Chemical Diversity from a Chinese Marine Red Alga, Symphyocladia latiuscula


 ,
, 
Abstract
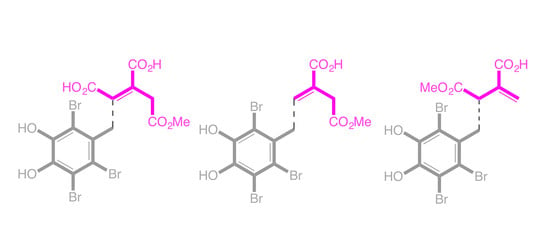
:
1. Introduction
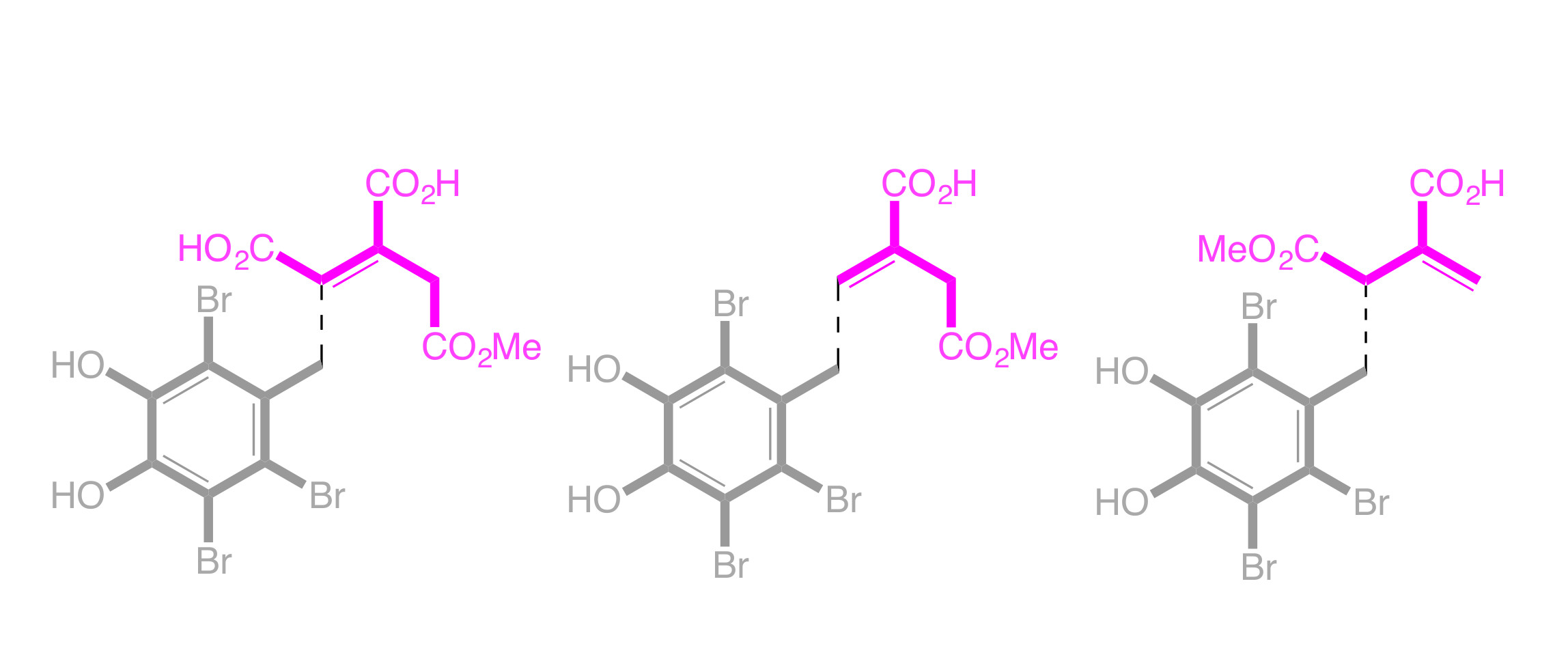
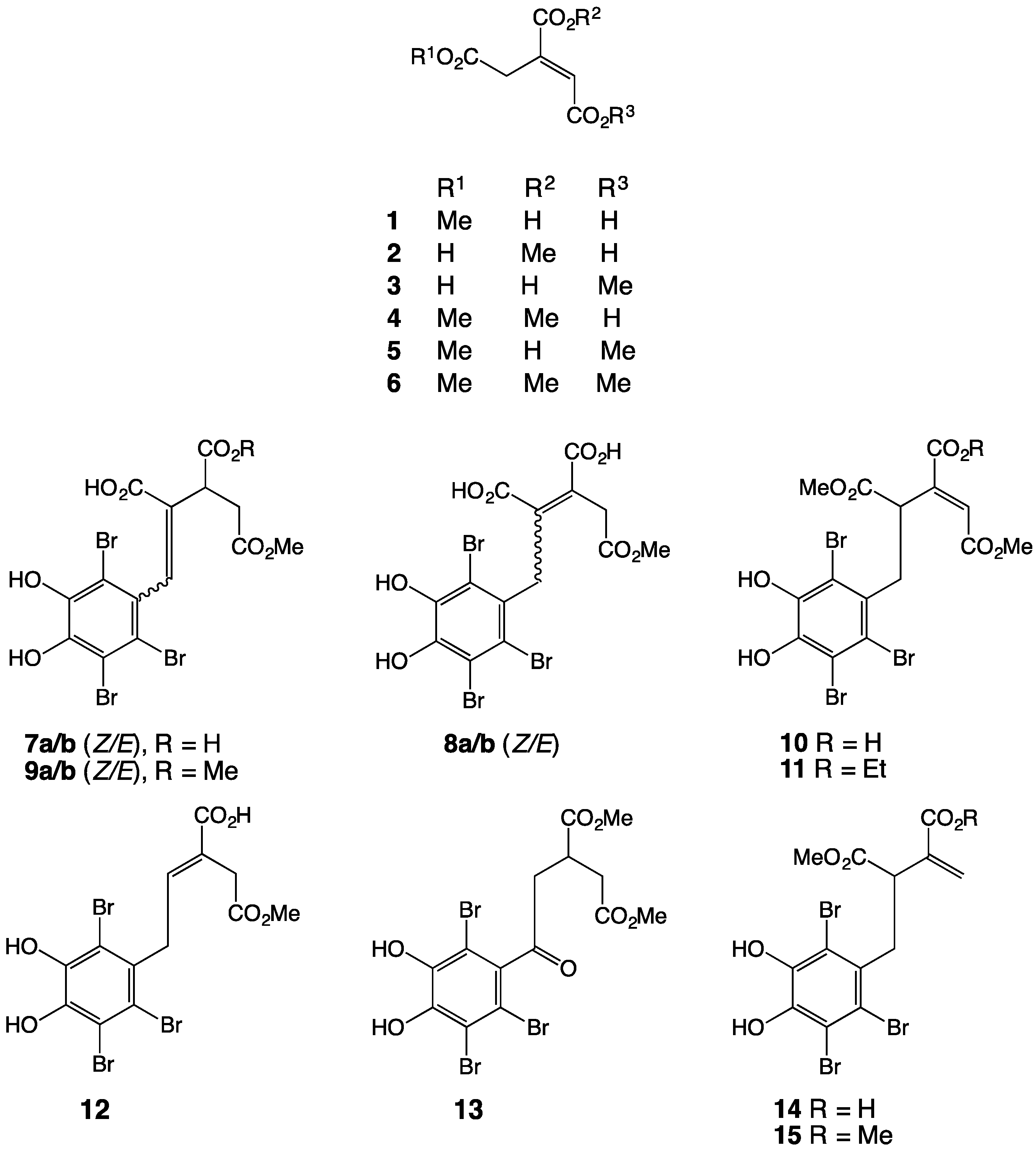
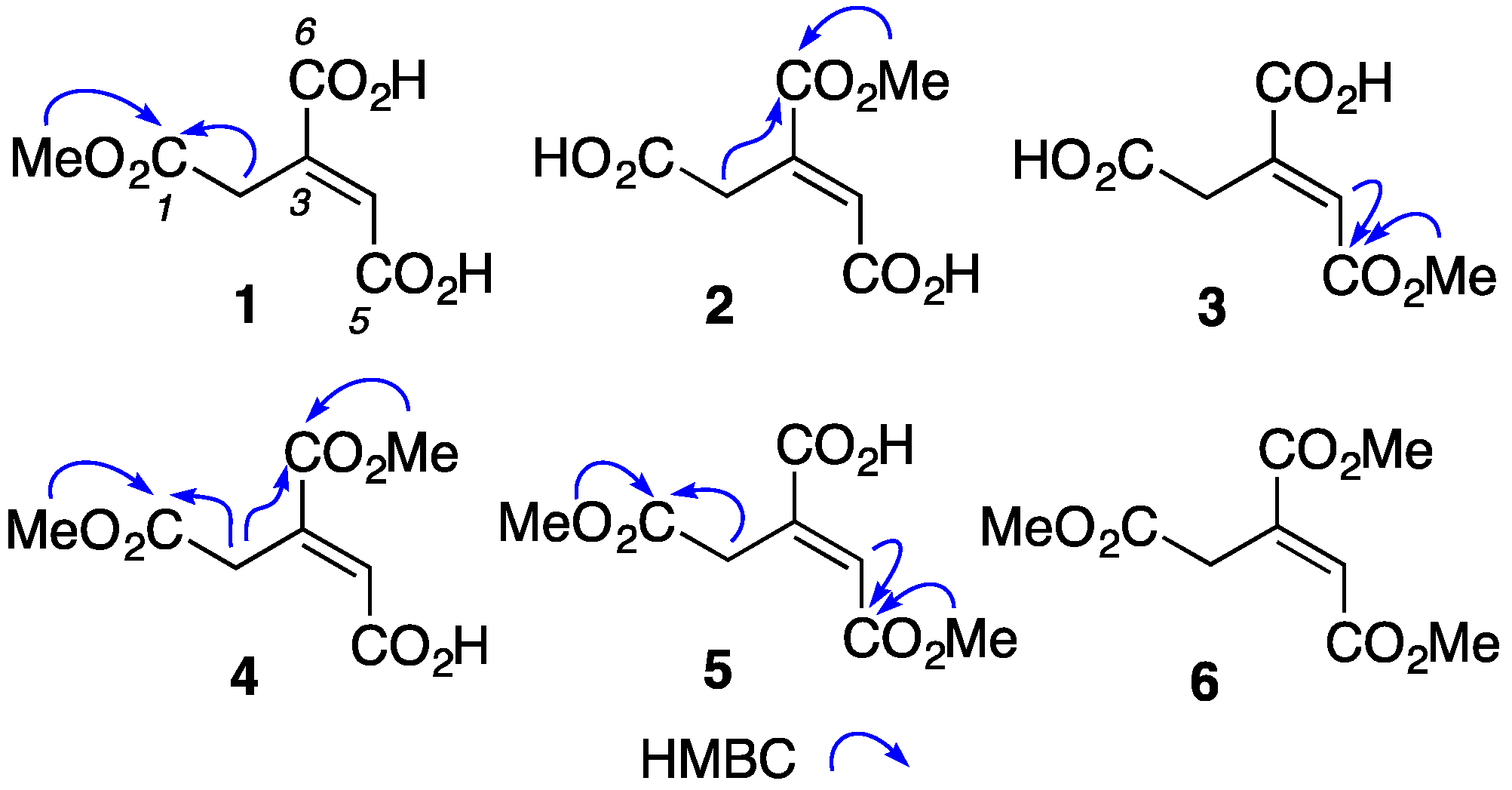
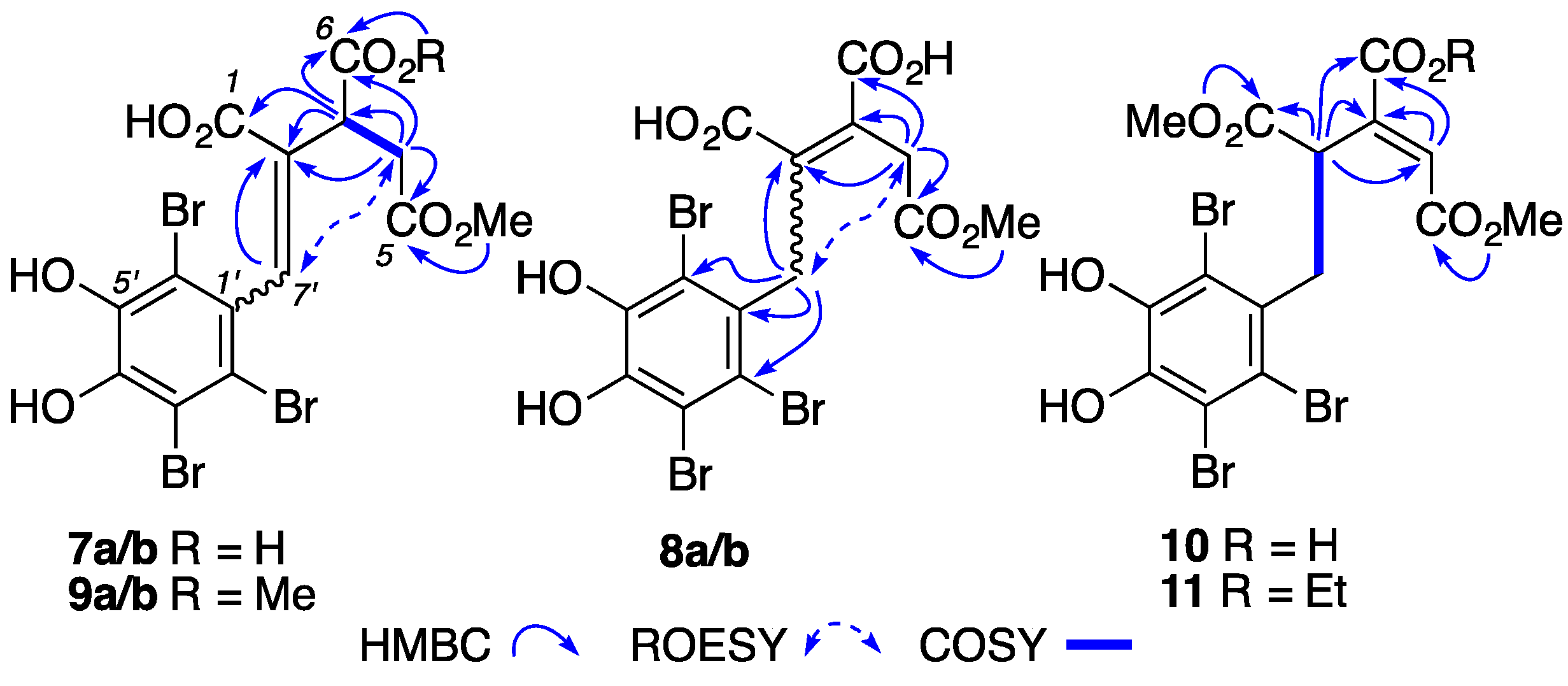
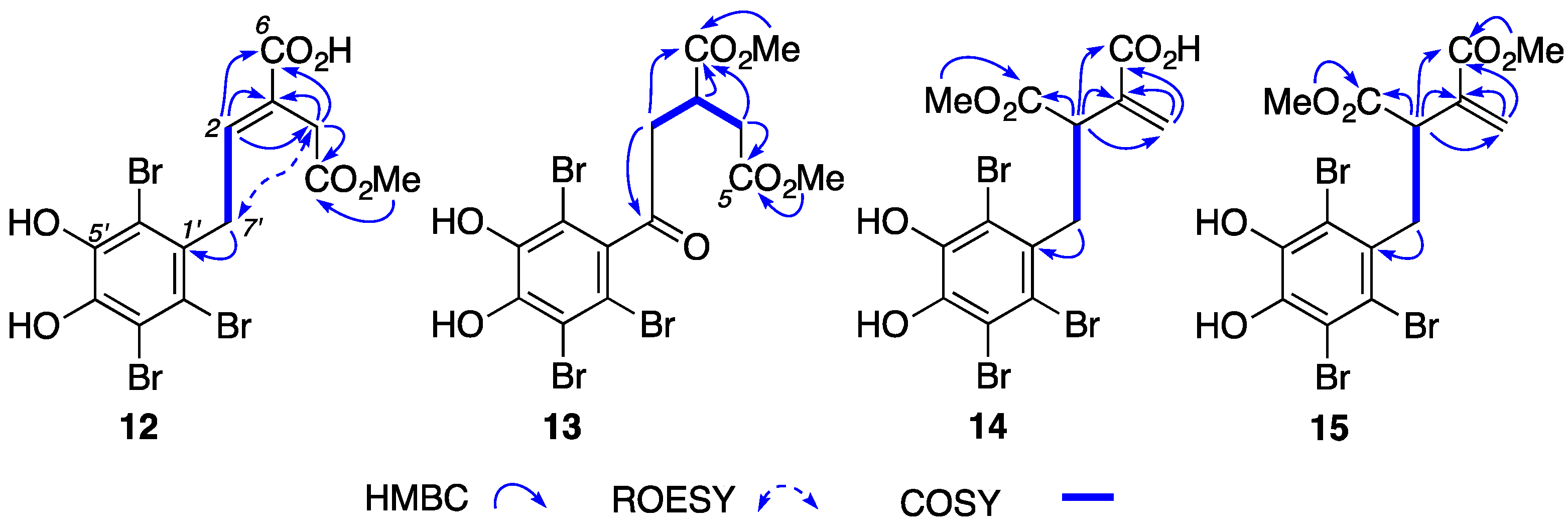
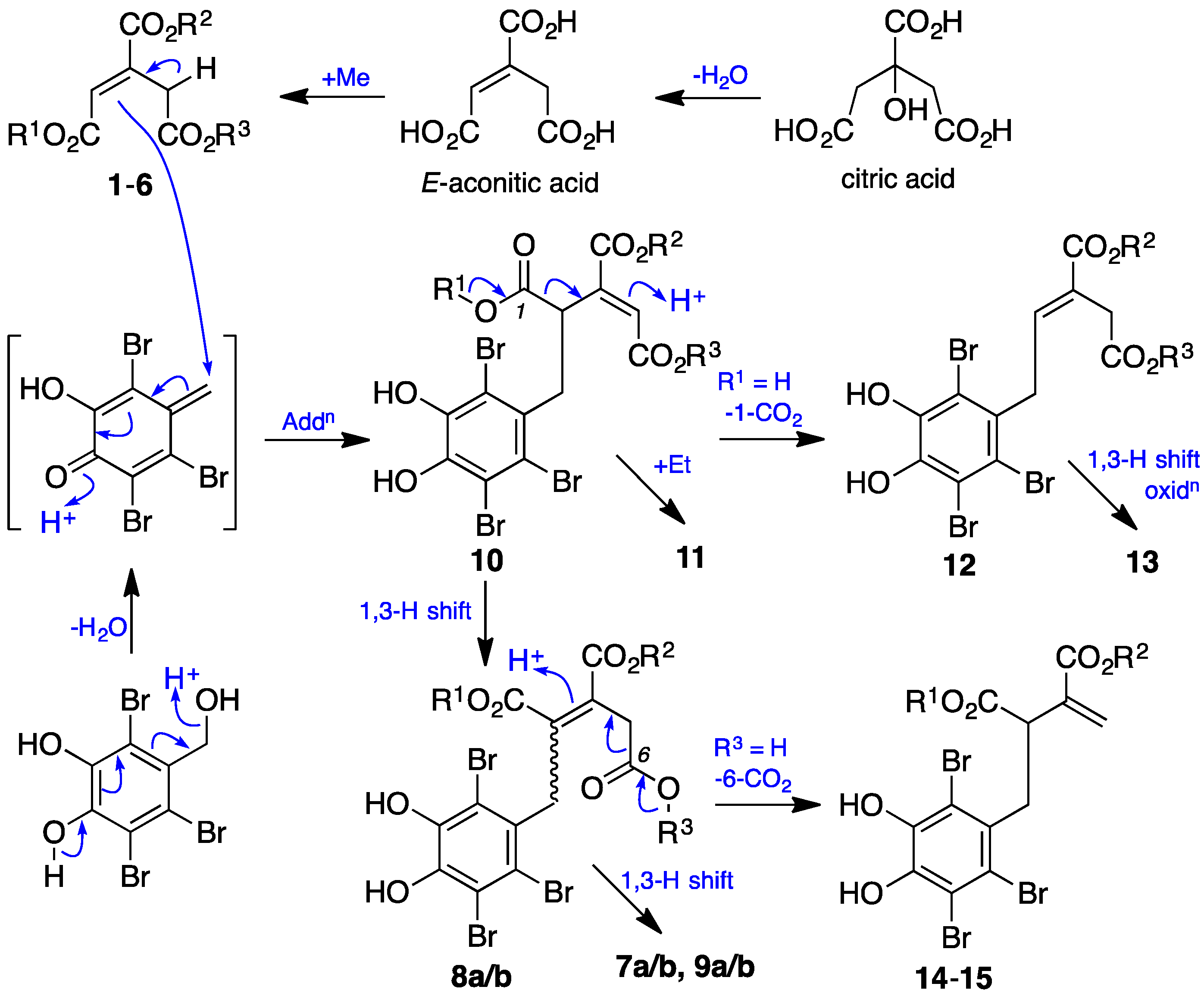
2. Results and Discussion
3. Materials and Methods
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Hansen, P.E.; Lin, X.K. Bromophenols in marine algae and their bioactivities. Mar. Drugs 2011, 9, 1273–1292. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, X.M.; Gao, L.X.; Cui, C.M.; Li, C.S.; Li, J.; Wang, B.G. Extraction and PTP1B inhibitory activity of bromophenols from the marine red alga Symphyocladia latiuscula. Chin. J. Oceanol. Limn. 2011, 29, 686–690. [Google Scholar] [CrossRef]
- Li, K.; Li, X.M.; Gloer, J.B.; Wang, B.G. New nitrogen-containing bromophenols from the marine red alga Rhodomela confervoides and their radical scavenging activity. Food Chem. 2012, 135, 868–872. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.K.; Liu, M. Bromophenols from marine algae with potential anti-diabetic activities. J. Ocean Univ. China 2012, 11, 533–538. [Google Scholar] [CrossRef]
- Olsen, E.K.; Hansen, E.; Isaksson, J.; Andersen, J.H. Antioxidant effect of four bromophenols from the red algae Vertebrata lanosa. Planta Med. 2012, 78, 1146. [Google Scholar] [CrossRef]
- Javan, A.J.; Javan, M.J.; Tehrani, Z.A. Theoretical investigation on antioxidant activity of bromophenols from the marine red alga Rhodomela confervoides: H-atom vs. electron transfer mechanism. J. Agric. Food Chem. 2013, 61, 1534–1541. [Google Scholar] [CrossRef] [PubMed]
- Olsen, E.K.; Hansen, E.; Isaksson, J.; Andersen, J.H. Cellular antioxidant effect of four bromophenols from the red algae, Vertebrata lanosa. Mar. Drugs 2013, 11, 2769–2784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Yin, L.; Gao, L.; Gao, J.; Chen, J.; Li, J.; Song, F. Two new bromophenols with radical scavenging activity from marine red alga Symphyocladia latiuscula. Mar. Drugs 2013, 11, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Yin, L.; Gao, J.; Gao, L.; Song, F. Antifungal bromophenols from marine red alga Symphyocladia latiuscula. Chem. Biodivers. 2014, 11, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Kurata, K.; Amiya, T. Bis(2,3,6-tribromo-4,5-dihydroxybenzyl) ether from the red alga, Symphyocladia latiuscula. Phytochemistry 1980, 19, 141–142. [Google Scholar] [CrossRef]
- Xu, X.; Song, F.; Fan, X.; Fang, N.; Shi, J. A novel bromophenol from marine red alga Symphyocladia latiuscula. Chem. Nat. Compd. 2009, 45, 811–813. [Google Scholar] [CrossRef]
- Xu, X.; Piggott, A.M.; Yin, L.; Capon, R.J.; Song, F. Symphyocladins A–G: Bromophenol adducts from a Chinese marine red alga, Symphyocladia latiuscula. Tetrahedron Lett. 2012, 53, 2103–2106. [Google Scholar] [CrossRef]
- Xu, X.; Yin, L.; Wang, Y.; Wang, S.; Song, F. A new bromobenzyl methyl sulphoxide from marine red alga Symphyocladia latiuscula. Nat. Prod. Res. 2013, 27, 723–726. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Kurokawa, M.; Shiraki, K.; Nakamura, N.; Choi, J.S.; Hattori, M. Antiviral activity of the marine alga Symphyocladia latiuscula against herpes simplex virus (HSV-1) in vitro and its therapeutic efficacy against HSV-1 infection in mice. Biol. Pharm. Bull. 2005, 28, 2258–2262. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Park, S.E.; Hossain, M.A.; Kim, M.Y.; Kim, M.N.; Chung, H.Y.; Choi, J.S.; Yoo, Y.H.; Kim, N.D. 2,3,6-Tribromo-4,5-dihydroxybenzyl methyl ether induces growth inhibition and apoptosis in MCF-7 human breast cancer cells. Arch. Pharm. Res. 2007, 30, 1132–1137. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.S.; Park, H.J.; Jung, H.A.; Chung, H.Y.; Jung, J.H.; Choi, W.C. A cyclohexanonyl bromophenol from the red alga Symphyocladia latiuscula. J. Nat. Prod. 2000, 63, 1705–1706. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.J.; Li, X.M.; Wang, B.G. Highly brominated mono-and bis-phenols from the marine red alga Symphyocladia latiuscula with radical-scavenging activity. J. Nat. Prod. 2007, 70, 1210–1213. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Okada, Y.; Shi, H.B.; Wang, Y.Q.; Okuyama, T. Structures and aldose reductase inhibitory effects of bromophenols from the red alga Symphyocladia latiuscula. J. Nat. Prod. 2005, 68, 620–622. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.J.; Oh, M.Y.; Jin, D.H.; Hong, Y.K. Identification of a Taq DNA polymerase inhibitor from the red seaweed Symphyocladia latiuscula. J. Environ. Biol. 2008, 29, 475–478. [Google Scholar] [PubMed]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. ORTEP-3 for Windows-a version of ORTEP-III with a Graphical User Interface (GUI). J. Appl. Crystallogr. 1997, 30, 565. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Spek, A.L. PLATON, An integrated tool for the analysis of the results of a single crystal structure determination. Acta Crystallogr. Sect A 1990, 46, C34. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 7a a | 7b a | 8a b | 8b b | 9a c | 9b c |
|---|---|---|---|---|---|---|
| 3 | 3.74, m | 3.74, m | 3.50, m | 3.50, m | ||
| 4a | 3.17, m | 3.17, m | 3.65, s | 3.19, s | 2.98, dd e | 2.976, dd e |
| 4b | 2.490, dd d | 2.489, dd d | 2.41, dd f | 2.40, dd f | ||
| 5-OCH3 | 3.55, s | 3.55, s | 3.71, s | 3.51, s | 3.52, s | 3.52, s |
| 6-OCH3 | 3.541, s | 3.535, s | ||||
| 7′ | 7.544, s | 7.538, s | 4.22, s | 4.34, s | 7.391, s | 7.388, s |
| Position | 7a a | 7b a | 8a b | 8b b | 9a c | 9b c |
|---|---|---|---|---|---|---|
| 1 | 167.18, C | 167.18, C | 170.3, C | 166.5, C | 166.8, C | 166.8, C |
| 2 | 135.46, C | 135.34, C | 145.0, C | 144.4, C | 133.5, C | 133.5, C |
| 3 | 41.39, CH | 41.37, CH | 127.4, C | 137.8, C | 40.2, CH | 40.2, CH |
| 4 | 35.40, CH2 | 35.35, CH2 | 35.0, CH2 | 29.5, CH2 | 33.9, CH2 | 33.9, CH2 |
| 5 | 172.80, C | 172.76, C | 168.7, C | 166.8, C | 171.5, C | 171.5, C |
| 6 | 172.54, C | 172.49, C | 172.4, C | 169.0, C | 171.5, C | 171.5, C |
| 5-OCH3 | 51.87, CH3 | 51.83, CH3 | 52.8, CH3 | 53.1, CH3 | 52.0, CH3 | 52.0, CH3 |
| 6-OCH3 | 51.6, CH3 | 51.6, CH3 | ||||
| 1′ | 129.79, C | 129.76, C | 128.8, C | 127.9, C | 128.07, C | 128.07, C |
| 2′ | 115.38, C | 115.23, C | 118.9, C | 118.4, C | 113.9, C | 113.9, C |
| 3′ | 113.98, C | 113.63, C | 114.8, C | 114.2, C | 113.7, C | 113.6, C |
| 4′ | 144.17, C | 144.10, C | 146.2, C | 145.5, C | 143.9, C | 143.8, C |
| 5′ | 145.36, C | 145.32, C | 145.6, C | 144.8, C | 145.2, C | 145.0, C |
| 6′ | 110.71, C | 110.67, C | 114.7, C | 114.3, C | 110.7, C | 110.6, C |
| 7′ | 141.52, C | 141.48, C | 41.1, CH | 35.9, CH | 140.7, C | 140.7, C |
| Position | 10 a δH, m (J in Hz) | 11 b δH, m (J in Hz) |
|---|---|---|
| 2 | 4.98, dd (11.4, 3.0) | 4.98, dd (11.4, 3.0) |
| 4 | 6.76, s | 6.73, s |
| 1-OCH3 | 3.66, s | 3.70, s |
| 5-OCH3 | 3.44, s | 3.45, s |
| 6-OCH2CH3 | 4.26, br q (7.2) | |
| 6-OCH2CH3 | 1.31, t (7.2) | |
| 7′a | 3.87, dd (14.4, 3.0) | 3.81, dd (14.4, 3.0) |
| 7′b | 3.61, dd (14.4, 11.4) | 3.56, dd (14.4, 11.4) |
| Position | 12 a δH, m (J in Hz) | 13 b δH, m (J in Hz) | 14 b δH, m (J in Hz) | 15 a δH, m (J in Hz) |
|---|---|---|---|---|
| 2a | 6.79, br t (6.6) | 3.35, m | 3.75, dd (9.0, 6.0) | 3.79, dd (9.6, 5.4) |
| 2b | 3.27, dd (20.4, 7.8) | |||
| 3 | 3.34, m | |||
| 4a | 3.60, br s | 2.86, dd (17.4, 7.2) | 6.22. d (1.2) | 6.17, d (1.2) |
| 4b | 2.73, dd (17.4, 6.0) | 5.44, br s | 5.52, s | |
| 1-OCH3 | 3.64, s | 3.62, s | ||
| 5-OCH3 | 3.66, s | 3.68, s | ||
| 6-OCH3 | 3.71, s | 3.71, s | ||
| 7′a | 4.05, s | 3.65, dd (13.8, 6.0) | 3.69, dd (14.4, 5.4) | |
| 7′b | 3.54, dd (13.8, 9.0) | 3.56, dd (14.4, 9.6) |
| Position | 10 a δC, Type | 11 b δC, Type | 12 a δC, Type | 13 b δC, Type | 14 b δC, Type | 15 a δC, Type |
|---|---|---|---|---|---|---|
| 1 | 171.9, C | 173.5, C | 174.5, C | 172.4, C | ||
| 2 | 43.0, C | 43.7, C | 141.3, C | 44.9, CH2 | 48.8, CH | 48.2, CH |
| 3 | 142.4, C | 142.4, C | 128.1, C | 37.4, CH | 139.3, C | 138.3, C |
| 4 | 130.5, CH | 131.3, CH | 33.2, CH2 | 35.6, CH2 | 129.1, CH2 | 128.7, CH2 |
| 5 | 165.8, C | 166.7, C | 171.3, C | 173.9, C | ||
| 6 | 167.0, C | 167.0, C | 167.9, C | 175.5, C | 169.2, C | 166.6, C |
| 1-OCH3 | 52.3, CH3 | 52.9, CH3 | 52.8, CH3 | 52.3, CH3 | ||
| 5-OCH3 | 52.1, CH3 | 52.5, CH3 | 52.0, CH3 | 52.5, CH3 | ||
| 6-OCH3 | 52.8, CH3 | 52.3, CH3 | ||||
| 6-OCH2CH3 | 63.2, C | |||||
| 6-OCH2CH3 | 14.5, CH3 | |||||
| 1′ | 130.7, C | 130.6, C | 131.0, C | 136.1, C | 131.4, C | 131.3, C |
| 2′ | 118.5, C | 118.8, C | 117.3, C | 114.5, C | 118.3, C | 118.0, C |
| 3′ | 113.7, C | 114.4, C | 114.0, C | 110.5, C | 114.5, C | 113.9, C |
| 4′ | 144.1, C | 145.1, C | 144.4, C | 147.3, C | 145.0, C | 144.0, C |
| 5′ | 143.8, C | 144.8, C | 144.3, C | 145.3, C | 144.8, C | 143.9, C |
| 6′ | 114.3, C | 114.8, C | 113.0, C | 106.3, C | 114.3, C | 113.8, C |
| 7′ | 39.0, CH2 | 39.4, CH2 | 39.0, CH2 | 202.2, C | 39.4, CH2 | 39.1, CH2 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, X.; Yang, H.; Khalil, Z.G.; Yin, L.; Xiao, X.; Neupane, P.; Bernhardt, P.V.; Salim, A.A.; Song, F.; Capon, R.J. Chemical Diversity from a Chinese Marine Red Alga, Symphyocladia latiuscula. Mar. Drugs 2017, 15, 374. https://doi.org/10.3390/md15120374
Xu X, Yang H, Khalil ZG, Yin L, Xiao X, Neupane P, Bernhardt PV, Salim AA, Song F, Capon RJ. Chemical Diversity from a Chinese Marine Red Alga, Symphyocladia latiuscula. Marine Drugs. 2017; 15(12):374. https://doi.org/10.3390/md15120374
Chicago/Turabian StyleXu, Xiuli, Haijin Yang, Zeinab G. Khalil, Liyuan Yin, Xue Xiao, Pratik Neupane, Paul V. Bernhardt, Angela A. Salim, Fuhang Song, and Robert J. Capon. 2017. "Chemical Diversity from a Chinese Marine Red Alga, Symphyocladia latiuscula" Marine Drugs 15, no. 12: 374. https://doi.org/10.3390/md15120374
APA StyleXu, X., Yang, H., Khalil, Z. G., Yin, L., Xiao, X., Neupane, P., Bernhardt, P. V., Salim, A. A., Song, F., & Capon, R. J. (2017). Chemical Diversity from a Chinese Marine Red Alga, Symphyocladia latiuscula. Marine Drugs, 15(12), 374. https://doi.org/10.3390/md15120374