Recent Advances in the Isolation, Synthesis and Biological Activity of Marine Guanidine Alkaloids



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
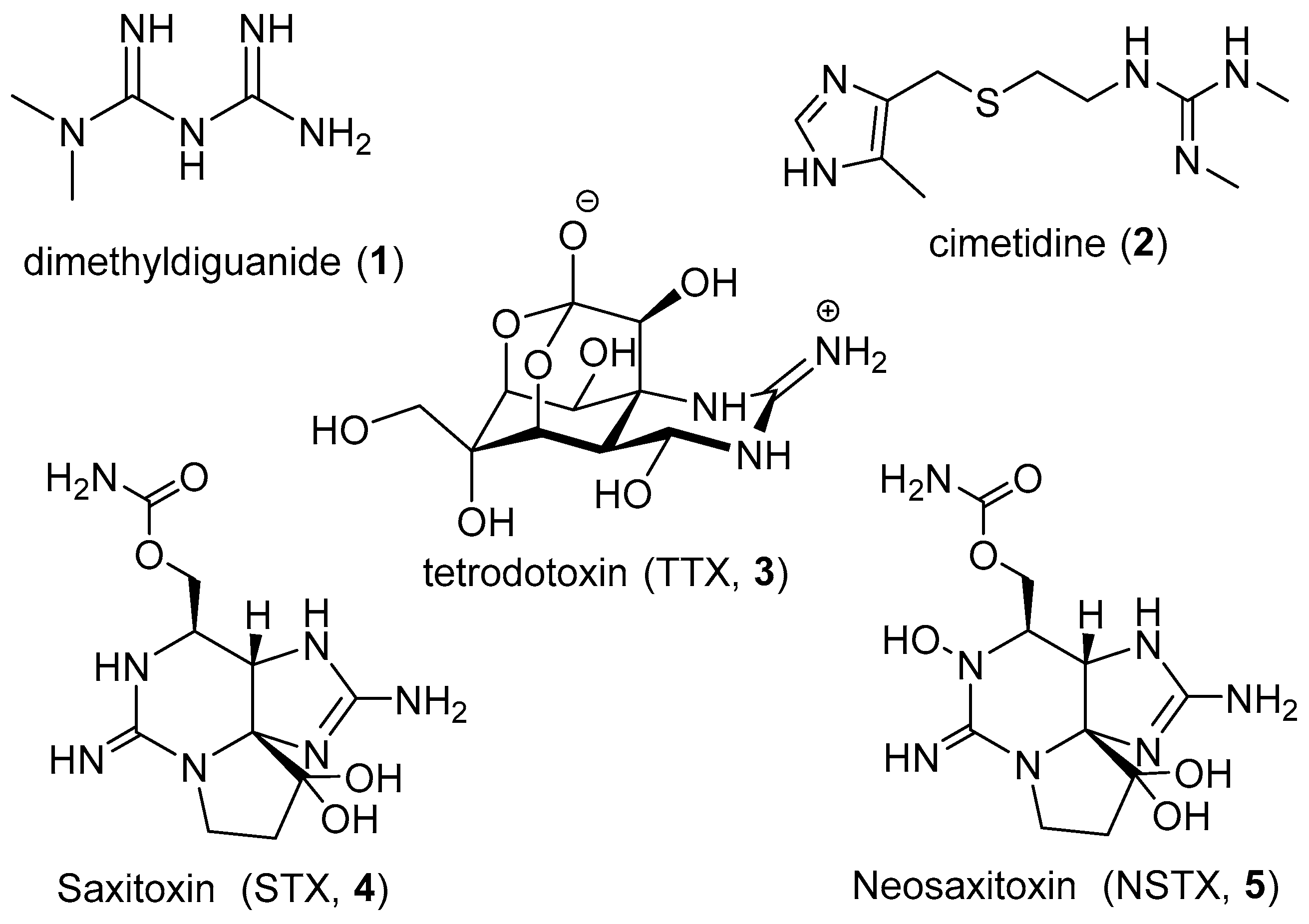
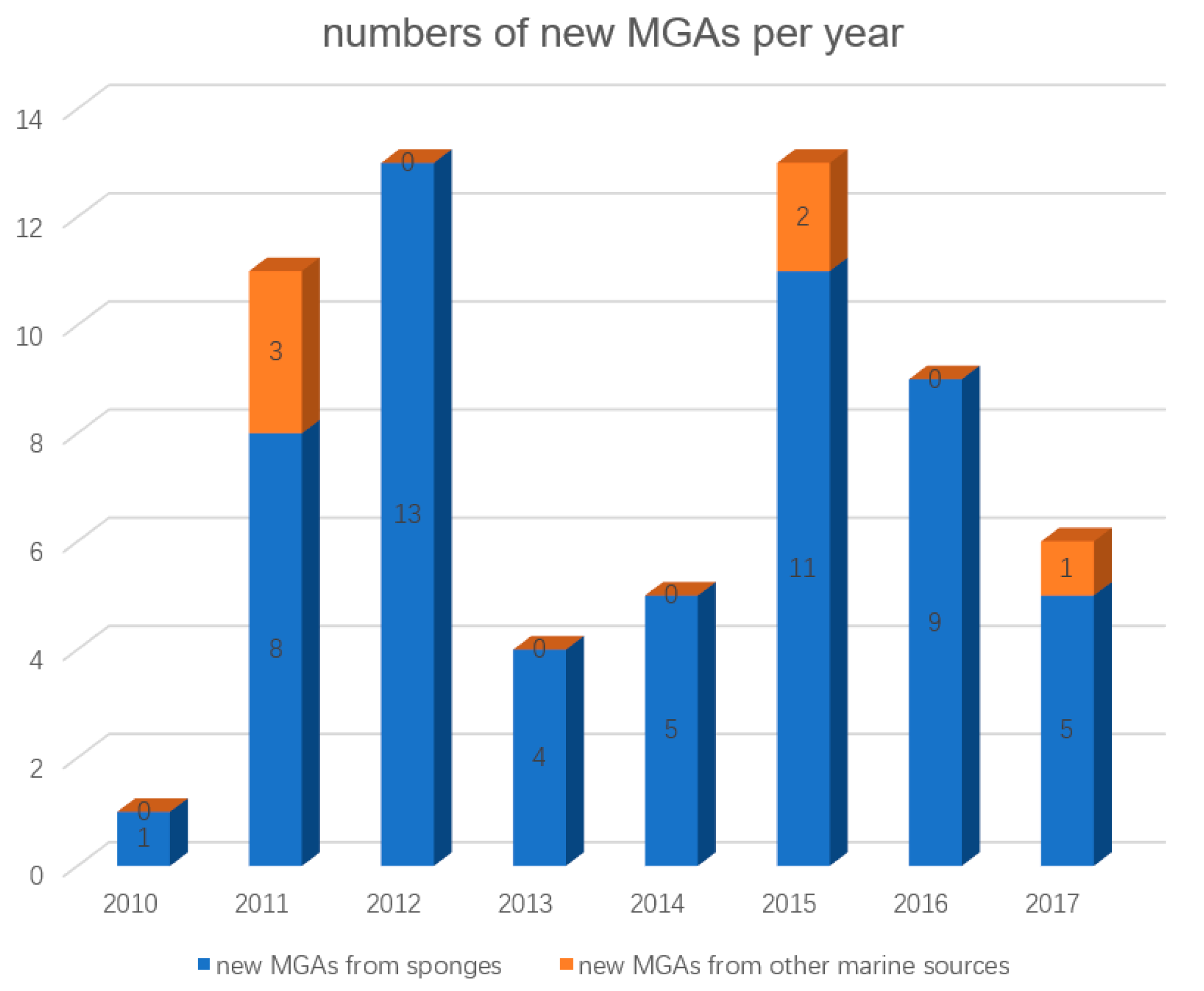
:1. Introduction
2. MGAs from Sponges
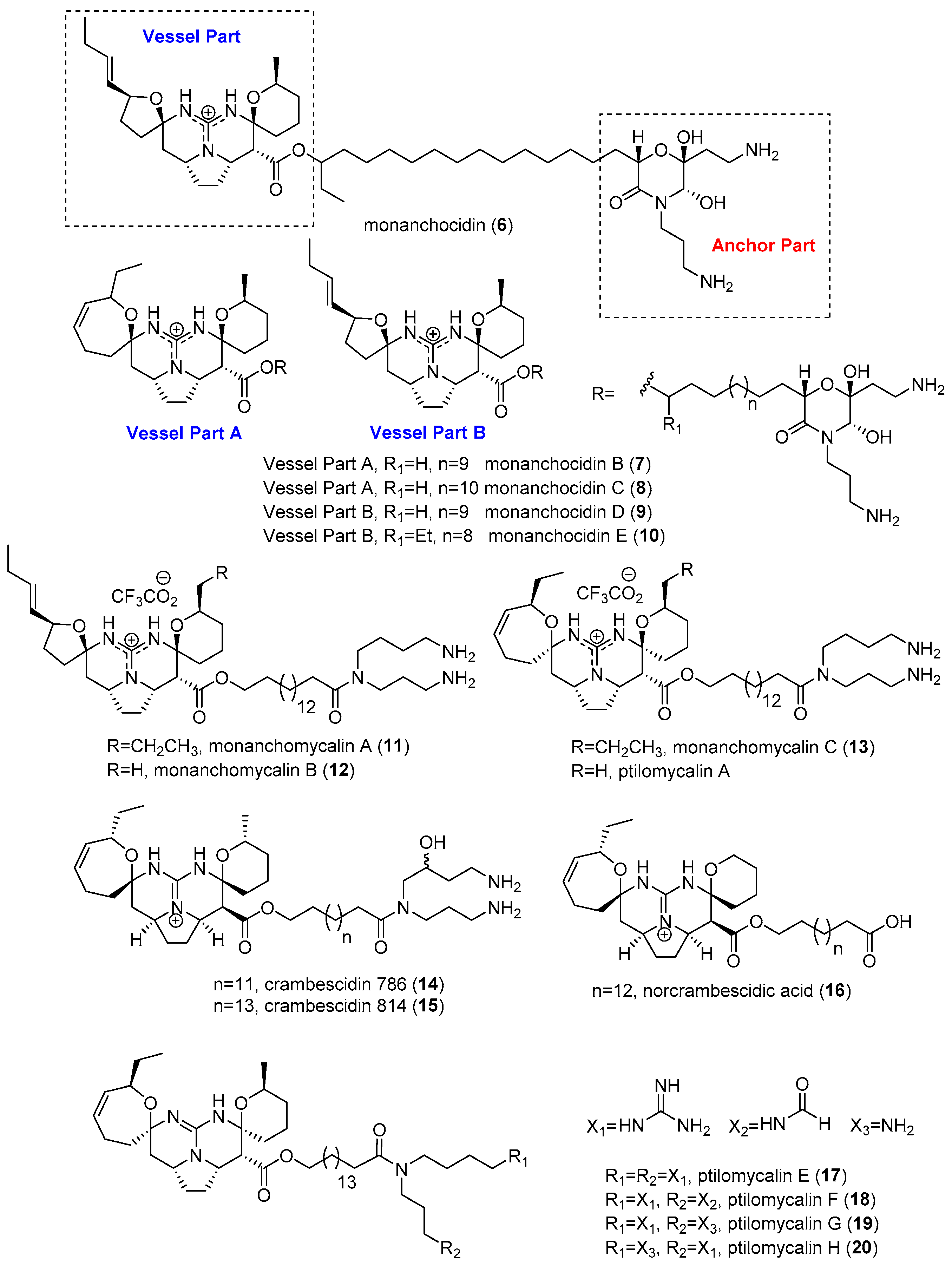
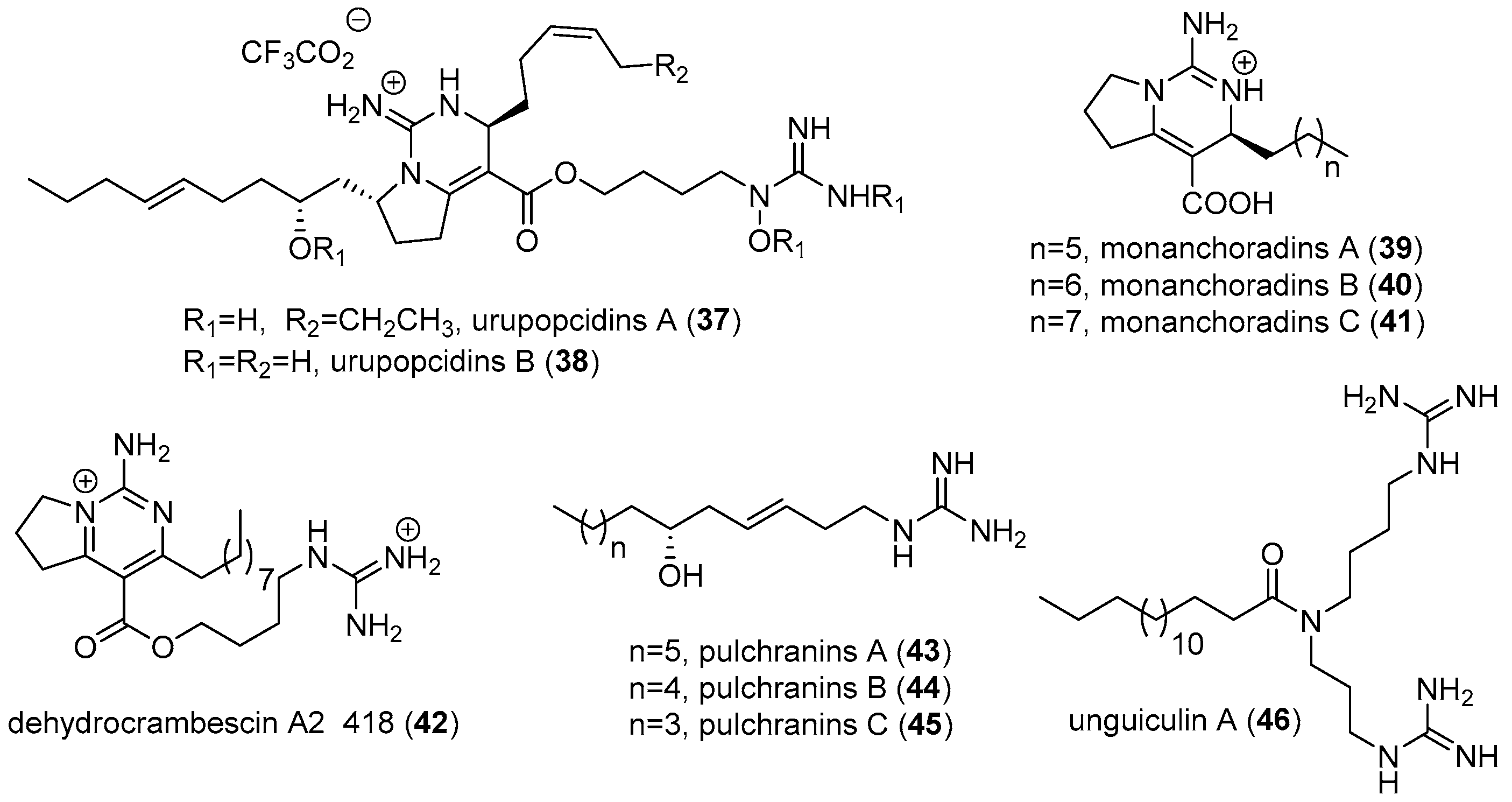
2.1. Monanchora
2.2. Crambe
2.3. Other Sponges
3. MGAs from Other Marine Sources
4. Synthetic Examples of Recently Discovered Bioactive MGAs
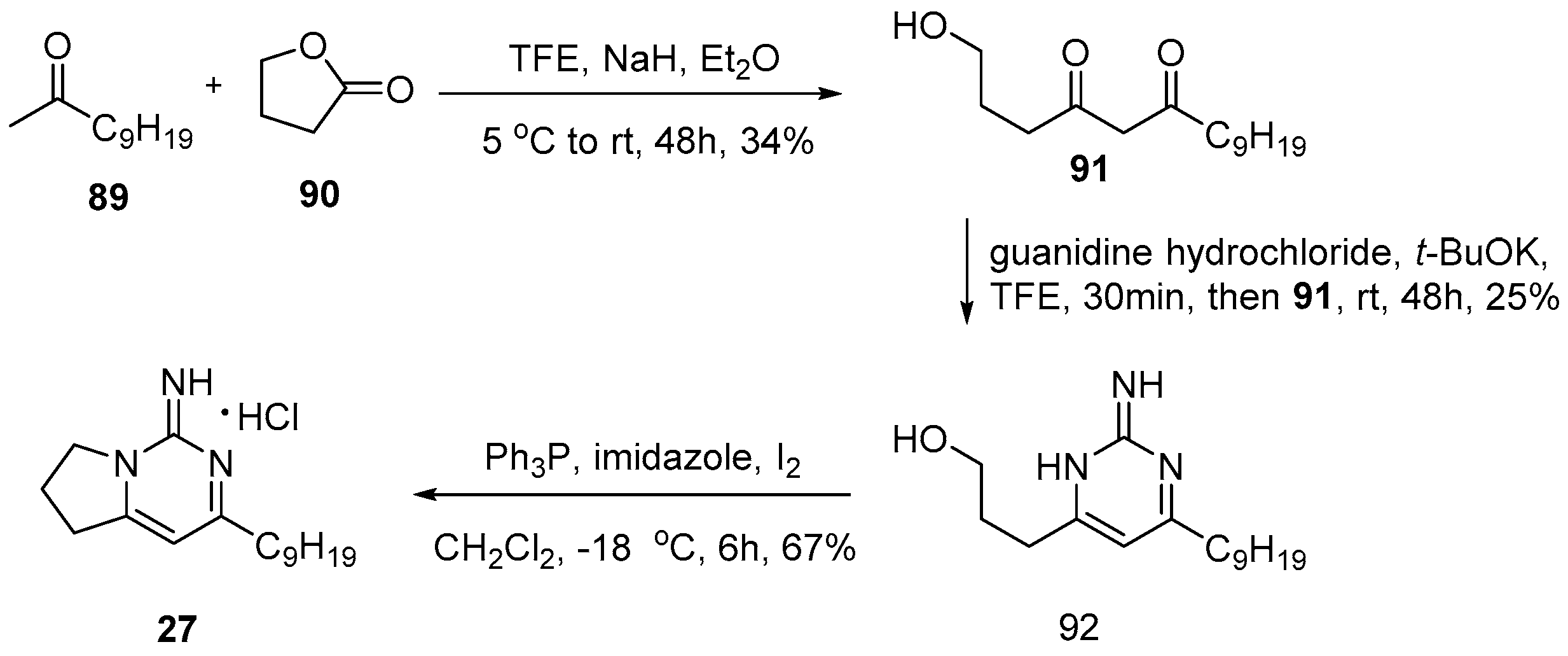
4.1. Monalidine A
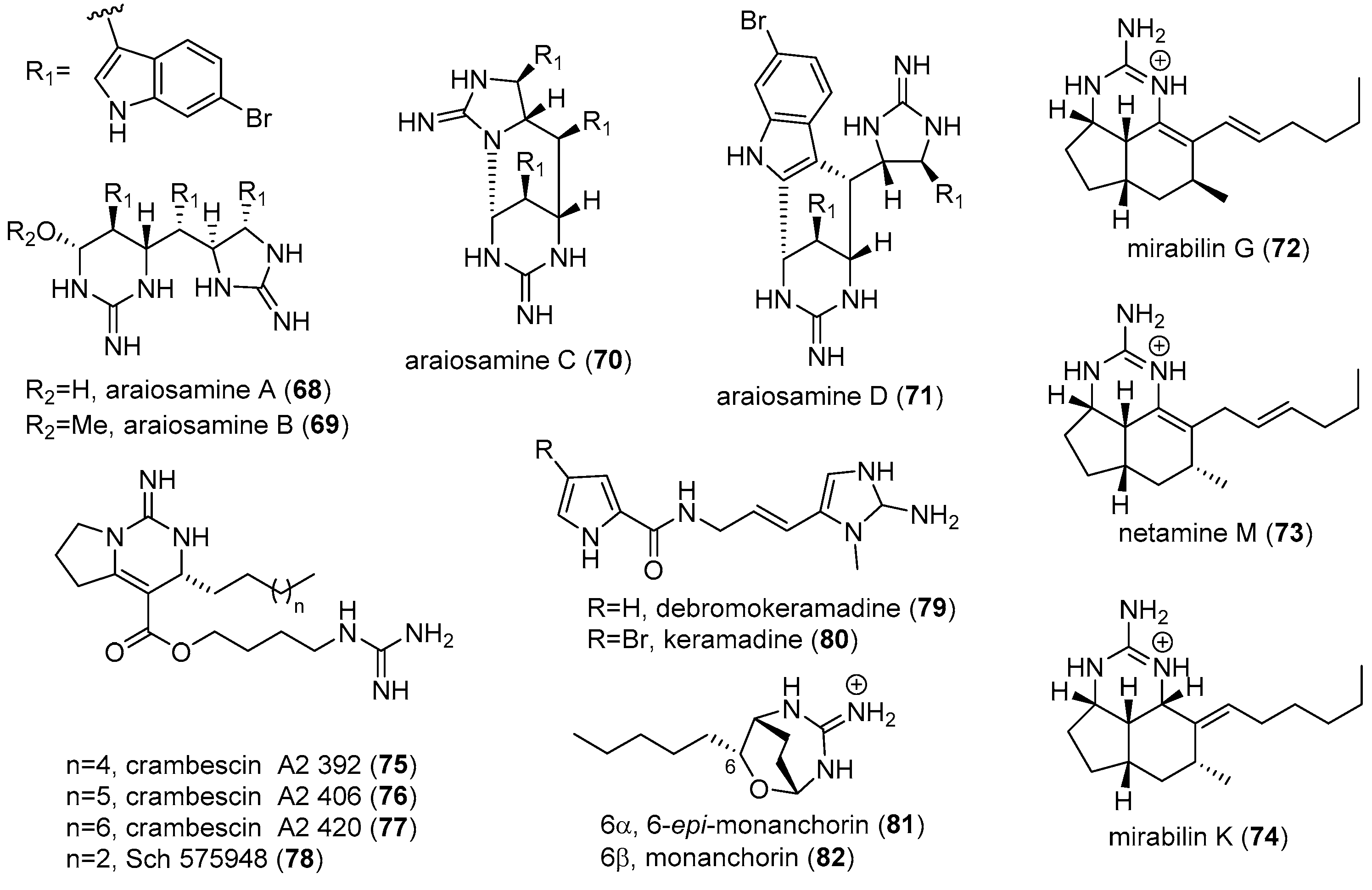
4.2. Araiosamines
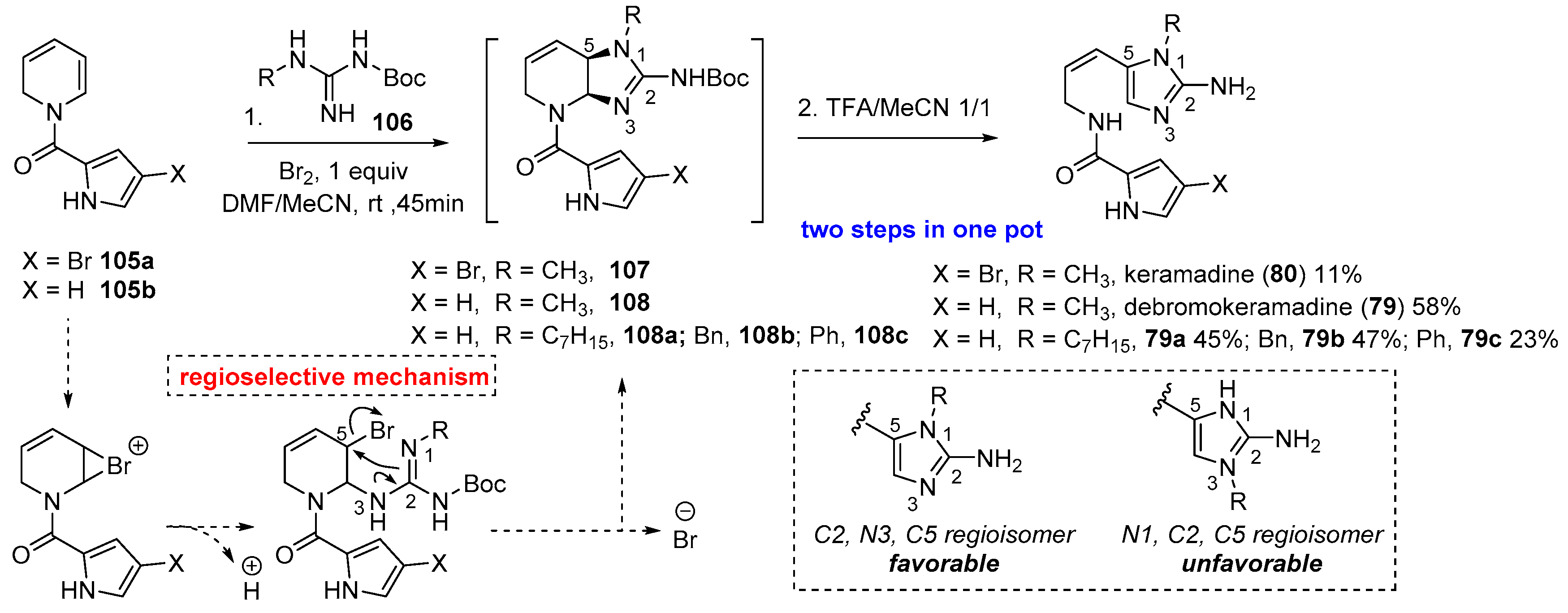
4.3. Keramadine and Debromokeramadine
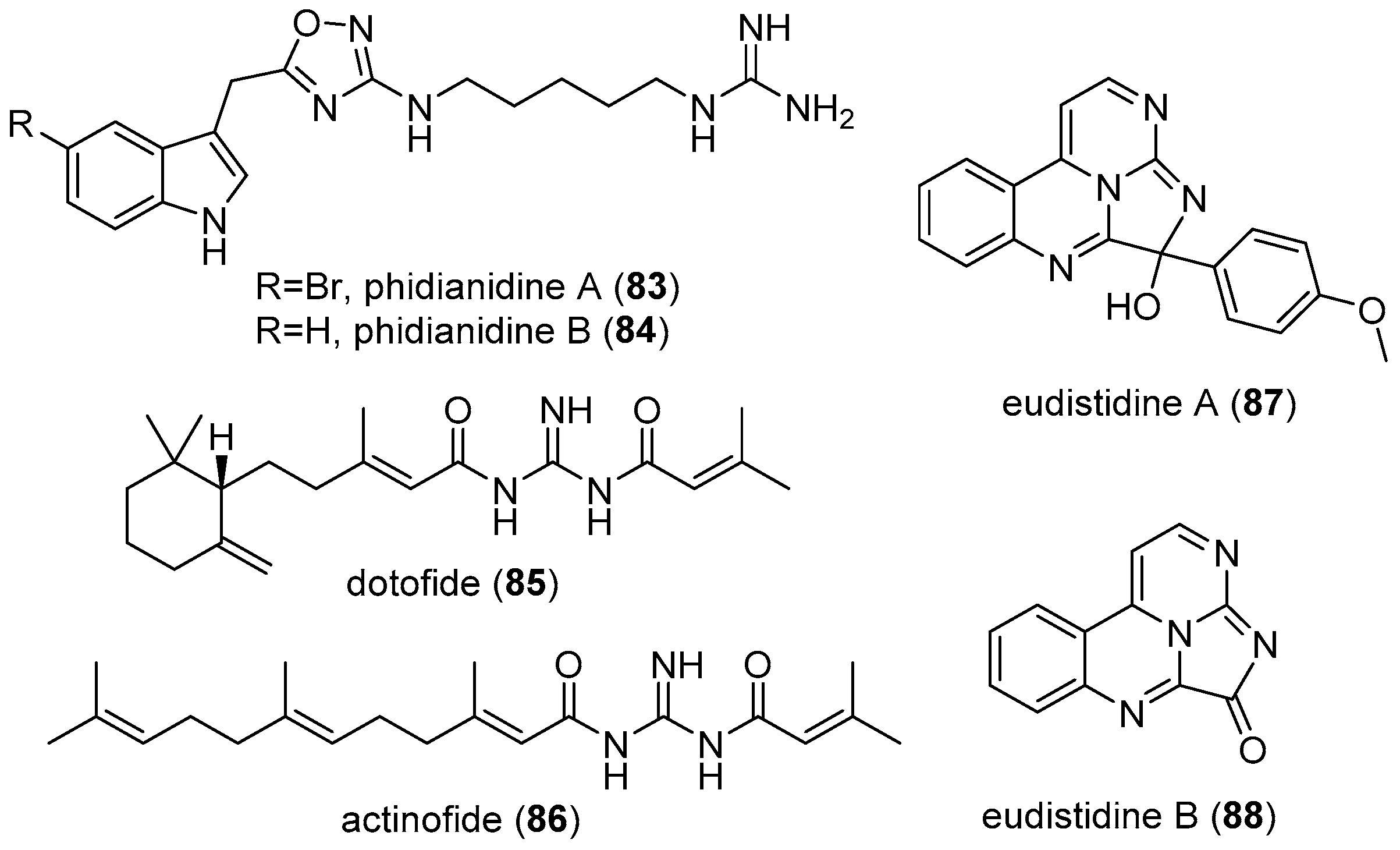
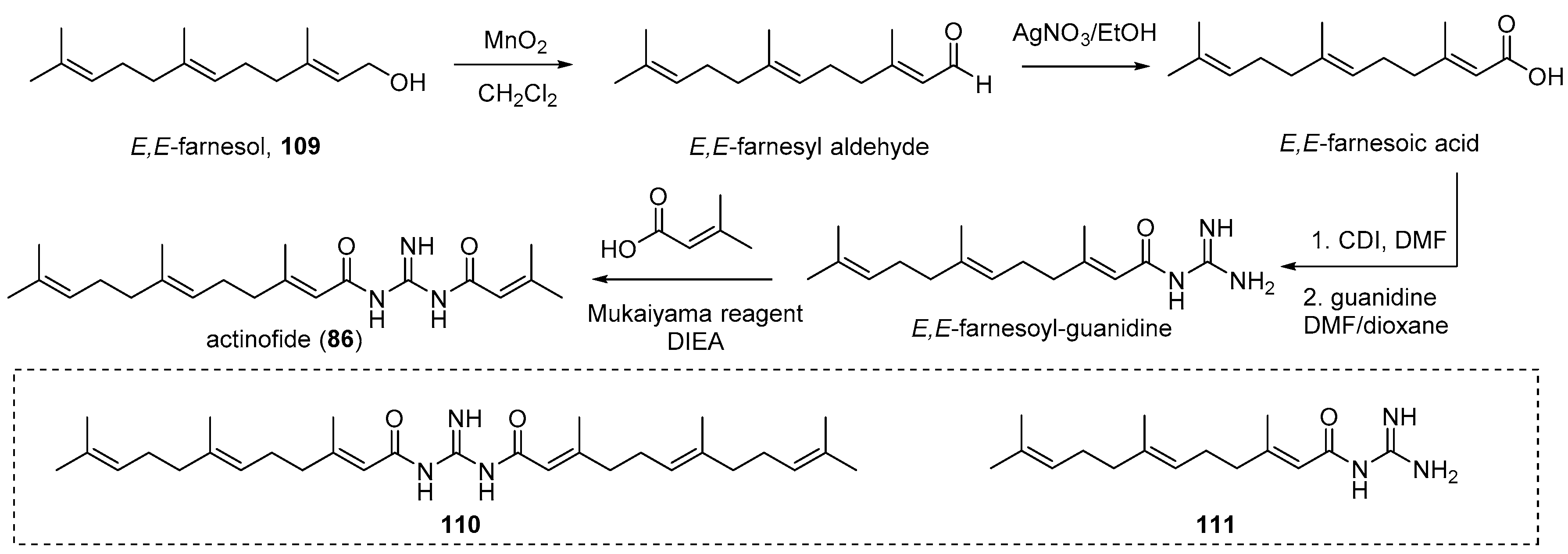
4.4. Actinofide
4.5. Phidianidines
5. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lindequist, U. Marine-derived pharmaceuticals—Challenges and opportunities. Biomol. Ther. 2016, 24, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Li, J.; Zhu, T.; Gu, Q.; Li, D. Advanced tools in marine natural drug discovery. Curr. Opin. Biotechnol. 2016, 42, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Gobbi, A.; Frenking, G. Y-conjugated compounds: The equilibrium geometries and electronic structures of guanidine, guanidinium cation, urea, and 1,1–diaminoethylene. J. Am. Chem. Soc. 1993, 115, 2362–2372. [Google Scholar] [CrossRef]
- Konrad, F.; Christoph, Z.; Heather, L.S.; Murray, G. Diprotected triflylguanidines: A new class of guanidinylation reagents. J. Org. Chem. 1998, 63, 3804–3805. [Google Scholar]
- Ma, Y.; De, S.; Chen, C. Syntheses of cyclic guanidine-containing natural products. Tetrahedron 2015, 71, 1145–1173. [Google Scholar] [CrossRef] [PubMed]
- Netz, N.; Opatz, T. Marine indole alkaloids. Mar. Drugs. 2015, 13, 4814–4914. [Google Scholar] [CrossRef] [PubMed]
- Berlinck, R.G.S. Natural guanidine derivatives. Nat. Prod. Rep. 2016, 12, 1347–1448. [Google Scholar]
- Bane, V.; Lehane, M.; Dikshit, M.; O’Riordan, A.; Furey, A. Tetrodotoxin: Chemistry, toxicity, source, distribution and detection. Toxins 2014, 6, 693–755. [Google Scholar] [CrossRef] [PubMed]
- Schuett, W.; Rapoport, H. Saxitoxin, the paralytic shellfish poison. Degradation to a pyrrolopyrimidine. J. Am. Chem. Soc. 1962, 84, 2266–2267. [Google Scholar] [CrossRef]
- Shimizu, Y.; Hsu, C.P.; Fallon, W.E.; Oshima, Y.; Miura, I.; Nakanishi, K. Structure of neosaxitoxin. J. Am. Chem. Soc. 1978, 100, 6791–6793. [Google Scholar] [CrossRef]
- Shi, Y.; Moazami, Y.; Pierce, J.G. Structure, synthesis and biological properties of the pentacyclic guanidinium alkaloids. Bioorg. Med. Chem. 2017, 25, 2817–2824. [Google Scholar] [CrossRef] [PubMed]
- Sfecci, E.; Lacour, T.; Amade, P.; Mehiri, M. Polycyclic guanidine alkaloids from Poecilosclerida marine sponges. Mar. Drugs 2016, 14, 77. [Google Scholar] [CrossRef] [PubMed]
- Guzii, A.G.; Makarieva, T.N.; Denisenko, V.A.; Dmitrenok, P.S.; Kuzmich, A.S.; Dyshlovoy, S.A.; Krasokhin, V.B.; Stonik, V.A. Monanchocidin: A new apoptosis-inducing polycyclic guanidine alkaloid from the marine sponge Monanchora pulchra. Org. Lett. 2010, 12, 4292–4295. [Google Scholar] [CrossRef] [PubMed]
- Makarieva, T.N.; Tabakmaher, K.M.; Guzii, A.G.; Denisenko, V.A.; Dmitrenok, P.S.; Shubina, L.K.; Kuzmich, A.S.; Lee, H.-S.; Stonik, V.A. Monanchocidins B-E: Polycyclic guanidine alkaloids with potent antileukemic activities from the sponge Monanchora pulchra. J. Nat. Prod. 2011, 74, 1952–1958. [Google Scholar] [CrossRef] [PubMed]
- Kashman, Y.; Hirsh, S.; McConnell, O.J.; Ohtani, I.; Kusumi, T.; Kakisawa, H. Ptilomycalin A: A novel polycyclic guanidine alkaloid of marine origin. J. Am. Chem. Soc. 1989, 111, 8925–8926. [Google Scholar] [CrossRef]
- Makarieva, T.N.; Tabakmaher, K.M.; Guzii, A.G.; Denisenko, V.A.; Dmitrenok, P.S.; Kuzmich, A.S.; Lee, H.-S.; Stonik, V.A. Monanchomycalins A and B, unusual guanidine alkaloids from the sponge Monanchora pulchra. Tetrahedron Lett. 2012, 53, 4228–4231. [Google Scholar] [CrossRef]
- Tabakmakhera, K.M.; Denisenkoa, V.A.; Guziia, A.G.; Dmitrenoka, P.S.; Dyshlovoya, S.A.; Leeb, H.S.; Makarievaa, T.N. Monanchomycalin C, a new Pentacyclic Guanidine Alkaloid from the Far-Eastern marine sponge Monanchora pulchra. Nat. Prod. Commun. 2013, 8, 1399–1402. [Google Scholar]
- El-Demerdash, A.; Moriou, C.; Martin, M.T.; Rodrigues-Stien, A.S.; Petek, S.; Demoy-Schneider, M.; Hall, K.; Hooper, J.N.A.; Debitus, C.; Al-Mourabit, A. Cytotoxic guanidine alkaloids from a French Polynesian Monanchora n. sp. sponge. J. Nat. Prod. 2016, 79, 1929–1937. [Google Scholar] [CrossRef] [PubMed]
- Campos, P.E.; Wolfender, J.L.; Queiroz, E.F.; Marcourt, L.; Al-Mourabit, A.; Frederich, M.; Bordignon, A.; De Voogd, N.; Illien, B.; Gauvin-Bialecki, A. Unguiculin A and ptilomycalins E-H, antimalarial guanidine alkaloids from the marine sponge Monanchora unguiculata. J. Nat. Prod. 2017, 80, 1404–1410. [Google Scholar] [CrossRef] [PubMed]
- Barrow, R.A.; Murray, L.M.; Lim, T.K.; Capon, R.J. Mirabilins (A–F): New alkaloids from a Southern Australian marine sponge, Arenochalina mirabilis. Aust. J. Chem. 1996, 49, 767–773. [Google Scholar]
- Ferreira, E.G.; Wilkea, D.V.; Jimenez, P.C.; de Oliveirac, J.R.; Pessoa, O.D.L.; Silveira, E.R.; Viana, F.A.; Pessoa, C.; de Moraes, M.O.; Hajdu, E.; et al. Guanidine alkaloids from Monanchora arbuscula: Chemistry and antitumor potential. Chem. Biodivers. 2011, 8, 1433–1445. [Google Scholar] [CrossRef]
- Patil, A.D.; Kumar, N.V.; Kokke, W.C.; Bean, M.F.; Freyer, A.J.; De Brosse, C.; Mai, S.; Truneh, A.; Carte, B. Novel alkaloids from the sponge Batzella sp.: Inhibitors of HIV gp120-Human CD4 binding. J. Org. Chem. 1995, 60, 1182–1188. [Google Scholar] [CrossRef]
- Arevabinia, C.; Crivelentia, Y.D.; de Abreua, M.H.; Bitencourta, T.A.; Santos, M.F.; Berlinck, R.G.; Hajdu, E.; Beleboni, R.O.; Fachin, A.L.; Marins, M. Antifungal activity of metabolites from the marine sponges Amphimedon sp. and Monanchora arbuscula against Aspergillus flavus Strains isolated from peanuts (Arachis hypogaea). Nat. Prod. Commun. 2014, 9, 33–36. [Google Scholar]
- Santos, M.F.; Harper, P.M.; Williams, D.E.; Mesquimar drugta, J.T.; Pint, E.G.; da Costa-Silva, T.A.; Hajdu, E.; Ferreira, A.G.; Santos, R.A.; Murphy, P.J. Anti-parasitic guanidine and pyrimidine alkaloids from the marine sponge Monanchora arbuscula. J. Nat. Prod. 2015, 78, 1101–1112. [Google Scholar] [CrossRef] [PubMed]
- Makarieva, T.N.; Ogurtsova, E.K.; Denisenko, V.A.; Dmitrenok, P.S.; Tabakmakher, K.M.; Guzii, A.G.; Pislyagin, E.A.; Es’kov, A.A.; Kozhemyako, V.B.; Aminin, D.L.; et al. Urupocidin A: A new, inducing iNOS expression bicyclic guanidine alkaloid from the marine sponge Monanchora pulchra. Org. Lett. 2014, 16, 4292–4295. [Google Scholar] [CrossRef] [PubMed]
- Guzii, A.G.; Makarieva, T.N.; Korolkova, Y.V.; Andreev, Y.A.; Mosharova, I.V.; Tabakmaher, K.M.; Denisenko, V.A.; Dmitrenok, P.S.; Ogurtsova, E.K.; Antonov, A.S.; et al. Pulchranin A, isolated from the Far-Eastern marine sponge, Monanchora pulchra: The first marine non-peptide inhibitor of TRPV-1 channels. Tetrahedron Lett. 2013, 54, 1247–1250. [Google Scholar] [CrossRef]
- Makarieva, T.N.; Ogurtsova, E.K.; Korolkova, Y.V.; Andreev, Y.A.; Mosharova, I.V.; Tabakmaher, K.M.; Guzii, A.G.; Denisenko, V.A.; Dmitrenok, P.S.; Lee, H.S.; et al. Pulchranins B and C, new acyclic guanidine alkaloids from the Far-Eastern marine sponge Monanchora pulchra. Nat. Prod. Commun. 2013, 8, 1229–1232. [Google Scholar] [PubMed]
- Berlinck, R.G.S.; Braekman, J.C.; Daloze, D.; Bruno, I.; Riccio, R.; Rogeau, D.; Amade, P. Crambines C1 and C2: Two further ichthyotoxic guanidine alkaloids from the sponge Crambe crambe. J. Nat. Prod. 1992, 55, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Berlinck, R.G.S.; Braekman, J.C.; Daloze, D.; Hallenga, K.; Ottinger, R.; Bruno, I.; Riccio, R. Two new guanidine alkaloids from the Mediterranean sponge Crambe crambe. Tetrahedron Lett. 1990, 31, 6531–6534. [Google Scholar] [CrossRef]
- Berlinck, R.G.; Braekman, J.C.; Daloze, D.; Bruno, I.; Riccio, R.; Ferri, S.; Spampinato, S.; Speroni, E. Polycyclic guanidine alkaloids from the marine sponge Crambe crambe and calcium channel blocker activity of crambescidin 816. J. Nat. Prod. 1993, 56, 1007–1015. [Google Scholar] [CrossRef] [PubMed]
- Jares-Erijman, E.A.; Sakai, R.; Rinehart, K.L. Crambescidins: New antiviral and cytotoxic compounds from the sponge Crambe crambe. J. Org. Chem. 1991, 56, 5712–5715. [Google Scholar] [CrossRef]
- Bondu, S.; Genta-Jouve, G.; Leiròs, M.; Vale, C.; Guigonis, J.-M.; Botana, L.M.; Thomas, O.P. Additional bioactive guanidine alkaloids from the Mediterranean sponge Crambe crambe. RSC Adv. 2012, 2, 2828–2835. [Google Scholar]
- Roel, M.; Rubiolo, J.A.; Ternon, E.; Thomas, O.P.; Vieytes, M.R.; Botana, L.M. Crambescin C1 exerts a cytoprotective effect on HepG2 cells through metallothionein induction. Mar. Drugs 2015, 13, 4633–4653. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Henriksen, N.M.; Skalicky, J.J.; Harper, M.K.; Cheatham, T.E., 3rd; Ireland, C.M.; Van Wagoner, R.M. Araiosamines A-D: Tris-bromoindole cyclic guanidine alkaloids from the marine sponge Clathria (Thalysias) araiosa. J. Org. Chem. 2011, 76, 5515–5523. [Google Scholar] [CrossRef] [PubMed]
- Grkovic, T.; Blees, J.S.; Bayer, M.M.; Colburn, N.H.; Thomas, C.L.; Henrich, C.J.; Peach, M.L.; McMahon, J.B.; Schmid, T.; Gustafson, K.R. Tricyclic guanidine alkaloids from the marine sponge Acanthella cavernosa that stabilize the tumor suppressor PDCD4. Mar. Drugs 2014, 12, 4593–4601. [Google Scholar] [CrossRef] [PubMed]
- Jamison, M.T.; Molinski, T.F. Antipodal crambescin A2 homologues from the marine sponge Pseudaxinella reticulata. antifungal structure-activity relationships. J. Nat. Prod. 2015, 78, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Schroif-Grégoire, C.; Appenzeller, J.; Debitus, C.; Zaparucha, A.; Al-Mourabit, A. Debromokeramadine from the marine sponge Agelas cf. mauritiana: Isolation and short regioselective and flexible synthesis. Tetrahedron 2015, 71, 3609–3613. [Google Scholar] [CrossRef]
- Nakamura, H.; Ohizumi, Y.; Kobayashi, J.; Hirata, Y. Physiologically active marine natural products from Porifera. III. Keramadine, a novel antagonist of serotonergic receptors isolated from the Okinawan sea sponge Agelas sp. Tetrahedron Lett. 1984, 25, 2475–2478. [Google Scholar] [CrossRef]
- Abdjul, D.B.; Yamazaki, H.; Kanno, S.; Takahashi, O.; Kirikoshi, R.; Ukai, K.; Namikoshi, M. Haliclonadiamine Derivatives and 6-epi-Monanchorin from the Marine Sponge Halichondria panicea Collected at Iriomote Island. J. Nat. Prod. 2016, 79, 1149–1154. [Google Scholar] [CrossRef] [PubMed]
- Carbone, M.; Li, Y.; Irace, C.; Mollo, E.; Castelluccio, F.; Di Pascale, A.; Cimino, G.; Santamaria, R.; Guo, Y.W.; Gavagnim, M. Structure and cytotoxicity of phidianidines A and B, first finding of 1,2,4-oxadiazole system in a marine natural product. Org. Lett. 2011, 13, 2516–2519. [Google Scholar] [CrossRef] [PubMed]
- Vitale, R.M.; Gatti, M.; Carbone, M.; Barbieri, F.; Felicita, V.; Gavagnin, M.; Florio, T.; Amodeo, P. Minimalist hybrid ligand/receptor-based pharmacophore model for CXCR4 applied to a small-library of marine natural products led to the identification of phidianidine a as a new CXCR4 ligand exhibiting antagonist activity. ACS Chem. Biol. 2013, 8, 2762–2770. [Google Scholar] [CrossRef] [PubMed]
- Putz, A.; Kehraus, S.; Díaz-Agras, G.; Wägele, H.; König, G.M. Dotofide, a guanidine-interrupted terpenoid from the marine slug Doto pinnatifida (Gastropoda, Nudibranchia. Eur. J. Org. Chem. 2011, 42, 3733–3737. [Google Scholar] [CrossRef]
- Carbone, M.; Ciavatta, M.L.; Mathieu, V.; Ingels, A.; Kiss, R.; Pascale, P.; Mollo, E.; Ungur, N.; Guo, Y.W.; Gavagnin, M. Marine terpenoid diacylguanidines: Structure, synthesis, and biological evaluation of naturally occurring actinofide and synthetic analogues. J. Nat. Prod. 2017, 80, 1339–1346. [Google Scholar] [CrossRef] [PubMed]
- Rinehart, K.L.; Holt, T.G.; Fregeau, N.L.; Stroh, J.G.; Keifer, P.A.; Sun, F.; Li, L.H.; Martin, D.G. Ecteinascidins 729, 743, 745, 759A, 759B, and 770: Potent antitumor agents from the Caribbean tunicate Ecteinascidia turbinata. J. Org. Chem. 1990, 55, 4512–4515. [Google Scholar] [CrossRef]
- Wright, A.E.; Forleo, D.A.; Gunawardana, G.P.; Gunasekera, S.P.; Koehn, F.E.; McConnell, O.J. Antitumor tetrahydroisoquinoline alkaloids from the colonial ascidian Ecteinascidia turbinata. J. Org. Chem. 1990, 55, 4508–4512. [Google Scholar] [CrossRef]
- Chan, S.T.; Patel, P.R.; Ransom, T.R.; Henrich, C.J.; McKee, T.C.; Goey, A.K.; Cook, K.M.; Figg, W.D.; McMahon, J.B.; Schnermann, M.J.; et al. Structural elucidation and synthesis of eudistidine A: An unusual polycyclic marine alkaloid that blocks interaction of the protein binding domains of p300 and HIF-1alpha. J. Am. Chem. Soc. 2015, 137, 5569–5575. [Google Scholar] [CrossRef] [PubMed]
- Hinman, A.; Du Bois, J. A stereoselective synthesis of (−)-Tetrodotoxin. J. Am. Chem. Soc. 2003, 125, 11510–11511. [Google Scholar] [CrossRef] [PubMed]
- Fleming, J.J.; McReynolds, M.D.; Du Bois, J. (+)-Saxitoxin: A first and second generation stereoselective synthesis. J. Am. Chem. Soc. 2007, 129, 9964–9975. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, K.; Hashimoto, Y. Synthesis of marine guanidine alkaloids and their application as chemical/biological tools. Chem. Rec. 2003, 3, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Feldman, K.S.; Ngernmeesri, P. Total synthesis of (+/−)-dragmacidin E: Problems solved and lessons learned. Synlett 2012, 23, 1882–1892. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Wang, X.; Wang, X.; Rodriguez, R.A.; Moore, C.E.; Gao, S.; Tan, S.; Ma, Y.; Rheingold, A.L.; Baran, P.S.; et al. Asymmetric syntheses of sceptrin and massadine and evidence for biosynthetic enantiodivergence. Science 2014, 346, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, R.A.; Barrios Steed, D.; Kawamata, Y.; Su, S.; Smith, P.A.; Steed, T.C.; Romesberg, F.E.; Baran, P.S. Axinellamines as broad-spectrum antibacterial agents: Scalable synthesis and biology. J. Am. Chem. Soc. 2014, 136, 15403–15413. [Google Scholar] [CrossRef] [PubMed]
- Tomoaki, M.; Keisuke, M.; Tatsuya, K.; Satoshi, Y.; Tohru, F. Total synthesis of (−)-tetrodotoxin and 11-nor TTX-6(R)-ol. Angew. Chem. Int. Ed. 2017, 56, 1549–1552. [Google Scholar]
- Tian, M.; Yan, M.; Baran, P.S. 11-Step total synthesis of araiosamines. J. Am. Chem. Soc. 2016, 138, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Snider, B.B. Synthesis of phidianidines A and B. J. Org. Chem. 2012, 77, 4832–4836. [Google Scholar] [CrossRef] [PubMed]
- Brogan, J.T.; Stoops, S.L.; Lindsley, C.W. Total synthesis and biological evaluation of phidianidines A and B uncovers unique pharmacological profiles at CNS targets. ACS Chem. Neurosci. 2012, 3, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Manzo, E.; Pagano, D.; Carbone, M.; Ciavatt, M.L.; Gavagnin, M. Synthesis of phidianidine B, a highly cytotoxic 1,2,4-oxadiazole marine metabolite. Arkivoc 2012, 2012, 220–228. [Google Scholar]
- Buchanan, J.C.; Petersen, B.P.; Chamberland, S. Concise total synthesis of phidianidine A and B. Tetrahedron Lett. 2013, 54, 6002–6004. [Google Scholar] [CrossRef]
- Jiang, C.S.; Fu, Y.; Zhang, L.; Gong, J.X.; Wang, Z.Z.; Xiao, W.; Zhang, H.Y.; Guo, Y.W. Synthesis and biological evaluation of novel marine-derived indole-based 1,2,4-oxadiazoles derivatives as multifunctional neuroprotective agents. Bioorg. Med. Chem. Lett. 2015, 25, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jiang, C.S.; Gao, L.X.; Gong, J.X.; Wang, Z.H.; Li, J.Y.; Li, J.; Li, X.W.; Guom, Y.W. Design, synthesis and in vitro activity of phidianidine B derivatives as novel PTP1B inhibitors with specific selectivity. Bioorg. Med. Chem. Lett. 2016, 26, 778–781. [Google Scholar] [CrossRef] [PubMed]
- Putra, M.Y.; Ianaro, A.; Panze, E.; Bavestrello, G.; Cerrano, C.; Fattorusso, E.; Taglialatela-Scafati, O. Sinulasulfoxide and sinulasulfone, sulfur-containing alkaloids from the Indonesian soft coral Sinularia sp. Tetrahedron Lett. 2012, 53, 3937–3939. [Google Scholar] [CrossRef]
- Rigano, D.; Sirignano, C.; Taglialatela-Scafati, O. The potential of natural products for targeting PPARα. Acta Pharm. Sin. B. 2017, 4, 427–438. [Google Scholar] [CrossRef] [PubMed]















© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Li, X.-W.; Guo, Y.-W. Recent Advances in the Isolation, Synthesis and Biological Activity of Marine Guanidine Alkaloids. Mar. Drugs 2017, 15, 324. https://doi.org/10.3390/md15100324
Liu J, Li X-W, Guo Y-W. Recent Advances in the Isolation, Synthesis and Biological Activity of Marine Guanidine Alkaloids. Marine Drugs. 2017; 15(10):324. https://doi.org/10.3390/md15100324
Chicago/Turabian StyleLiu, Jin, Xu-Wen Li, and Yue-Wei Guo. 2017. "Recent Advances in the Isolation, Synthesis and Biological Activity of Marine Guanidine Alkaloids" Marine Drugs 15, no. 10: 324. https://doi.org/10.3390/md15100324
APA StyleLiu, J., Li, X. -W., & Guo, Y. -W. (2017). Recent Advances in the Isolation, Synthesis and Biological Activity of Marine Guanidine Alkaloids. Marine Drugs, 15(10), 324. https://doi.org/10.3390/md15100324