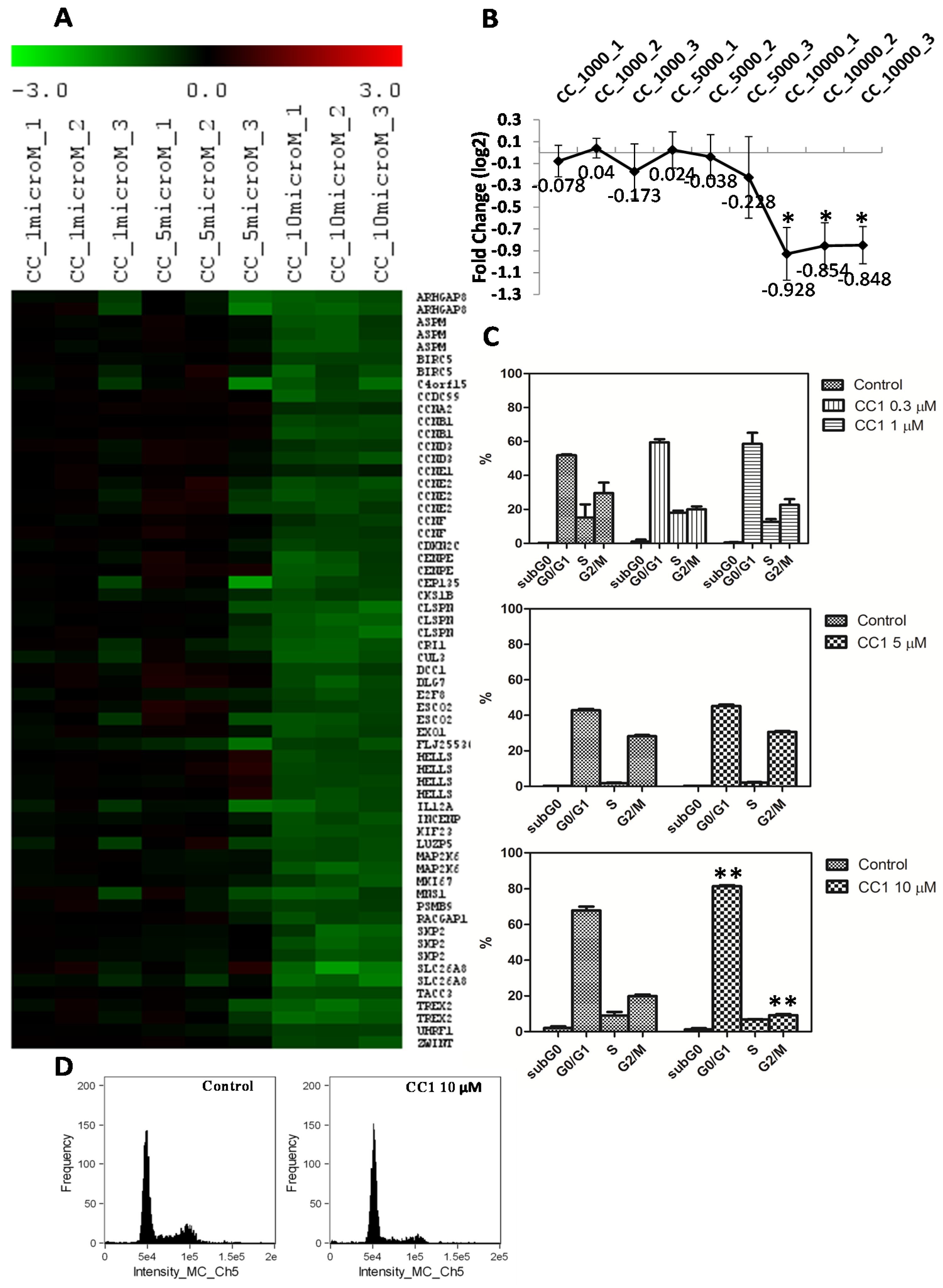
2.2. Transcriptional Alterations Induced by CC1 on HepG2 Cells


2.3. CC1 Arrests HepG2 Cell Cycle in G0/G1

2.4. CC1 Protects Cells against Oxidative Injury

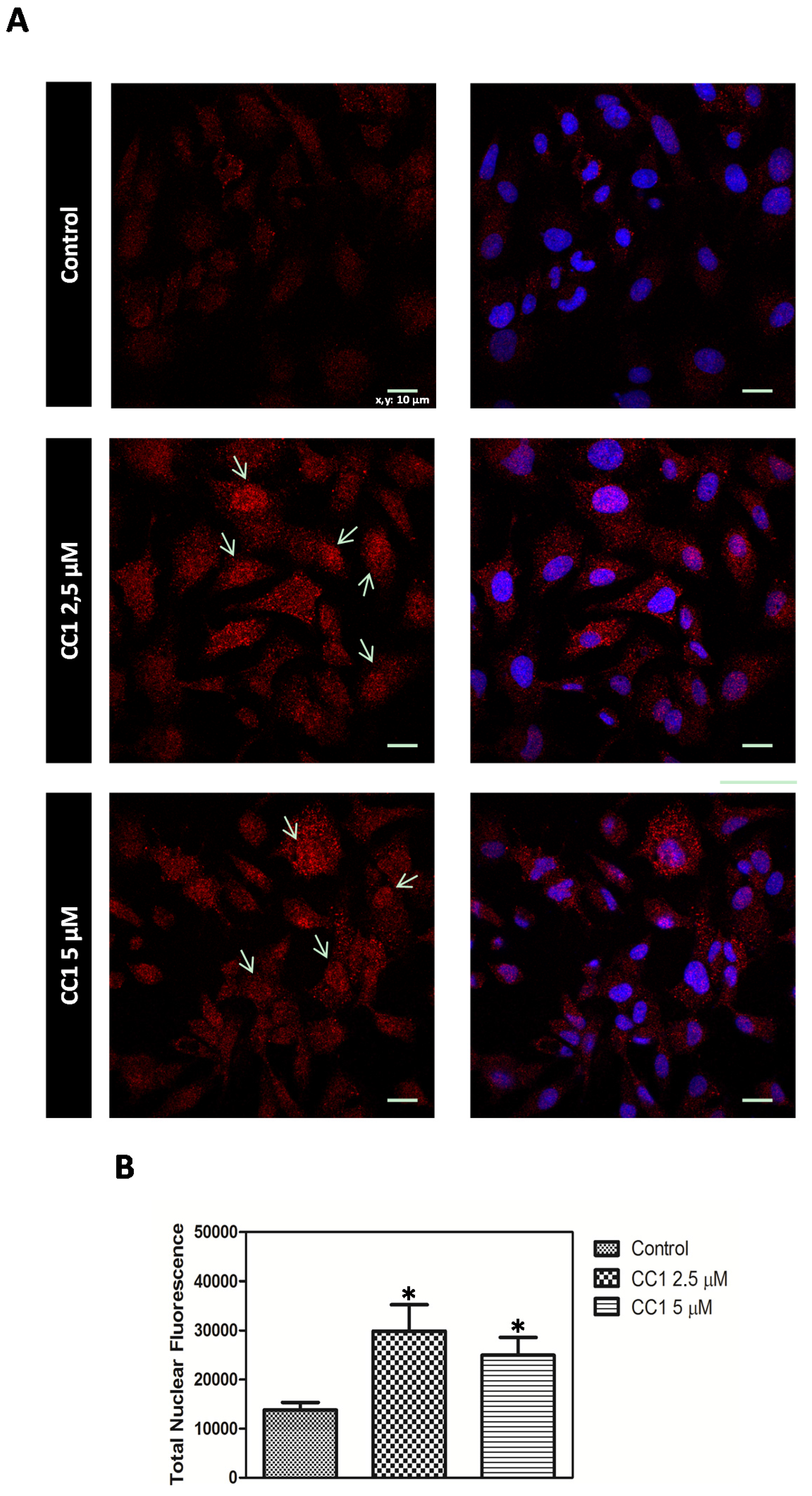
CC1 Increases Nuclear MTs Levels

3. Experimental Section
3.1. Reagents and Solutions
3.2. Crambescins Isolation
3.3. Cell Culture
3.4. Cell Viability Assays
3.5. Apoptosis Determination
3.6. Microarrays Assay and Analysis
3.7. Antioxidant Activity Assay
3.8. Glutathione Quantification
3.9. Western Blot Analysis
3.10. Cell Cycle Assay by Flow Cytometry
3.11. Confocal Microscopy
3.12. Statistics
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Riguera, R. Isolating bioactive compounds from marine organisms. J. Mar. Biotechnol. 1997, 5, 187–193. [Google Scholar]
- Leal, M.C.; Madeira, C.; Brandão, C.A.; Puga, J.; Calado, R. Bioprospecting of marine invertebrates for new natural products—A chemical and zoogeographical perspective. Molecules 2012, 17, 9842–9854. [Google Scholar] [CrossRef] [PubMed]
- Jha, R.K.; Zi-rong, X. Biomedical compounds from marine organisms. Mar. Drugs 2004, 2, 123–146. [Google Scholar] [CrossRef]
- Munro, M.H.G.; Blunt, J.W.; Dumdei, E.J.; Hickford, S.J.H.; Lill, R.E.; Li, S.; Battershill, C.N.; Duckworth, A.R. The discovery and development of marine compounds with pharmaceutical potential. J. Biotechnol. 1999, 70, 15–25. [Google Scholar] [CrossRef]
- Sagar, S.; Kaur, M.; Minneman, K.P. Antiviral lead compounds from marine sponges. Mar. Drugs 2010, 8, 2619–2638. [Google Scholar] [CrossRef] [PubMed]
- Haefner, B. Drugs from the deep: Marine natural products as drug candidates. Drug Discov. Today 2003, 8, 536–544. [Google Scholar] [CrossRef]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Product Rep. 2014, 31, 160–258. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.-P.; Yuan, J.; Sun, L.; She, Z.-G.; Wu, J.-H.; Lan, X.-J.; Zhu, X.; Lin, Y.-C.; Chen, S.-P. Statistical research on marine natural products based on data obtained between 1985 and 2008. Mar. Drugs 2011, 9, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, W.; Feeney, R.J. Contributions to the study of marine products. XXXII. The nucleosides of sponges 1.1. J. Org. Chem. 1951, 16, 981–987. [Google Scholar] [CrossRef]
- Thakur, N.L.; Müller, W.E. Biotechnological potential of marine sponges. Curr. Sci. 2004, 86, 1506–1512. [Google Scholar]
- Mayer, A.; Glaser, K.B.; Cuevas, C.; Jacobs, R.S.; Kem, W.; Little, R.D.; McIntosh, J.M.; Newman, D.J.; Potts, B.C.; Shuster, D.E. The odyssey of marine pharmaceuticals: A current pipeline perspective. Trends Pharmacol. Sci. 2010, 31, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Huyck, T.K.; Gradishar, W.; Manuguid, F.; Kirkpatrick, P. Eribulin mesylate. Nat. Rev. Drug Discov. 2011, 10, 173–174. [Google Scholar] [CrossRef] [PubMed]
- Berlinck, R.G.S.; Braekman, J.C.; Daloze, D.; Bruno, I.; Riccio, R.; Rogeau, D.; Amade, P. Crambines C1 and C2: Two further ichthyotoxic guanidine alkaloids from the sponge Crambe crambe. J. Nat. Products 1992, 55, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Berlinck, R.G.S.; Braekman, J.C.; Daloze, D.; Bruno, I.; Riccio, R.; Ferri, S.; Spampinato, S.; Speroni, E. Polycyclic guanidine alkaloids from the marine sponge Crambe crambe and Ca++ channel blocker activity of crambescidin 816. J. Nat. Products 1993, 56, 1007–1015. [Google Scholar] [CrossRef]
- Laville, R.M.; Thomas, O.P.; Berrué, F.; Marquez, D.; Vacelet, J.; Amade, P. Bioactive guanidine alkaloids from two Caribbean marine sponges. J. Nat. Products 2009, 72, 1589–1594. [Google Scholar] [CrossRef] [PubMed]
- Berlinck, R.G.S.; Braekman, J.C.; Daloze, D.; Hallenga, K.; Ottinger, R.; Bruno, I.; Riccio, R. Two new guanidine alkaloids from the Mediterranean sponge Crambe crambe. Tetrahedron Lett. 1990, 31, 6531–6534. [Google Scholar] [CrossRef]
- Bondu, S.; Genta-Jouve, G.; Leirόs, M.; Vale, C.; Guigonis, J.-M.; Botana, L.M.; Thomas, O.P. Additional bioactive guanidine alkaloids from the Mediterranean sponge Crambe crambe. RSC Adv. 2012, 2, 2828–2835. [Google Scholar] [CrossRef]
- Buscema, M.; van de Vyver, G. Cytotoxic rejection of xenografts between marine sponges. J. Exp. Zool. 1985, 235, 297–308. [Google Scholar] [CrossRef]
- Olszewski, A.; Sato, K.; Aron, Z.D.; Cohen, F.; Harris, A.; McDougall, B.R.; Robinson, W.E.; Overman, L.E.; Weiss, G.A. Guanidine alkaloid analogs as inhibitors of HIV-1 Nef interactions with p53, actin, and p56lck. Proc. Natl. Acad. Sci. USA 2004, 101, 14079–14084. [Google Scholar] [CrossRef] [PubMed]
- Lazaro, J.E.; Nitcheu, J.; Mahmoudi, N.; Ibana, J.A.; Mangalindan, G.C.; Black, G.P.; Howard-Jones, A.G.; Moore, C.G.; Thomas, D.A.; Mazier, D.; et al. Antimalarial activity of crambescidin 800 and synthetic analogues against liver and blood stage of Plasmodium sp. J. Antibiot. (Tokyo) 2006, 59, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Suna, H.; Aoki, S.; Setiawan, A.; Kobayashi, M. Crambescidin 800, a pentacyclic guanidine alkaloid, protects a mouse hippocampal cell line against glutamate-induced oxidative stress. J. Nat. Med. 2007, 61, 288–295. [Google Scholar] [CrossRef]
- Aoki, S.; Kong, D.; Matsui, K.; Kobayashi, M. Erythroid differentiation in K562 chronic myelogenous cells induced by crambescidin 800, a pentacyclic guanidine alkaloid. Anticancer Res. 2004, 24, 2325–2330. [Google Scholar] [PubMed]
- Rubiolo, J.A.; Ternon, E.; López-Alonso, H.; Thomas, O.P.; Vega, F.V.; Vieytes, M.R.; Botana, L.M. Crambescidin-816 acts as a fungicidal with more potency than crambescidin-800 and -830, inducing cell cycle arrest, increased cell size and apoptosis in Saccharomyces cerevisiae. Mar. Drugs 2013, 11, 4419–4434. [Google Scholar] [CrossRef] [PubMed]
- Rubiolo, J.; López-Alonso, H.; Roel, M.; Vieytes, M.; Thomas, O.; Ternon, E.; Vega, F.; Botana, L. Mechanism of cytotoxic action of crambescidin-816 on human liver-derived tumour cells. Br. J. Pharmacol. 2014, 171, 1655–1667. [Google Scholar] [CrossRef] [PubMed]
- Kagi, J.; Coombs, T.L.; Overnell, J.; Webb, M. Synthesis and function of metallothioneins. Nature 1981, 292, 495–496. [Google Scholar] [CrossRef] [PubMed]
- Nath, R.; Kambadur, R.; Gulati, S.; Paliwal, V.K.; Sharma, M. Molecular aspects, physiological function, and clinical significance of metallothioneins. Crit. Rev. Food Sci. Nutr. 1988, 27, 41–85. [Google Scholar] [CrossRef] [PubMed]
- Palmiter, R.D. The elusive function of metallothioneins. Proc. Natl. Acad. Sci. USA 1998, 95, 8428–8430. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Eddy, R.L.; Henry, W.M.; Haley, L.L.; Byers, M.G.; Shows, T.B. Human metallothionein genes are clustered on chromosome 16. Proc. Natl. Acad. Sci. USA 1984, 81, 5494–5498. [Google Scholar] [CrossRef] [PubMed]
- West, A.; Stallings, R.; Hildebrand, C.; Chiu, R.; Karin, M.; Richards, R. Human metallothionein genes: Structure of the functional locus at 16q13. Genomics 1990, 8, 513–518. [Google Scholar] [CrossRef]
- Palmiter, R.D.; Findley, S.D.; Whitmore, T.E.; Durnam, D.M. MT-III, a brain-specific member of the metallothionein gene family. Proc. Natl. Acad. Sci. USA 1992, 89, 6333–6337. [Google Scholar] [CrossRef] [PubMed]
- Quaife, C.J.; Findley, S.D.; Erickson, J.C.; Froelick, G.J.; Kelly, E.J.; Zambrowicz, B.P.; Palmiter, R.D. Induction of a new metallothionein isoform (MT-IV) occurs during differentiation of stratified squamous epithelia. Biochemistry 1994, 33, 7250–7259. [Google Scholar] [CrossRef] [PubMed]
- Haq, F.; Mahoney, M.; Koropatnick, J. Signaling events for metallothionein induction. Mutat. Res. 2003, 533, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Sadhu, C.; Gedamu, L. Regulation of human metallothionein (MT) genes. Differential expression of MTI-F, MTI-G, and MTII-A genes in the hepatoblastoma cell line (HepG2). J. Biol. Chem. 1988, 263, 2679–2684. [Google Scholar] [PubMed]
- Tao, X.; Zheng, J.M.; Xu, A.M.; Chen, X.F.; Zhang, S.H. Downregulated expression of metallothionein and its clinicopathological significance in hepatocellular carcinoma. Hepatol. Res. 2007, 37, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Sadhu, C.; Gedamu, L. Metal-specific posttranscriptional control of human metallothionein genes. Mol. Cell. Biol. 1989, 9, 5738–5741. [Google Scholar] [PubMed]
- Jahroudi, N.; Foster, R.; Price-Haughey, J.; Beitel, G.; Gedamu, L. Cell-type specific and differential regulation of the human metallothionein genes. Correlation with DNA methylation and chromatin structure. J. Biol. Chem. 1990, 265, 6506–6511. [Google Scholar] [PubMed]
- Bauman, J.; Liu, J.; Liu, Y.; Klaassen, C. Increase in metallothionein produced by chemicals that induce oxidative stress. Toxicol. Appl. Pharmacol. 1991, 110, 347–354. [Google Scholar] [CrossRef]
- Bauman, J.W.; Madhu, C.; McKim, J.M., Jr.; Liu, Y.; Klaassen, C.D. Induction of hepatic metallothionein by paraquat. Toxicol. Appl. Pharmacol. 1992, 117, 233–241. [Google Scholar] [CrossRef]
- Schwarz, M.A.; Lazo, J.S.; Yalowich, J.C.; Allen, W.P.; Whitmore, M.; Bergonia, H.A.; Tzeng, E.; Billiar, T.R.; Robbins, P.D.; Lancaster, J.R. Metallothionein protects against the cytotoxic and DNA-damaging effects of nitric oxide. Proc. Natl. Acad. Sci. USA 1995, 92, 4452–4456. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, M.; Nomura, T.; Sato, F.; Mimata, H. Metallothionein is up-regulated under hypoxia and promotes the survival of human prostate cancer cells. Oncol. Rep. 2007, 18, 1145–1153. [Google Scholar] [PubMed]
- Robbins, A.; Stout, C. X-ray structure of metallothionein. Methods Enzymol. 1991, 205, 485–502. [Google Scholar] [PubMed]
- Otvos, J.D.; Armitage, I.M. Structure of the metal clusters in rabbit liver metallothionein. Proc. Natl. Acad. Sci. USA 1980, 77, 7094–7098. [Google Scholar] [CrossRef] [PubMed]
- Maret, W.; Vallee, B.L. Thiolate ligands in metallothionein confer redox activity on zinc clusters. Proc. Natl. Acad. Sci. USA 1998, 95, 3478–3482. [Google Scholar] [CrossRef] [PubMed]
- Janssen, Y.; Van Houten, B.; Borm, P.; Mossman, B. Cell and tissue responses to oxidative damage. Lab. Investig. 1993, 69, 261–274. [Google Scholar] [PubMed]
- Cai, L.; Klein, J.B.; Kang, Y.J. Metallothionein inhibits peroxynitrite-induced DNA and lipoprotein damage. J. Biol. Chem. 2000, 275, 38957–38960. [Google Scholar] [CrossRef] [PubMed]
- Kumari, M.R.; Hiramatsu, M.; Ebadi, M. Free radical scavenging actions of metallothionein isoforms I and II. Free Radic. Res. 1998, 29, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Rusnak, J.M.; Hoyt, D.G.; Settineri, C.E.; Pitt, B.R.; Lazo, J.S. Enhanced apoptosis in metallothionein null cells. Mol. Pharmacol. 1997, 52, 195–201. [Google Scholar] [PubMed]
- Lazo, J.S.; Kondo, Y.; Dellapiazza, D.; Michalska, A.E.; Choo, K.A.; Pitt, B.R. Enhanced sensitivity to oxidative stress in cultured embryonic cells from transgenic mice deficient in metallothionein I and II genes. J. Biol. Chem. 1995, 270, 5506–5510. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Liu, J.; Liu, Y.; Klaassen, C.D. Hepatocytes from metallothionein-I and II knock-out mice are sensitive to cadmium- and tert-butylhydroperoxide-induced cytotoxicity. Toxicol. Lett. 1996, 87, 139–145. [Google Scholar] [CrossRef]
- Lazo, J.S.; Kuo, S.-M.; Woo, E.S.; Pitt, B.R. The protein thiol metallothionein as an antioxidant and protectant against antineoplastic drugs. Chem. Biol. Interact. 1998, 111, 255–262. [Google Scholar] [CrossRef]
- Ebadi, M.; Leuschen, M.; El Refaey, H.; Hamada, F.; Rojas, P. The antioxidant properties of zinc and metallothionein. Neurochem. Int. 1996, 29, 159–166. [Google Scholar] [CrossRef]
- Oshima, Y.; Fujio, Y.; Nakanishi, T.; Itoh, N.; Yamamoto, Y.; Negoro, S.; Tanaka, K.; Kishimoto, T.; Kawase, I.; Azuma, J. STAT3 mediates cardioprotection against ischemia/reperfusion injury through metallothionein induction in the heart. Cardiovasc. Res. 2005, 65, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Koff, A.; Giordano, A.; Desai, D.; Yamashita, K.; Harper, J.W.; Elledge, S.; Nishimoto, T.; Morgan, D.O.; Franza, B.R.; Roberts, J.M. Formation and activation of a cyclin E-cdk2 complex during the G1 phase of the human cell cycle. Science 1992, 257, 1689–1694. [Google Scholar] [CrossRef] [PubMed]
- Bates, S.; Bonetta, L.; MacAllan, D.; Parry, D.; Holder, A.; Dickson, C.; Peters, G. CDK6 (PLSTIRE) and CDK4 (PSK-J3) are a distinct subset of the cyclin-dependent kinases that associate with cyclin D1. Oncogene 1994, 9, 71–79. [Google Scholar] [PubMed]
- Méplan, C.; Richard, M.-J.; Hainaut, P. Metalloregulation of the tumor suppressor protein p53: Zinc mediates the renaturation of p53 after exposure to metal chelators in vitro and in intact cells. Oncogene 2000, 19, 5227–5236. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Cherian, M. Potential role of p53 on metallothionein induction in human epithelial breast cancer cells. Br. J. Cancer 2002, 87, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Andrews, G.K. Regulation of metallothionein gene expression by oxidative stress and metal ions. Biochem. Pharmacol. 2000, 59, 95–104. [Google Scholar] [CrossRef]
- Nguyen, T.; Sherratt, P.J.; Pickett, C.B. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 233–260. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [PubMed]
- Viña, J.; Sáez, G.; Viña, J. The physiological functions of glutathione. In Handbook of Free Radicals and Antioxidants in Biomedicine; CRC Press: Boca Raton, FL, USA, 1989; Volume 2, pp. 121–132. [Google Scholar]
- Sies, H. Glutathione and its role in cellular functions. Free Radic. Biol. Med. 1999, 27, 916–921. [Google Scholar] [CrossRef]
- Haïdara, K.; Moffatt, P.; Denizeau, F. Metallothionein induction attenuates the effects of glutathione depletors in rat hepatocytes. Toxicol. Sci. 1999, 49, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.M.; George Cherian, M. Protective roles of metallothionein and glutathione in hepatotoxicity of cadmium. Toxicology 1992, 72, 281–290. [Google Scholar] [CrossRef]
- Samson, S.L.-A.; Gedamu, L. Molecular analyses of metallothionein gene regulation. Prog. Nucleic Acid Res. Mol. Biol. 1997, 59, 257–288. [Google Scholar]
- Zalups, R.K.; Koropatnick, J. Temporal changes in metallothionein gene transcription in rat kidney and liver: Relationship to content of mercury and metallothionein protein. J. Pharmacol. Exp. Ther. 2000, 295, 74–82. [Google Scholar] [PubMed]
- Nagel, W.W.; Vallee, B.L. Cell cycle regulation of metallothionein in human colonic cancer cells. Proc. Natl. Acad. Sci. USA 1995, 92, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, K.; Jacob, S.T. Regulation of metallothionein gene expression. Prog. Nucleic Acid Res. Mol. Biol. 2000, 66, 357–384. [Google Scholar]
- Cherian, M.; Apostolova, M. Nuclear localization of metallothionein during cell proliferation and differentiation. Cell. Mol. Biol. (Noisy-le-grand) 2000, 46, 347–356. [Google Scholar]
- Cherian, M.G. The significance of the nuclear and cytoplasmic localization of metallothionein in human liver and tumor cells. Environ. Health Perspect. 1994, 102, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Yukihisa, T.; Yasumitsu, O.; Kenji, I.; Kazuo, T.S. Role of metallothionein in the cell cycle: Protection against the retardation of cell proliferation by endogenous reactive oxygen species. J. Health Sci. 2004, 50, 154–158. [Google Scholar] [CrossRef]
- Woo, E.S.; Lazo, J.S. Nucleocytoplasmic functionality of metallothionein. Cancer Res. 1997, 57, 4236–4241. [Google Scholar] [PubMed]
- Ogra, Y.; Onishi, S.; Kajiwara, A.; Hara, A.; Suzuki, K.T. Enhancement of nuclear localization of metallothionein by nitric oxide. J. Health Sci. 2008, 54, 339–342. [Google Scholar] [CrossRef]
- Saeed, A.I.; Bhagabati, N.K.; Braisted, J.C.; Liang, W.; Sharov, V.; Howe, E.A.; Li, J.; Thiagarajan, M.; White, J.A.; Quackenbush, J. TM4 microarray software suite. Methods Enzymol. 2006, 411, 134–193. [Google Scholar] [PubMed]
- Saeed, A.; Sharov, V.; White, J.; Li, J.; Liang, W.; Bhagabati, N.; Braisted, J.; Klapa, M.; Currier, T.; Thiagarajan, M. TM4: A free, open-source system for microarray data management and analysis. Biotechniques 2003, 34, 374–378. [Google Scholar] [PubMed]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roel, M.; Rubiolo, J.A.; Ternon, E.; Thomas, O.P.; Vieytes, M.R.; Botana, L.M. Crambescin C1 Exerts a Cytoprotective Effect on HepG2 Cells through Metallothionein Induction. Mar. Drugs 2015, 13, 4633-4653. https://doi.org/10.3390/md13084633
Roel M, Rubiolo JA, Ternon E, Thomas OP, Vieytes MR, Botana LM. Crambescin C1 Exerts a Cytoprotective Effect on HepG2 Cells through Metallothionein Induction. Marine Drugs. 2015; 13(8):4633-4653. https://doi.org/10.3390/md13084633
Chicago/Turabian StyleRoel, María, Juan A. Rubiolo, Eva Ternon, Olivier P. Thomas, Mercedes R. Vieytes, and Luis M. Botana. 2015. "Crambescin C1 Exerts a Cytoprotective Effect on HepG2 Cells through Metallothionein Induction" Marine Drugs 13, no. 8: 4633-4653. https://doi.org/10.3390/md13084633
APA StyleRoel, M., Rubiolo, J. A., Ternon, E., Thomas, O. P., Vieytes, M. R., & Botana, L. M. (2015). Crambescin C1 Exerts a Cytoprotective Effect on HepG2 Cells through Metallothionein Induction. Marine Drugs, 13(8), 4633-4653. https://doi.org/10.3390/md13084633