Quantification of Representative Ciguatoxins in the Pacific Using Quantitative Nuclear Magnetic Resonance Spectroscopy

Abstract
:1. Introduction
2. Results
2.1. Determination of the Residual Proton Content of Pyridine-d5 and Purity Calculation
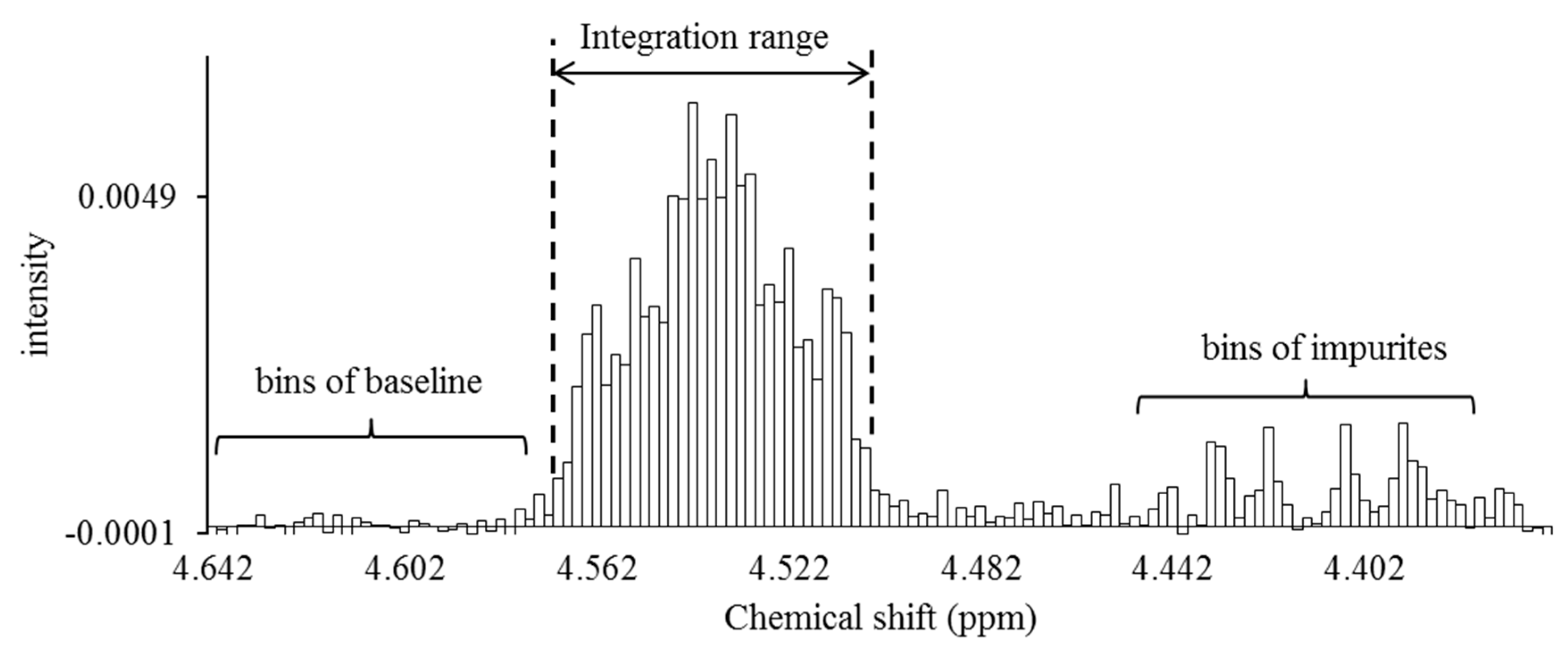
2.2. Quantification of Respective CTXs
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Apparatus
4.3. Determination of the Residual Proton Content in Pyridine-d5
4.4. Preparation of Test Solutions of CTX
4.5. 1H-NMR Measurements
4.6. Data Processing
4.7. Calculation of CTXs Content
5. Conclusions
Acknowledgments
Author Contributions
Conflict of interest
References
- Yasumoto, T. The chemistry and biological function of natural marine toxins. Chem. Rec. 2001, 1, 228–242. [Google Scholar] [CrossRef] [PubMed]
- Yasumoto, T. Chemistry, etiology, and food chain dynamics of marine toxins. Proc. Jpn. Acad. Ser. B 2005, 81, 43–51. [Google Scholar] [CrossRef]
- Yasumoto, T.; Inoue, A.; Bagnis, R.; Garcon, M. Ecological survey on a dinoflagellate possibly responsible for the induction of ciguatera. Bull. Jpn. Soc. Sci. Fish. 1979, 45, 395–399. [Google Scholar] [CrossRef]
- Yogi, K.; Oshiro, N.; Inafuku, Y.; Hirama, M.; Yasumoto, T. Detailed LC-MS/MS analysis of ciguatoxins revealing distinct regional and species characteristics in fish and causative alga from the Pacific. Anal. Chem. 2011, 83, 8886–8891. [Google Scholar] [CrossRef] [PubMed]
- Ikehara, T.; Kuniyoshi, K.; Oshiro, N.; Yasumoto, T. Biooxidation of ciguatoxins leads to species-specific toxin profiles. Toxins 2017, 9, 205. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Fish and Fishery Products Hazards and Control Guidance, 4th ed. Available online: http://www:fda.gov/food/guidanceregulation/guidancedocumentsregulatoryinformation/seafood/ucm2018426.htm (accessed on 15 January 2014).
- Saito, T.; Nakaie, S.; Kinoshita, M.; Ihara, T.; Kinugasa, S.; Nomura, A.; Maeda, T. Practical guide for accurate quantitative solution state NMR analysis. Metrologia 2004, 41, 213–218. [Google Scholar] [CrossRef]
- Saito, T.; Ihara, T.; Koike, M.; Kinugasa, S.; Fujimine, Y.; Nose, K.; Hirai, T. A new traceability scheme for the development of international system-traceable persistent organic pollutant reference materials by quantitative nuclear magnetic resonance. Accred. Qual. Assur. 2009, 14, 79–86. [Google Scholar] [CrossRef]
- Murata, M.; Legrand, A.M.; Ishibashi, Y.; Fukui, M.; Yasumoto, T. Structures and configurations of ciguatoxin from the moray eel Gymnothorax javanicus and its likely precursor from the dinoflagellate Gambierdiscus toxicus. J. Am. Chem. Soc. 1990, 112, 4380–4386. [Google Scholar] [CrossRef]
- Satake, M.; Murata, M.; Yasumoto, T. The structure of CTX3C, a ciguatoxin congener isolated from cultured Gambierdiscus toxicus. Tetrahedron Lett. 1993, 34, 1975–1978. [Google Scholar] [CrossRef]
- Kato, T.; Saito, M.; Nagae, M.; Fujita, K.; Watai, M.; Igarashi, T.; Yasumoto, T.; Inagaki, M. Absolute quantification of lipophilic shellfish toxins by quantitative nuclear magnetic resonance using removable internal reference substance with SI traceability. Anal. Sci. 2016, 32, 729–734. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Signals | Assign of Signals | S/N | Signal Intensity | Quantitative Value (mg) |
|---|---|---|---|---|---|
| CTX1B | A | Hydroxyl group 2 | 28 | - | 0.1374 ± 0.0012 |
| B | Olefin 3, 4 | 68 | 0.25 | 0.1381 ± 0.0003 | |
| deoxyCTX1B | C | Hydroxyl group 2 | 11 | - | 0.0590 ± 0.0017 |
| D | Olefin 3, 4 | 48 | 0.11 | 0.0586 ± 0.0003 | |
| CTX4A | E | Hydroxyl group 47 | 14 | - | 0.0832 ± 0.0007 |
| F | 1,3-diene 3 | 20 | 0.075 | 0.0794 ± 0.0018 | |
| G | 1,3-diene 2 | 20 | - | 0.0743 ± 0.0017 |
| Compounds | Signals | Assign of Signals | S/N | Signal Intensity | Quantitative Value (mg) |
|---|---|---|---|---|---|
| CTX3C | H | Hydroxyl group 44 | 81 | 0.40 | 0.4060 ± 0.0038 |
| I | Olefin 2, 3, 13, 14, 18, 19, 23, 24 | - | - | 0.4408 ± 0.0018 | |
| J | 17-H | 37 | - | 0.4243 ± 0.0067 | |
| 51OHCTX3C | K | 17-H | <10 | 0.013 | 0.0134 ± 0.0002 |
| L | 38, 42-H | 25 | - | 0.01089 | |
| M | 22, 25-H | 10 | - | 0.01230 | |
| N | 17-H | 10 | - | 0.01085 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kato, T.; Yasumoto, T. Quantification of Representative Ciguatoxins in the Pacific Using Quantitative Nuclear Magnetic Resonance Spectroscopy. Mar. Drugs 2017, 15, 309. https://doi.org/10.3390/md15100309
Kato T, Yasumoto T. Quantification of Representative Ciguatoxins in the Pacific Using Quantitative Nuclear Magnetic Resonance Spectroscopy. Marine Drugs. 2017; 15(10):309. https://doi.org/10.3390/md15100309
Chicago/Turabian StyleKato, Tsuyoshi, and Takeshi Yasumoto. 2017. "Quantification of Representative Ciguatoxins in the Pacific Using Quantitative Nuclear Magnetic Resonance Spectroscopy" Marine Drugs 15, no. 10: 309. https://doi.org/10.3390/md15100309
APA StyleKato, T., & Yasumoto, T. (2017). Quantification of Representative Ciguatoxins in the Pacific Using Quantitative Nuclear Magnetic Resonance Spectroscopy. Marine Drugs, 15(10), 309. https://doi.org/10.3390/md15100309