
Discovery Strategies of Bioactive Compounds Synthesized by Nonribosomal Peptide Synthetases and Type-I Polyketide Synthases Derived from Marine Microbiomes



Abstract
:1. Introduction
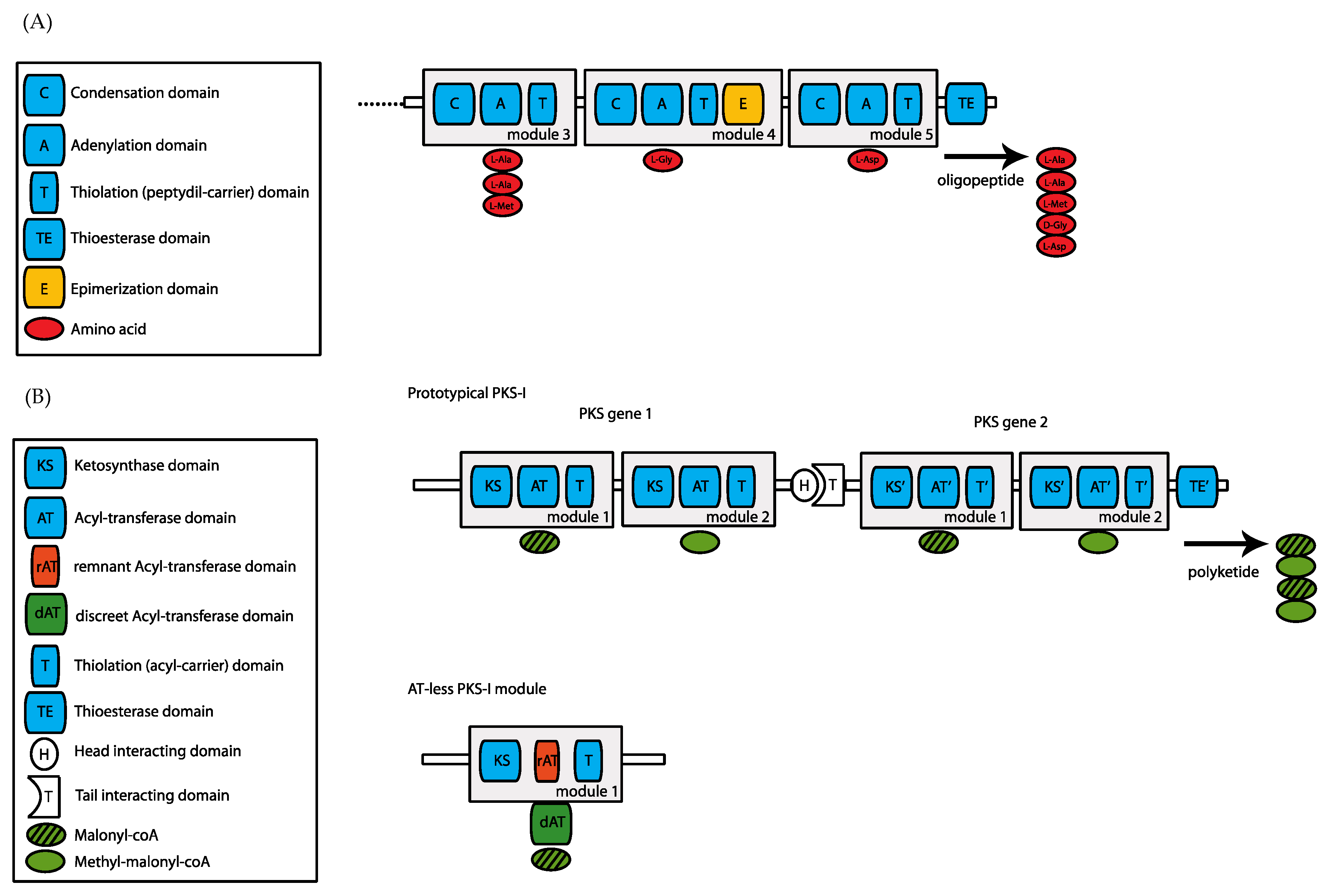
2. Evolution of Nonribosomal Peptide Synthetases (NRPSes) and Type-I Polyketide Synthases (PKSes)
3. Bioinformatics Tools for the Discovery of Nonribosomal Peptides (NRPs) and Polyketides (PKs)
4. Discovery of NRPSes and Type-I PKSes Derived from Marine Microbiomes through Genome Mining
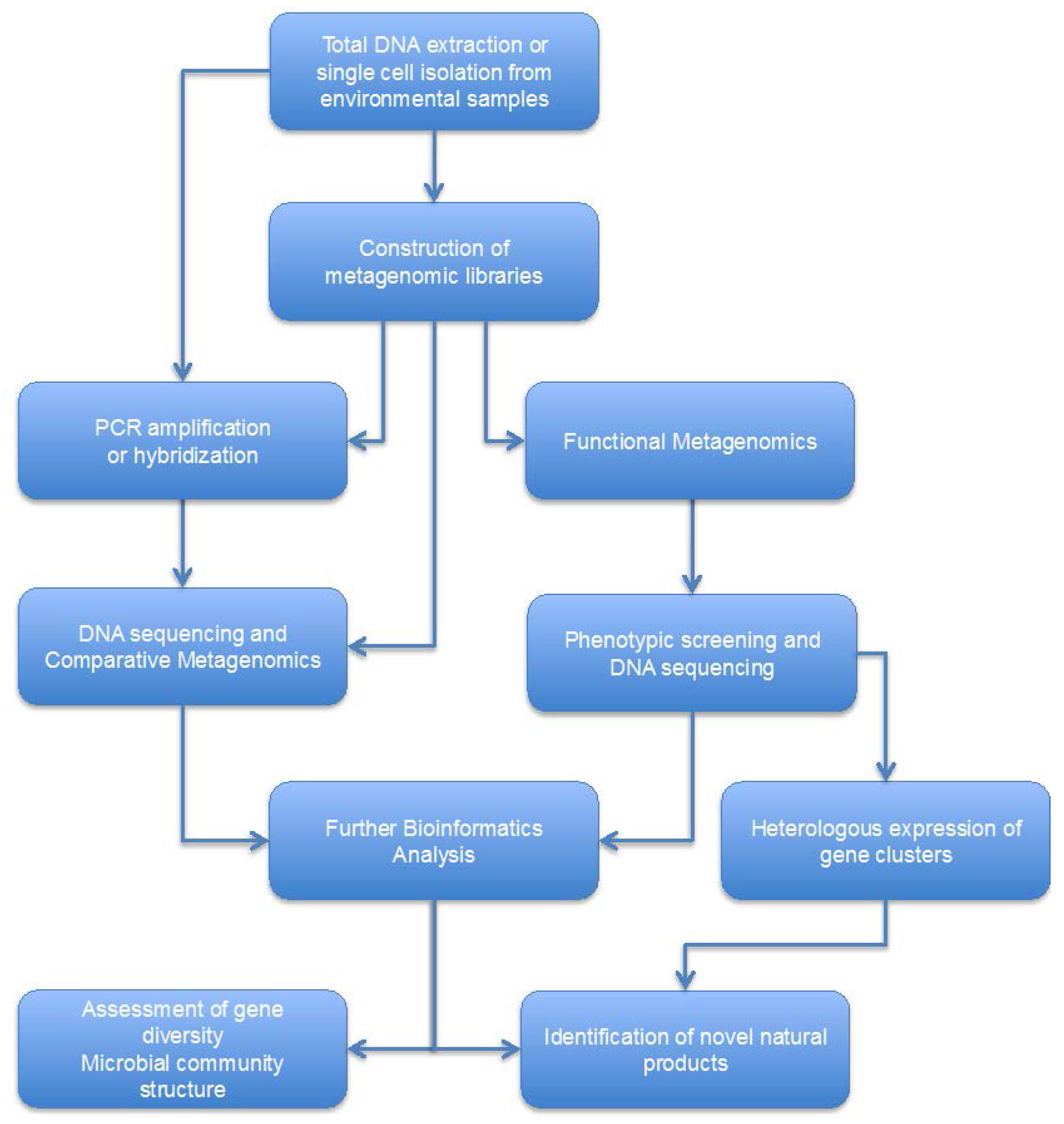
5. Discovery of NRPSes and Type-I PKSes Derived from Marine Microbiomes through Metagenomics
6. Concluding Remarks
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Reen, J.F.; Gutierrez-Barranquero, J.A.; Dobson, A.D.; Adams, C.; O’Gara, F. Emerging concepts promising new horizons for marine biodiscovery and synthetic biology. Mar. Drugs 2015, 13, 2924–2954. [Google Scholar] [CrossRef] [PubMed]
- Trindade, M.; van Zyl, L.J.; Navarro-Fernandez, J.; Abd Elfrazak, A. Targeted metagenomics as a tool to tap into marine natural product diversity for the discovery and production of drug candidates. Front. Microbiol. 2015, 6, 890. [Google Scholar] [CrossRef] [PubMed]
- Vartoukian, S.; Palmer, R.; Wade, W. Strategies for culture of “unculturable” bacteria. FEMS Microbiol. Lett. 2010, 309, 1–7. [Google Scholar] [PubMed]
- Nagarajan, M.; Rajesh Kumar, R.; Meenakshi Sundaram, K.; Sundaram, M. Marine biotechnology: Potentials of marine microbes and algae with reference to pharmacological and commercial values. In Plant Biology and Biotechnology (Plant. Genomics and Biotechnology); Bahadur, B., Venkat , M., Sahijram, L., Krishnamurthy, K.V., Eds.; Springer: New Delhi, (India), 2015; Volume II, pp. 685–723. [Google Scholar]
- Kleigrewe, K.; Gerwick, L.; Sherman, D.H.; Gerwick, W.H. Unique marine derived cyanobacterial biosynthetic genes for chemical diversity. Nat. Prod. Rep. 2016, 33, 348–364. [Google Scholar] [CrossRef] [PubMed]
- Desriac, F.; Jegou, C.; Balnois, E.; Brillet, B.; Le Chevalier, P.; Fleury, Y. Antimicrobial peptides from marine proteobacteria. Mar. Drugs 2013, 11, 3632–3660. [Google Scholar] [CrossRef] [PubMed]
- Nikolouli, K.; Mossialos, D. Bioactive compounds synthesized by non-ribosomal synthetases and type-I polyketide synthases discovered through genome-mining and metagenomics. Biotechnol. Lett. 2012, 34, 1393–1403. [Google Scholar] [CrossRef] [PubMed]
- Amoutzias, G.D.; van de Peer, Y.; Mossialos, D. Evolution and taxonomic distribution of nonribosomal peptide and polyketide synthases. Future Microbiol. 2008, 3, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Mossialos, D.; Amoutzias, D. Siderophores in fluorescent pseudomonads: New tricks from an old dog. Future Microbiol. 2007, 2, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Strieker, M.; Tanovic, A.; Marahiel, M.A. Nonribosomal peptide synthetases: Structures and dynamics. Curr. Opin. Struct. Biol. 2010, 20, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Von Döhren, H. Biochemistry and general genetics of nonribosomal peptide synthetases in fungi. Adv. Biochem. Eng. Biotechnol. 2004, 88, 217–264. [Google Scholar] [PubMed]
- Stachelhaus, T.; Walsh, C.T. Mutational analysis of the epimerization domain in the initiation module PheATE of gramicidin S synthetase. Biochemistry 2000, 39, 5775–5787. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Chen, M.; Sánchez, C.; Shen, B. An oxidation domain in the BlmIII non-ribosomal peptide synthetase probably catalyzing thiazole formation in the biosynthesis of the anti-tumor drug bleomycin in Streptomyces verticillus ATCC15003. FEMS Microbiol. Lett. 2000, 189, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Trauger, J.W.; Kohli, R.M.; Mootz, H.D.; Marahiel, M.A.; Walsh, C.T. Peptide cyclization catalysed by the thioesterase domain of tyrocidine synthetase. Nature 2000, 407, 215–218. [Google Scholar] [PubMed]
- Tanovic, A.; Samel, S.A.; Essen, L.O.; Marahiel, M.A. Crystal structure of the termination module of a nonribosomal peptide synthetase. Science 2008, 321, 659–663. [Google Scholar] [CrossRef] [PubMed]
- Kopp, F.; Marahiel, M.A. Macrocyclization strategies in polyketide and nonribosomal peptide biosynthesis. Nat. Prod. Rep. 2007, 24, 735–749. [Google Scholar] [CrossRef] [PubMed]
- Hertweck, C. Decoding and reprogramming complex polyketide assembly lines: Prospects for synthetic biology. Trends Biochem. Sci. 2015, 40, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Helfrich, E.J.; Piel, J. Biosynthesis of polyketides by trans-AT polyketide synthases. Nat. Prod. Rep. 2016, 33, 231–316. [Google Scholar] [CrossRef] [PubMed]
- Till, M.; Race, P.R. Progress challenges and opportunities for the re-engineering of trans-AT polyketide synthases. Biotechnol. Lett. 2014, 36, 877–888. [Google Scholar] [CrossRef] [PubMed]
- Donadio, S.; Monciardini, P.; Sosio, M. Polyketide synthases and nonribosomal peptide synthatases: The emerging view from bacterial genomics. Nat. Prod. Rep. 2007, 24, 1073–1109. [Google Scholar] [CrossRef] [PubMed]
- Mocibob, M.; Ivic, N.; Bilokapic, S.; Maier, T.; Luic, M.; Ban, N.; Weygand-Durasevic, I. Homologs of aminoacyl-tRNA synthetases acylate carrier proteins and provide a link between ribosomal and nonribosomal peptide synthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 14585–14590. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ntai, I.; Kelleher, N.L.; Walsh, C.T. tRNA-dependent peptide bond formation by the transferase PacB in biosynthesis of the pacidamycin group of pentapeptidyl nucleoside antibiotics. Proc. Natl. Acad. Sci. USA 2011, 108, 12249–12253. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Fewer, D.P.; Holm, L.; Rouhiainen, L.; Sivonen, K. Atlas of nonribosomal peptide and polyketide biosynthetic pathways reveals common occurrence of nonmodular enzymes. Proc. Natl. Acad. Sci. USA 2014, 111, 9259–9264. [Google Scholar] [CrossRef] [PubMed]
- Jenke-Kodama, H.; Dittmann, E. Evolution of metabolic diversity: Insights from microbial polyketide synthases. Phytochemistry 2009, 70, 1858–1866. [Google Scholar] [CrossRef] [PubMed]
- Van Belkum, M.J.; Martin-Visscher, L.A.; Vederas, J.C. Structure and genetics of circular bacteriocins. Trends Microbiol. 2011, 19, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Fischbach, M.; Walsh, C.T.; Clardy, J. The evolution of gene collectives: How natural selection drives chemical innovation. Proc. Natl. Acad. Sci. USA 2008, 105, 4601–4608. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S.C.; Kunze, B.; Höfle, G.; Silakowski, B.; Scharfe, M.; Blöcker, H.; Müller, R. Structure and biosynthesis of myxochromides S1–3 in Stigmatella aurantiaca: Evidence for an iterative bacterial type-I polyketide synthase and for module skipping in nonribosomal peptide synthesis. ChemBioChem 2005, 6, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Jenke-Kodama, H.; Sandmann, A.; Müller, R.; Dittmann, E. Evolutionary implications of bacterial polyketide synthases. Mol. Biol. Evol. 2005, 22, 2027–2039. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S.C.; Meiser, P.; Binz, T.M.; Mahmud, T.; Müller, R. Nonribosomal peptide biosynthesis: Point mutations and module skipping lead to chemical diversity. Angew. Chem. Int. Ed. Engl. 2006, 45, 2296–2301. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.L.; Cheng, Y.Q.; Shen, B. Polyketide chain skipping mechanism in the biosynthesis of the hybrid nonribosomal peptide-polyketide antitumor antibiotic leinamycin in Streptomyces atroolivaceus S-140. J. Nat. Prod. 2006, 69, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Slot, J.C.; Rokas, A. Horizontal transfer of a large and highly toxic secondary metabolic gene cluster between fungi. Curr. Biol. 2011, 21, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Piel, J.; Hui, D.; Fusetani, N.; Matsunaga, S. Targeting modular polyketide synthases with iteratively acting acyltransferases from metagenomes of uncultured bacterial consortia. Environ. Microbiol. 2004, 6, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Calteau, A.; Fewer, D.P.; Latifi, A.; Coursin, T.; Laurent, T.; Jokela, J.; Kerfeld, C.A.; Sivonen, K.; Piel, J.; Gugger, M. Phylum-wide comparative genomics unravel the diversity of secondary metabolism in Cyanobacteria. BMC Genom. 2014, 15, 977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziemert, N.; Lechner, A.; Wietz, M.; Millan-Aguinaga, N.; Chavarria, K.L.; Jensen, P.R. Diversity and evolution of secondary metabolism in the marine actinomycete genus Salinospora. Proc. Natl. Acad. Sci. USA 2014, 111, 1130–1139. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.R.; Chavarria, K.L.; Fenical, W.; Moore, B.S.; Ziemert, N. Challenges and triumphs to genomics-based natural product discovery. J. Ind. Microbiol. Biotechnol. 2014, 41, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Wong, F.T.; Khosla, C. Combinatorial biosynthesis of polyketides—A perspective. Curr. Opin. Chem. Biol. 2012, 16, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.; Fyans, J.K.; Zhuo, Y.; Micklefield, J. Recent advances in engineering nonribosomal peptide assembly lines. Nat. Prod. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Weissman, K.J. Genetic engineering of modular PKSs: From combinatorial biosynthesis to synthetic biology. Nat. Prod. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Calcott, M.J.; Ackerley, D.F. Genetic manipulation of non-ribosomal peptide synthetases to generate novel bioactive peptide products. Biotechnol. Lett. 2014, 36, 2407–2416. [Google Scholar] [CrossRef] [PubMed]
- Rui, Z.; Zhang, W. Engineering biosynthesis of non-ribosomal peptides and polyketides by directed evolution. Curr. Top. Med. Chem. 2015, in press. [Google Scholar] [CrossRef]
- Loman, N.J.; Pallen, M.J. Twenty years of bacterial genome sequencing. Nat. Rev. Microbiol. 2015, 13, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Didelot, X.; Bowden, R.; Wilson, D.J.; Peto, T.E.; Crook, D.W. Transforming clinical microbiology with bacterial genome sequencing. Nat. Rev. Genet. 2012, 13, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Niedringhaus, T.P.; Milanova, D.; Kerby, M.B.; Snyder, M.P.; Barron, A.E. Landscape of next-generation sequencing technologies. Anal. Chem. 2011, 83, 4327–4341. [Google Scholar] [CrossRef] [PubMed]
- Kodama, Y.; Shumway, M.; Leinonen, R. The Sequence Read Archive: Explosive growth of sequencing data. Nucleic Acid Res. 2012, 40, D54–D56. [Google Scholar] [CrossRef] [PubMed]
- Reddy, T.B.K.; Thomas, A.D.; Stamatis, D.; Bertsch, J.; Isbandi, M.; Jansson, J.; Mallajosyula, J.; Pagani, I.; Lobos, E.A.; Kyrpides, N.C. The genomes online database (GOLD) v.5: A metadata management system based on a four level (meta)genome project classification. Nucleic Acid Res. 2015, 43, D1099–D1106. [Google Scholar] [PubMed]
- Markowitz, V.M.; Chen, I.M.; Palaniapan, K.; Chu, K.; Szeto, E.; Pillay, M.; Ratner, A.; Huang, J.; Woyke, T.; Huntemann, M.; et al. IMG4 version of the integrated microbial genomes comparative analysis system. Nucleic Acid Res. 2014, 42, D560–D567. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, V.M.; Chen, I.M.; Chu, K.; Szeto, E.; Palaniapan, K.; Pillay, M.; Ratner, A.; Huang, J.; Pagani, I.; Tringe, S.; et al. IMG/M 4 version of the integrated metagenome comparative analysis system. Nucleic Acid Res. 2014, 42, D568–D573. [Google Scholar] [CrossRef] [PubMed]
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.M.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A.; et al. The metagenomics RAST server—A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 2008, 9, 386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, S.; Chen, J.; Li, W.; Altintas, I.; Lin, A.; Peltier, S.; Stocks, K.; Allen, E.E.; Ellisman, M.; Grether, J.; et al. Community cyberinfrastructure for advanced microbial ecology research and analysis: The CAMERA resource. Nucleic Acid Res. 2011, 39, D546–D551. [Google Scholar] [CrossRef] [PubMed]
- Boddy, C.N. Bioinformatics tools for genome mining of polyketide and non-ribosomal peptides. J. Ind. Microbiol. Biotechnol. 2013, 41, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acid Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Weber, T.; Blin, K.; Duddela, S.; Krug, D.; Kim, H.U.; Bruccoleri, R.; Lee, S.Y.; Fischbach, M.A.; Müller, R.; Wohlleben, W.; et al. AntiSMASH 3.0—A comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acid Res. 2015, 43, W237–W243. [Google Scholar] [CrossRef] [PubMed]
- Cimermancic, P.; Medema, M.H.; Claesen, J.; Kurita, K.; Wieland Brown, L.C.; Mavrommatis, K.; Pati, A.; Godfrey, P.A.; Koehrsen, M.; Clardy, J.; et al. Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic clusters. Cell 2014, 158, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Ziemert, N.; Podell, S.; Penn, K.; Badger, J.H.; Allen, E.; Jensen, P.R. The natural product domain seeker NaPDoS: A phylogeny-based bioinformatics tool to classify secondary metabolite gene diversity. PLoS ONE 2012, 7, e34064. [Google Scholar] [CrossRef] [PubMed]
- Li, M.H.T.; Ung, P.M.U.; Zajkowski, J.; Garneau-Tsodikova, S.; Sherman, D.H. Automated genome mining for natural products. BMC Bioinform. 2009, 10, 185. [Google Scholar] [CrossRef] [PubMed]
- Rottig, M.; Medema, M.H.; Blin, K.; Weber, T.; Rausch, C.; Kohlbacher, O. NRPSpredictor2—A web server for predicting NRPS adenylation domain specificity. Nucleic Acid Res. 2011, 39, W362–W367. [Google Scholar] [CrossRef] [PubMed]
- Anand, S.; Prasad, M.V.; Yadav, G.; Kumar, N.; Shehara, J.; Ansari, M.Z.; Mohanty, D. SBSPKS: Structure based sequence analysis of polyketide synthases. Nucleic Acids Res. 2010, 38, W487–W496. [Google Scholar] [CrossRef] [PubMed]
- Khayatt, B.I.; Overmars, L.; Siezen, R.J.; Francke, C. Classification of the adenylation and acyl-transferase activity of NRPS and PKS systems using ensembles of substrate specific hidden Masrkov models. PLoS ONE 2013, 8, e62136. [Google Scholar]
- Flissi, A.; Dufresner, Y.; Michalik, J.; Tonon, L.; Janot, S.; Noe, L.; Jacques, P.; Leclère, V.; Pupin, M. Norine, the knowledgebase dedicated ton non-ribosomal peptides, is now open to crowdsourcing. Nucleic Acids Res. 2016, 44, D1113–D1118. [Google Scholar] [CrossRef] [PubMed]
- Caboche, S.; Pupin, M.; Leclère, V.; Fontaine, A.; Jacques, P.; Kucherov, G. NORINE: A database of nonribosomal peptides. Nucleic Acids Res. 2008, 36, D326–D331. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.R.; Boddy, C.N. ClusterMine360: A database of microbial PKS/NRPS biosynthesis. Nucleic Acids Res. 2013, 41, D402–D407. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sivonen, K.; Fewer, D.P. Genomic insights into the distribution, genetic diversity and evolution of polyketide synthases and nonribosomal peptide synthetases. Curr. Opin. Genet. Dev. 2015, 35, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Aigle, B.; Lautru, S.; Spiteller, D.; Dickschat, J.S.; Challis, G.L.; Leblond, P.; Pernodet, J.L. Genome mining of Streptomyces ambofaciens. J. Ind. Microbiol. Biotechnol. 2014, 41, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, B.O.; van Lanen, S.G.; Baltz, R.H. Microbial genome mining for accelerated natural products discovery: Is a renaissance in the making? J. Ind. Microbiol. Biotechnol. 2014, 41, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Zerikly, M.; Challis, G.L. Strategies for the discovery of new natural products by genome mining. Chembiochem 2009, 10, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Challis, G.L. Genome mining for novel natural product discovery. J. Med. Chem. 2008, 51, 2618–2628. [Google Scholar] [CrossRef] [PubMed]
- Abdelmohsen, U.R.; Bayer, K.; Hentschel, U. Diversity, abundance and natural products of marinse-sponge-associated actinomycetes. Nat. Prod. Rep. 2014, 31, 381–399. [Google Scholar] [CrossRef] [PubMed]
- Hentschel, U.; Piel, J.; Degnan, S.M.; Taylor, M.W. Genomic insights into marine sponge microbiome. Nat. Rev. Microbiol. 2012, 10, 641–654. [Google Scholar] [CrossRef] [PubMed]
- Still, P.C.; Johnson, T.A.; Theodore, C.M.; Loveridge, S.T.; Crews, P. Scrutinizing the scaffolds of marine biosynthetics from different source organisms: Gram-negative cultured bacterial products enter center stage. J. Nat. Prod. 2014, 77, 690–702. [Google Scholar] [CrossRef] [PubMed]
- Mansson, M.; Gram, L.; Larsen, T.O. Production of bioasctive secondary metabolites by marine vibrionaceae. Mar. Drugs 2011, 9, 1440–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, H.; Sonnenschein, E.C.; Melchiorsen, J.; Gram, L. Genome mining reveals unlocked bioactive potential of marine Gram-negative bacteria. BMC Genom. 2015, 16, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.Y.; Sanchez, L.M.; Rath, C.M.; Liu, X.; Boudreau, P.D.; Bruns, N.; Glukhov, E.; Wodtke, A.; de Felicio, R.; Fenner, A.; et al. Molecular networking as a dereplication strategy. J. Nat. Prod. 2013, 76, 1686–1699. [Google Scholar] [CrossRef] [PubMed]
- Duncan, K.R.; Crüsemann, M.; Lechner, A.; Sarkar, A.; Li, J.; Ziemert, N.; Wang, M.; Bandeira, N.; Moore, B.S.; Dorrestein, P.C.; et al. Molecular networking and pattern-based genome mining improves discovery of biosynthetic gene clusters and their products from Salinispora species. Chem. Biol. 2015, 22, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Lombó, F.; Velasco, A.; Castro, A.; de la Calle, F.; Braña, A.F.; Sánchez-Puelles, J.M.; Méndez, C.; Salas, J.A. Deciphering the biosynthesis pathway of the antitumor thiocoraline from a marine actinomycete and its expression in two Streptomyces species. Chem. Biol. Chem. 2006, 7, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Schäberle, T.F.; Goralski, E.; Neu, E.; Erol, O.; Hölzl, G.; Dörmann, P.; Bierbaum, G.; König, G.M. Marine myxobacteria as a source of antibiotics-comparison of physiology, polyketide-type genes and antibiotic production of three new isolates of Enhygromyxa salina. Mar. Drugs 2010, 8, 2466–2479. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Tomura, T.; Sato, J.; Iizuka, T.; Fudou, R.; Ojika, M. Isolation and biosynthetic analysis of haliamide, a new PKS-NRPS hybrid metabolite from the marine myxobacterium Haliangium ochraceum. Molecules 2016, 21, 59. [Google Scholar] [CrossRef] [PubMed]
- Udwary, D.W.; Zeigler, L.; Asolkar, R.N.; Singan, V.; Lapidus, A.; Fenical, W.; Jensen, P.R.; Moore, B.S. Genome sequencing reveals complex secondary metabolome in the marine actinomycete Salinispora tropica. Proc. Natl. Acad. Sci. USA 2007, 104, 10376–10381. [Google Scholar] [CrossRef] [PubMed]
- Skellam, E.J.; Stewart, A.K.; Strangman, W.K.; Wright, J.L. Identification of micromonolactam, a new polyene macrocyclic lactam from two marine Micromonospora strains using chemical and molecular methods: Clarification of the biosynthetic pathway from a glutamate starter unit. J. Antibiot. 2013, 66, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Eustáquio, A.S.; Nam, S.J.; Penn, K.; Lechner, A.; Wilson, M.C.; Fenical, W.; Jensen, P.R.; Moore, B.S. The discovery of salinosporamide K from the marine bacterium “Salinispora pacifica” by genome mining gives insight into pathway evolution. ChemBioChem 2011, 12, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Reen, J.F.; Romano, S.; Dobson, A.D.; O’Gara, F. The sound of silence: Activating silent biosynthetic gene clusters in marine microorganisms. Mar. Drugs 2015, 13, 4754–4783. [Google Scholar] [CrossRef] [PubMed]
- Rutledge, P.J.; Challis, G.L. Discovery of microbial natural products by activation of silent biosynthetic genes clusters. Nat. Rev. Microbiol. 2015, 13, 509–523. [Google Scholar] [CrossRef] [PubMed]
- Romano, S.; Schultz-Vogt, H.N.; Gonzalez, J.M.; Bondarev, V. Phosphate limitation induces drastic physiological changes, virulence-related gene expression and secondary metabolite production in Pseudovibrio sp. Strain FO-BEG1. Appl. Environ. Microbiol. 2015, 81, 3518–3528. [Google Scholar] [CrossRef] [PubMed]
- Shang, Z.; Li, X.M.; Li, C.S.; Wang, B.G. Diverse secondary metabolites produced by marine-derived fungus Nigrospora sp. MA75 on various culture media. Chem. Biodivers. 2012, 9, 1338–1348. [Google Scholar] [CrossRef] [PubMed]
- Zan, J.; Liu, Y.; Fuqua, C.; Hill, R.T. Acyl-homoserine lactone quorum sensing in the Roseobacter clade. Int. J. Mol. Sci. 2014, 15, 654–669. [Google Scholar] [CrossRef] [PubMed]
- Wietz, M.; Duncan, K.; Patin, N.V.; Jensen, P.R. Antagonistic interactions mediated by marine bacteria: The role of small molecules. J. Chem. Ecol. 2013, 39, 879–891. [Google Scholar] [CrossRef] [PubMed]
- Della Sala, G.; Hochmuth, T.; Costantino, V.; Teta, R.; Gerwick, W.; Gerwick, L.; Piel, J.; Mangoni, A. Polyketide genes in the marine sponge Plakortis simplex: A new group of mono-modular type I polyketide synthases from sponge symbionts. Environ. Microbiol. 2013, 5, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Russo, P.; Kisialiou, A.; Lamonaca, P.; Moroni, R.; Prinzi, G.; Fini, M. New drugs from marine organisms in Alzheimer’s Disease. Mar. Drugs 2015, 14, 5. [Google Scholar] [CrossRef] [PubMed]
- Della Sala, G.; Hochmuth, T.; Teta, R.; Costantino, V.; Mangoni, A. Polyketide synthases from the marine sponge Plakortis halichondrioides: A metagenomic update. Mar. Drugs 2014, 12, 5425–5440. [Google Scholar] [CrossRef] [PubMed]
- Schofield, M.M.; Jain, S.; Porat, D.; Dick, G.J.; Sherman, D.H. Identification and analysis of the bacterial endosymbiont specialized for production of the chemotherapeutic natural product ET-743. Environ. Microbiol. 2015, 17, 3964–3975. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.K.; Allen, S.W.; Lim, G.E.; Anderson, C.M.; Haygood, M.G. Evidence for the biosynthesis of bryostatins by the bacterial symbiont “Candidatus Endobugula sertula” of the bryozoan Bugula neritina. Appl. Environ. Microbiol. 2001, 67, 4531–4537. [Google Scholar] [CrossRef] [PubMed]
- Vartoukian, S.R.; Adamowska, A.; Lawlor, M.; Moazzez, R.; Dewhirst, F.E.; Wade, W.G. In vitro cultivation of “unculturable” oral bacteria facilitated by community culture and media supplementation with siderophores. PLoS ONE 2016, 11, e0146926. [Google Scholar] [CrossRef] [PubMed]
- Stewart, E.J. Growing unculturable bacteria. J. Bacteriol. 2012, 194, 4151–4160. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.; Zengler, K. Tapping into microbial diversity. Nat. Rev. Microbiol. 2004, 2, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.; Epstein, S.; D’Onofrio, A.; Ling, L.L. Uncultured microorganisms as a source of secondary metabolites. J. Antibiot. 2010, 63, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Ling, L.L.; Schneider, T.; Peoples, A.J.; Spoering, A.L.; Engels, I.; Conlon, B.P.; Mueller, A.; Schäberle, T.F.; Hughes, D.E.; Epstein, S.; et al. A new antibiotic kills pathogens without detectable resistance. Nature 2015, 517, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Nichols, D.; Cahoon, N.; Trakhtenberg, E.M.; Pham, L.; Mehta, A.; Belanger, A.; Kanigan, T.; Lewis, K.; Epstein, S.S. Use of ichip for high-throughput in situ cultivation of “uncultivable” microbial species. Appl. Environ. Microbiol. 2010, 76, 2445–2450. [Google Scholar] [CrossRef] [PubMed]
- Aoi, Y.; Kinoshita, T.; Hata, T.; Ohta, H.; Obokata, H.; Tsuneda, S. Hollow-fiber membrane chamber as a device for in situ environmental cultivation. Appl. Environ. Microbiol. 2009, 75, 3826–3833. [Google Scholar] [CrossRef] [PubMed]
- Kaeberlein, T.; Lewis, K.; Epstein, S.S. Isolating “uncultivable” microorganisms in pure culture in a simulated natural environment. Science 2002, 296, 1127–1129. [Google Scholar] [CrossRef] [PubMed]
- Garza, D.R.; Dutilh, B.E. From cultured to uncultured genome sequences: Metagenomics and modeling microbial ecosystems. Cell. Mol. Life Sci. 2015, 72, 4287–4308. [Google Scholar] [CrossRef] [PubMed]
- Escobar-Zepeda, A.; Vera-Ponce de León, A.; Sanchez-Flores, A. The road to metagenomics: From microbiology to DNA sequencing technologies and bioinformatics. Front. Genet. 2015, 6, 348. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, J.; Flemer, B.; Jackson, S.A.; Lejon, D.P.; Morrissey, J.P.; O’Gara, F.; Dobson, A.D. Marine metagenomics: New tools for the study and exploitation of marine microbial metabolism. Mar. Drugs 2010, 8, 608–628. [Google Scholar] [CrossRef] [PubMed]
- Simon, C.; Daniel, R. Achievements and new knowledge unraveled by metagenomic approaches. Appl. Microbiol. Biotechnol. 2009, 85, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Blainey, P.C. The future is now: Single-cell genomics of bacteria and archaea. FEMS Microbiol. Rev. 2013, 37, 407–427. [Google Scholar] [CrossRef] [PubMed]
- Ekkers, D.M.; Cretoiu, M.S.; Kielak, A.M.; Elsas, J.D. The great screen anomaly—A new frontier in product discovery through functional metagenomics. Appl. Microbiol. Biotechnol. 2012, 93, 1005–1020. [Google Scholar] [CrossRef] [PubMed]
- Craig, J.W.; Chang, F.Y.; Kim, J.H.; Obiajulu, S.C.; Brady, S.F. Expanding small-molecule functional metagenomics through parallel screening of broad-host-range cosmid environmental DNA libraries in diverse proteobacteria. Appl. Environ. Microbiol. 2010, 76, 1633–1641. [Google Scholar] [CrossRef] [PubMed]
- Chistoserdova, L. Recent progress and new challenges in metagenomics for biotechnology. Biotechnol. Lett. 2010, 32, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Schmeisser, C.; Steele, H.; Streit, W.R. Metagenomics, biotechnology with non-culturable microbes. Appl. Microbiol. Biotechnol. 2007, 75, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Bayer, K.; Scheuemayer, M.; Fieseler, L.; Hentschel, U. Genomic mining for novel FADH2–dependent halogenases in marine sponge-associated microbial consortia. Mar. Biotechnol. 2013, 15, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, S.; Tsai, P.; Bell, J.; Fromont, J.; Ilan, M.; Lindquist, N.; Perez, T.; Rodrigo, A.; Schupp, P.J.; Vacelet, J.; et al. Assessing the complex sponge microbiota: Core, variable and species-specific bacterial communities in marine sponges. ISME J. 2012, 6, 564–576. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Dong, J.D.; Yang, J.; Luo, X.M.; Zhang, S. Detection of polyketide synthase and nonribosomal peptide synthetase biosynthetic genes from antimicrobial coral-associated actinomycetes. Antonie Van Leeuwenhoek 2014, 106, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Woodhouse, J.N.; Fan, L.; Brown, M.V.; Thomas, T.; Neilan, B.A. Deep sequencing of non-ribosomal peptide synthetases and polyketide synthases from the microbiomes of Australian marine sponges. ISME J. 2013, 7, 1842–1851. [Google Scholar] [CrossRef] [PubMed]
- Pimentel-Elardo, S.M.; Grozdanov, L.; Proksch, S.; Hentschel, U. Diversity of nonribosomal peptide synthetase genes in the microbial metagenomes of marine sponges. Mar. Drugs 2012, 10, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, M.; Waggoner, L.E.; Liu, H.; Sudek, S.; Allen, S.; Anderson, C.M.; Sherman, D.H.; Haygood, M. bryA: An unusual modular polyketide synthase gene from the uncultivated bacterial symbiont of the marine bryozoan Bugula neritina. Chem. Biol. 2004, 11, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- Rath, C.M.; Janto, B.; Earl, J.; Ahmed, A.; Hu, F.Z.; Hiller, L.; Dahlgren, M.; Kreft, R.; Yu, F.; Wolff, J.J.; et al. Meta-omic characterization of the marine invertebrate microbial consortium that produces the chemotherapeutic natural product ET-743. ACS Chem. Biol. 2011, 6, 1244–1256. [Google Scholar] [CrossRef] [PubMed]
- Piel, J. A polyketide synthase-peptide synthetase gene cluster from an uncultured bacterial symbiont of Paederus beetles. Proc. Natl. Acad. Sci. USA 2002, 99, 14002–14007. [Google Scholar] [CrossRef] [PubMed]
- Piel, J.; Hui, D.; Wen, G.; Butzke, D.; Platzer, M.; Fusetani, N.; Matsunaga, S. Antitumor polyketide biosynthesis by an uncultivated bacterial symbiont of the marine sponge Theonella swinhoei. Proc. Natl. Acad. Sci. USA 2004, 101, 16222–16227. [Google Scholar] [CrossRef] [PubMed]
- Thornburg, C.C.; Cowley, E.S.; Sikorska, J.; Shaala, L.A.; Ishmael, J.E.; Youssef, D.T.; McPhail, K.L. Apratoxin H and apratoxin A sulfoxide from the Red Sea cyanobacterium Moorea producens. J. Nat. Prod. 2013, 76, 1781–1788. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.Y.; Liu, Y.; Luesch, H. Systematic chemical mutagenesis identifies a potent novel apratoxin A/E hybrid with improved in vivo antitumor activity. ACS Med. Chem. Lett. 2011, 2, 861–865. [Google Scholar] [CrossRef] [PubMed]
- Grindberg, R.V.; Ishoey, T.; Brinza, D.; Esquenazi, E.; Coates, R.C.; Liu, W.T.; Gerwick, L.; Dorrestein, P.C.; Pevzner, P.; Lasken, R.; et al. Single cell genome amplification accelerates identification of the apratoxin biosynthetic pathway from a complex microbial assemblage. PLoS ONE 2011, 6, e18565. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Species | C and KS Domains | C Domains | Total NRPS and PKS Proteins | NRPS (or NRPS/PKS Hybrid) Proteins |
|---|---|---|---|---|
| Acaryochloris marina MBIC11017 | 10 | 10 | 6 | 6 |
| Alteromonas macleodii AltDE1 | 16 | 16 | 10 | 10 |
| Alteromonas macleodii str. “Ionian Sea U4” | 16 | 16 | 10 | 10 |
| Alteromonas macleodii str. “Ionian Sea U7” | 16 | 16 | 10 | 10 |
| Alteromonas macleodii str. “Ionian Sea U8” | 16 | 16 | 10 | 10 |
| Alteromonas macleodii str. “Ionian Sea UM7” | 16 | 16 | 10 | 10 |
| Haliangium ochraceum DSM 14365 | 41 | 24 | 15 | 9 |
| Halomonas elongata DSM 2581 | 4 | 4 | 2 | 2 |
| Marinomonas mediterranea MMB-1 | 14 | 14 | 4 | 4 |
| Marinomonas posidonica IVIA-Po-181 | 3 | 3 | 1 | 1 |
| Marinomonas sp. MWYL1 | 18 | 18 | 8 | 8 |
| Mycobacterium marinum M | 73 | 64 | 24 | 21 |
| Nostoc azollae 0708 | 3 | 3 | 1 | 1 |
| Nostoc punctiforme PCC 73102 | 61 | 52 | 29 | 25 |
| Nostoc sp. PCC 7107 | 9 | 7 | 7 | 7 |
| Nostoc sp. PCC 7120 | 18 | 17 | 10 | 10 |
| Nostoc sp. PCC 7524 | 3 | 1 | 2 | 1 |
| Oscillatoria acuminata PCC 6304 | 14 | 14 | 10 | 10 |
| Oscillatoria nigro-viridis PCC 7112 | 16 | 16 | 12 | 12 |
| Phaeobacter gallaeciensis 2.10 | 1 | 1 | 1 | 1 |
| Phaeobacter gallaeciensis DSM 26640 | 1 | 1 | 1 | 1 |
| Phaeobacter inhibens DSM 17395 | 1 | 1 | 1 | 1 |
| Photobacterium profundum SS9 | 9 | 8 | 4 | 4 |
| Planctomyces brasiliensis DSM 5305 | 2 | 0 | 1 | 0 |
| Planctomyces limnophilus DSM 3776 | 2 | 0 | 1 | 0 |
| Prochlorococcus marinus str. MIT 9303 | 1 | 1 | 1 | 1 |
| Pseudovibrio sp. FO-BEG1 | 9 | 7 | 5 | 5 |
| Salinispora arenicola CNS-205 | 42 | 25 | 21 | 16 |
| Salinispora tropica CNB-440 | 28 | 16 | 19 | 15 |
| Synechococcus sp. PCC 7502 | 2 | 2 | 1 | 1 |
| Vibrio alginolyticus NBRC 15630 = ATCC 17749 | 3 | 3 | 1 | 1 |
| Vibrio anguillarum 775 | 3 | 3 | 2 | 2 |
| Vibrio campbellii ATCC BAA-1116 | 9 | 9 | 7 | 7 |
| Vibrio cholerae IEC224 | 5 | 5 | 2 | 2 |
| Vibrio cholerae LMA3984-4 | 5 | 5 | 2 | 2 |
| Vibrio cholerae M66-2 | 5 | 5 | 2 | 2 |
| Vibrio cholerae MJ-1236 | 5 | 5 | 2 | 2 |
| Vibrio cholerae O1 biovar El Tor str. N16961 | 5 | 5 | 2 | 2 |
| Vibrio cholerae O1 str. 2010EL-1786 | 5 | 5 | 2 | 2 |
| Vibrio cholerae O395 | 10 | 10 | 4 | 4 |
| Vibrio furnissii NCTC 11218 | 5 | 5 | 3 | 3 |
| Vibrio nigripulchritudo | 25 | 24 | 12 | 12 |
| Vibrio sp. EJY3 | 1 | 1 | 1 | 1 |
| Vibrio vulnificus CMCP6 | 4 | 4 | 3 | 3 |
| Vibrio vulnificus MO6-24/O | 4 | 4 | 3 | 3 |
| Vibrio vulnificus YJ016 | 4 | 4 | 3 | 3 |
| Total | 563 | 486 | 288 | 263 |
| Compound | Enzyme | Discovering Approach | Microbial Source | Mode of Action | Reference |
|---|---|---|---|---|---|
| Haliamide | PKS-NRPS | Genome mining | Haliangium ochraceum | Cytotoxic | [71] |
| Salinosporamide K | NRPS | Genome mining | Salinispora pacifica | Antitumor | [74] |
| Retimycin A | NRPS | Genome mining | Sallinispora arenicola | Antitumor | [68] |
| Salinilactam A | PKS | Genome mining | Salinispora tropica | Antibiotic | [72] |
| ET-743 | NRPS | Metagenomics | “Candidatus Endoecteinascidia frumentensis” | Antitumor | [86] |
| Pederin | PKS | Metagenomics | Paederus fuscipes metagenome | Antitumor | [87] |
| Bryostatin | PKS | Metagenomics | Candidatus Endobugula sertula | Antitumor | [88] |
| Apratoxin A | PKS-NRPS | Metagenomics | Lyngbya bouillonii | Antitumor | [89] |
| Onnamide | PKS-NRPS | Metagenomics | Theonella swinhoei metagenome | Antitumor | [90] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amoutzias, G.D.; Chaliotis, A.; Mossialos, D. Discovery Strategies of Bioactive Compounds Synthesized by Nonribosomal Peptide Synthetases and Type-I Polyketide Synthases Derived from Marine Microbiomes. Mar. Drugs 2016, 14, 80. https://doi.org/10.3390/md14040080
Amoutzias GD, Chaliotis A, Mossialos D. Discovery Strategies of Bioactive Compounds Synthesized by Nonribosomal Peptide Synthetases and Type-I Polyketide Synthases Derived from Marine Microbiomes. Marine Drugs. 2016; 14(4):80. https://doi.org/10.3390/md14040080
Chicago/Turabian StyleAmoutzias, Grigoris D., Anargyros Chaliotis, and Dimitris Mossialos. 2016. "Discovery Strategies of Bioactive Compounds Synthesized by Nonribosomal Peptide Synthetases and Type-I Polyketide Synthases Derived from Marine Microbiomes" Marine Drugs 14, no. 4: 80. https://doi.org/10.3390/md14040080
APA StyleAmoutzias, G. D., Chaliotis, A., & Mossialos, D. (2016). Discovery Strategies of Bioactive Compounds Synthesized by Nonribosomal Peptide Synthetases and Type-I Polyketide Synthases Derived from Marine Microbiomes. Marine Drugs, 14(4), 80. https://doi.org/10.3390/md14040080