
Two Isomeric C16 Oxo-Fatty Acids from the Diatom Chaetoceros karianus Show Dual Agonist Activity towards Human Peroxisome Proliferator-Activated Receptors (PPARs) α/γ


 ,
,
Abstract
:1. Introduction
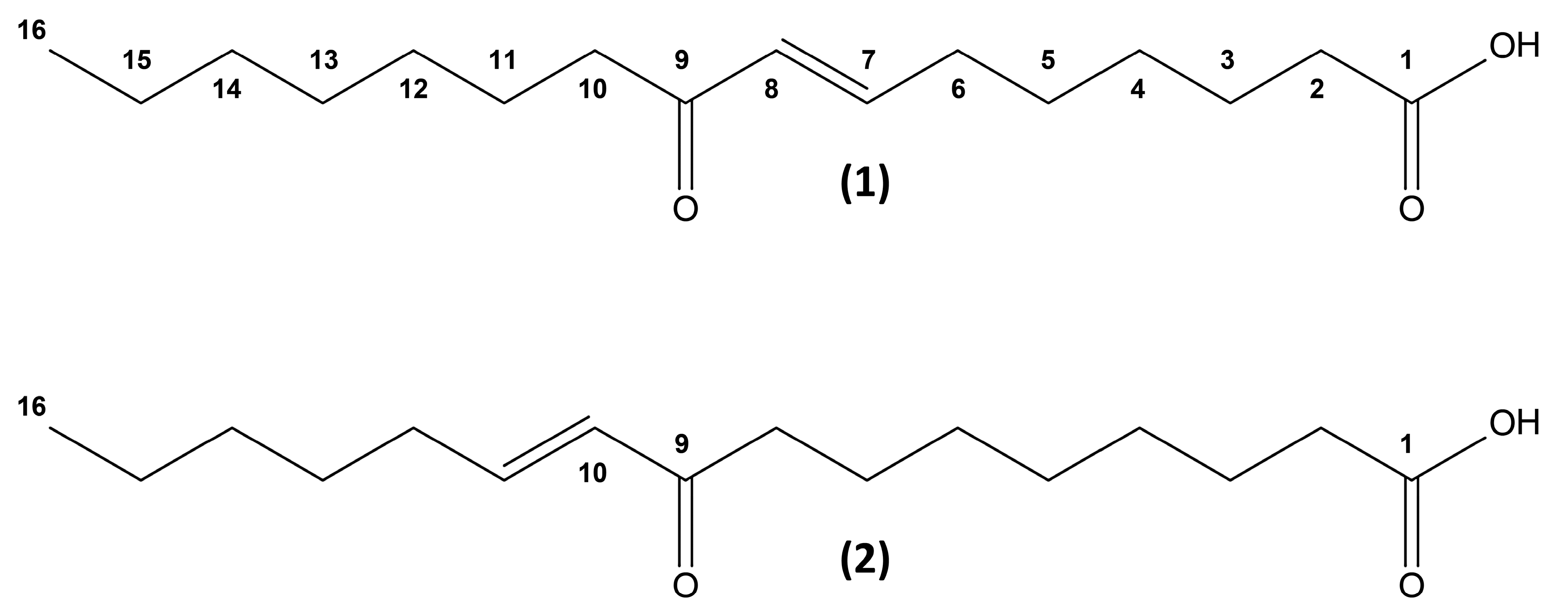
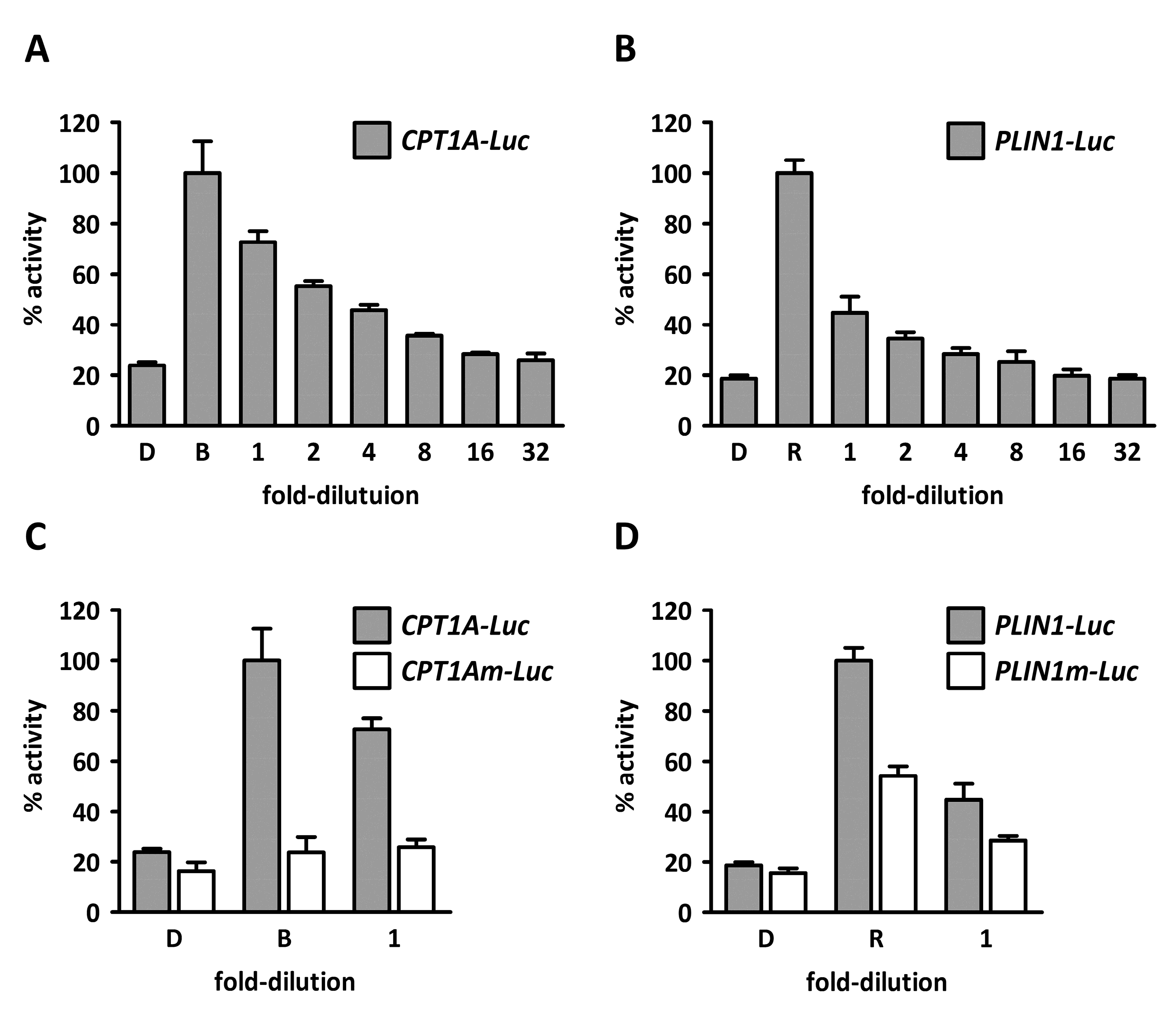
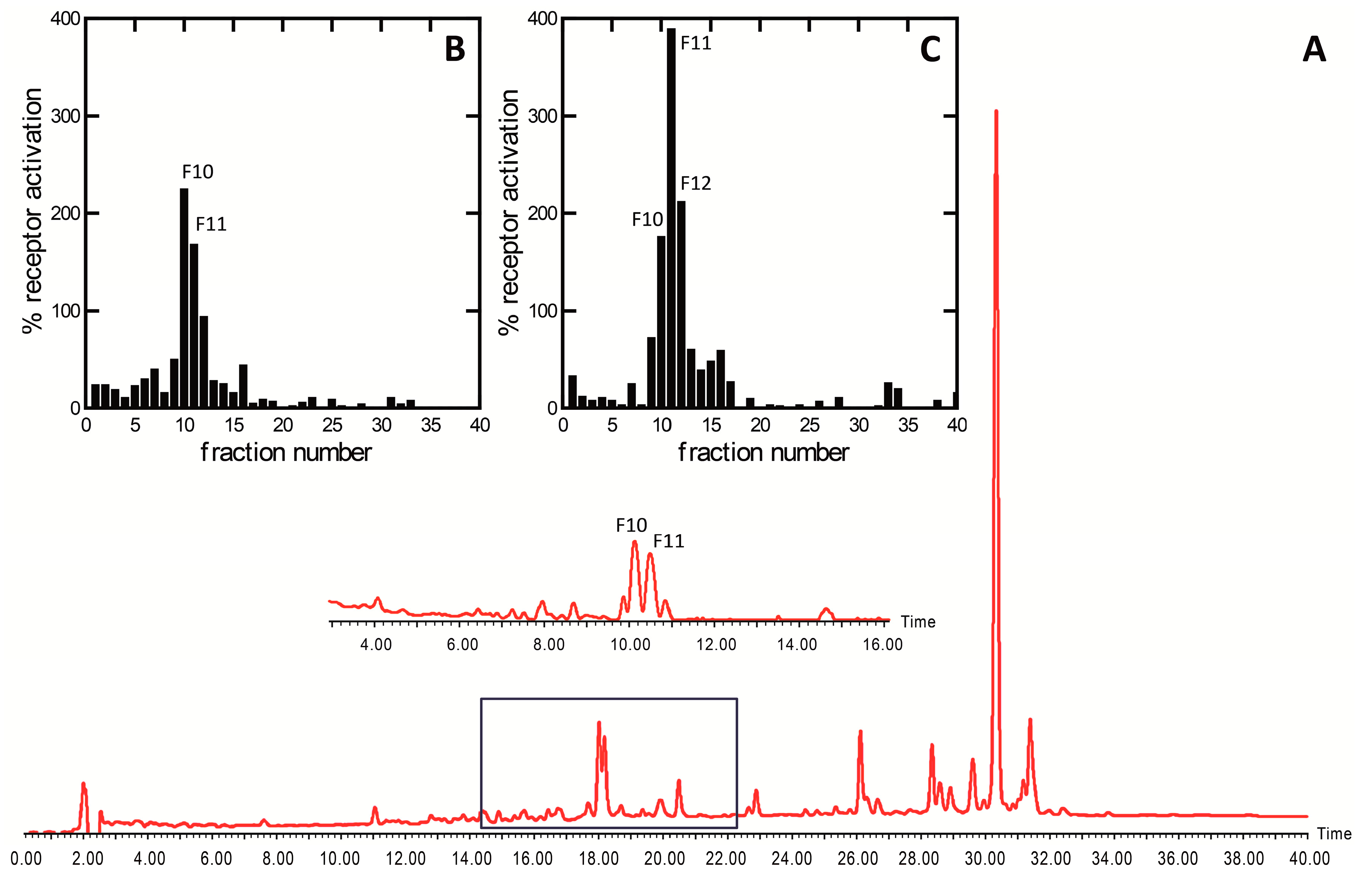
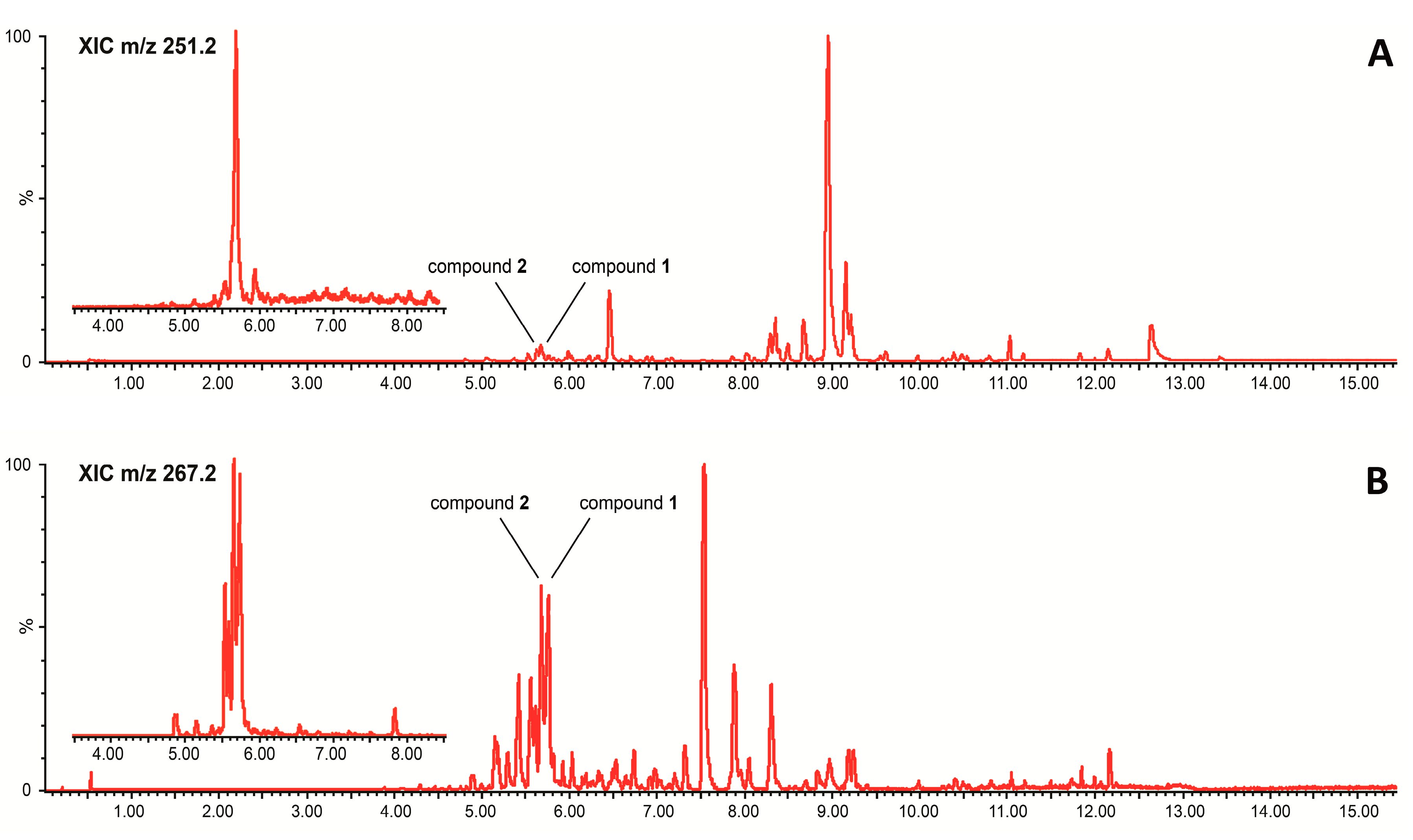
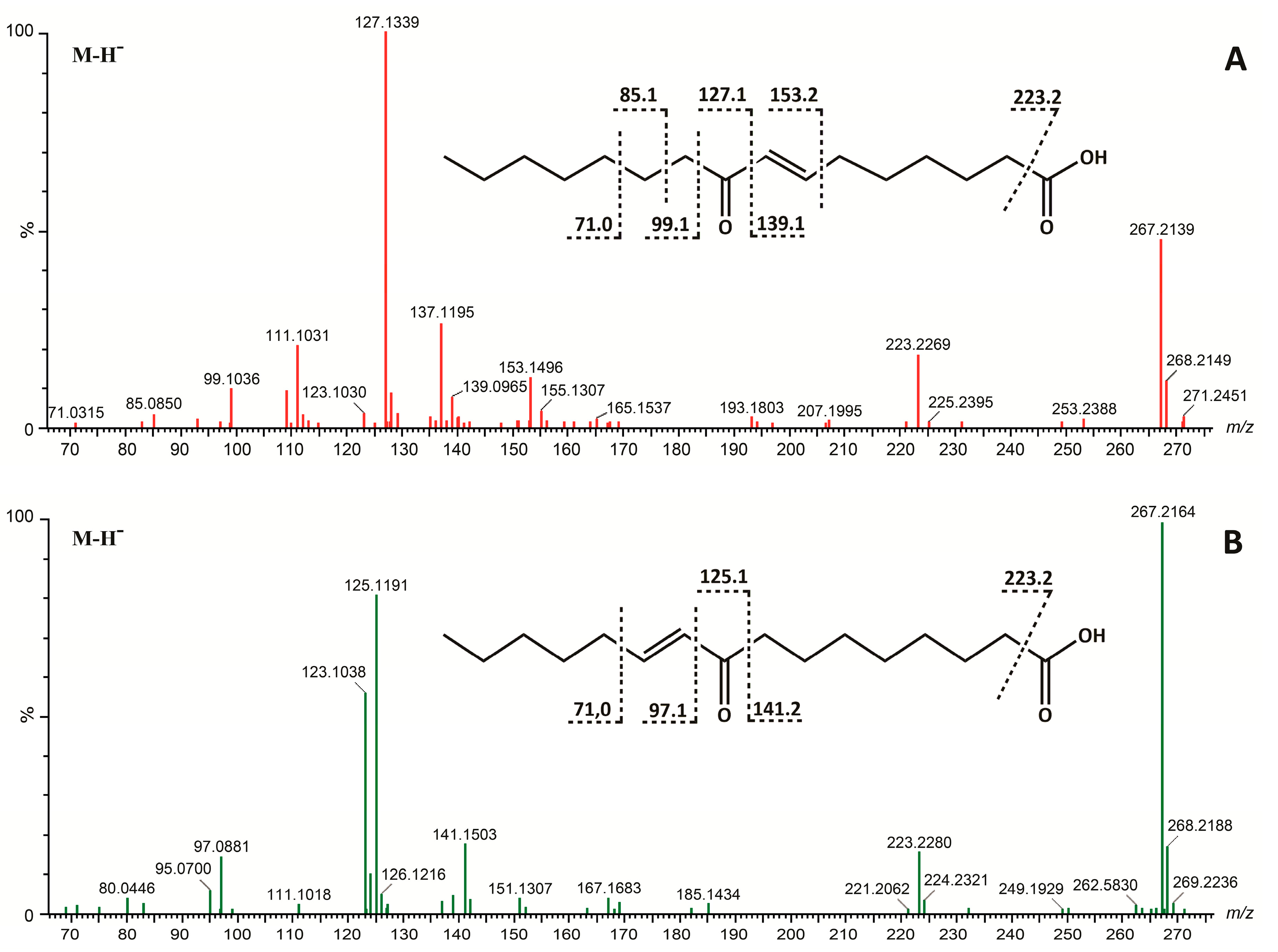
2. Results and Discussion
3. Materials and Methods
3.1. Biological Material
3.2. Bioassay-Guided Isolation Platform
3.2.1. General Extract Preparation
3.2.2. Specific Extract Preparation for Bioassay-Guided Fractionation
3.2.3. Semi-Preparative High-Performance Liquid Chromatography (Semiprep HPLC-PDA)
3.2.4. Plasmids
3.2.5. Cell Culture, Transfection, and Luciferase Assays
3.3. Structure Elucidation Platform
3.3.1. Ultra-Performance Liquid Chromatography Coupled to Data-Independent Mass Spectrometry (UPLC-MSe)
3.3.2. Ultra-Performance Liquid Chromatography Coupled to Photodiode Array Detection (UPLC-PDA)
3.3.3. NMR Spectroscopy
3.4. Metabolomics
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ferre, P. The biology of peroxisome proliferator-activated receptors: Relationship with lipid metabolism and insulin sensitivity. Diabetes 2004, 53 (Suppl. 1), S43–S50. [Google Scholar] [CrossRef] [PubMed]
- Kliewer, S.A.; Sundseth, S.S.; Jones, S.A.; Brown, P.J.; Wisely, G.B.; Koble, C.S.; Devchand, P.; Wahli, W.; Willson, T.M.; Lenhard, J.M.; et al. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors α and γ. Proc. Natl. Acad. Sci. USA 1997, 94, 4318–4323. [Google Scholar] [CrossRef] [PubMed]
- Itoh, T.; Fairall, L.; Amin, K.; Inaba, Y.; Szanto, A.; Balint, B.L.; Nagy, L.; Yamamoto, K.; Schwabe, J.W. Structural basis for the activation of PPARγ by oxidized fatty acids. Nat. Struct. Mol. Biol. 2008, 15, 924–931. [Google Scholar] [CrossRef] [PubMed]
- Nolte, R.T.; Wisely, G.B.; Westin, S.; Cobb, J.E.; Lambert, M.H.; Kurokawa, R.; Rosenfeld, M.G.; Willson, T.M.; Glass, C.K.; Milburn, M.V. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-γ. Nature 1998, 395, 137–143. [Google Scholar] [PubMed]
- Auboeuf, D.; Rieusset, J.; Fajas, L.; Vallier, P.; Frering, V.; Riou, J.P.; Staels, B.; Auwerx, J.; Laville, M.; Vidal, H. Tissue Distribution and Quantification of the Expression of mRNAs of Peroxisome Proliferator-Activated Receptors and Liver X Receptor-α in Humans: No Alteration in Adipose Tissue of Obese and NIDDM Patients. Diabetes 1997, 46, 1319–1327. [Google Scholar] [CrossRef] [PubMed]
- Chawla, A.; Schwarz, E.J.; Dimaculangan, D.D.; Lazar, M.A. Peroxisome proliferator-activated receptor (PPAR) gamma: Adipose-predominant expression and induction early in adipocyte differentiation. Endocrinology 1994, 135, 798–800. [Google Scholar] [CrossRef] [PubMed]
- Dalen, K.T.; Schoonjans, K.; Ulven, S.M.; Weedon-Fekjaer, M.S.; Bentzen, T.G.; Koutnikova, H.; Auwerx, J.; Nebbl, H.I. Adipose tissue expression of the lipid droplet-associating proteins S3-12 and perilipin is controlled by peroxisome proliferator-activated receptor-γ. Diabetes 2004, 53, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Tontonoz, P.; Hu, E.; Spiegelman, B.M. Stimulation of adipogenesis in fibroblasts by PPARγ2, a lipid-activated transcription factor. Cell 1994, 79, 1147–1156. [Google Scholar] [CrossRef]
- He, W.; Barak, Y.; Hevener, A.; Olson, P.; Liao, D.; Le, J.; Nelson, M.; Ong, E.; Olefsky, J.M.; Evans, R.M. Adipose-specific peroxisome proliferator-activated receptor γ knockout causes insulin resistance in fat and liver but not in muscle. Proc. Natl. Acad. Sci. USA 2003, 100, 15712–15717. [Google Scholar] [CrossRef] [PubMed]
- Barrero, M.J.; Camarero, N.; Marrero, P.F.; Haro, D. Control of human carnitine palmitoyltransferase II gene transcription by peroxisome proliferator-activated receptor through a partially conserved peroxisome proliferator-responsive element. Biochem. J. 2003, 369, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Mandard, S.; Muller, M.; Kersten, S. Peroxisome proliferator-activated receptor alpha target genes. Cell. Mol. Life Sci. 2004, 61, 393–416. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sztalryd, C. Oxidative tissue: Perilipin 5 links storage with the furnace. Trends Endocrinol. Metab. 2011, 22, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-α: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Lagathu, C.; Yvan-Charvet, L.; Bastard, J.P.; Maachi, M.; Quignard-Boulange, A.; Capeau, J.; Caron, M. Long-term treatment with interleukin-1β induces insulin resistance in murine and human adipocytes. Diabetologia 2006, 49, 2162–2173. [Google Scholar] [CrossRef] [PubMed]
- Steppan, C.M.; Bailey, S.T.; Bhat, S.; Brown, E.J.; Banerjee, R.R.; Wright, C.M.; Patel, H.R.; Ahima, R.S.; Lazar, M.A. The hormone resistin links obesity to diabetes. Nature 2001, 409, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Bluher, M. Adipose tissue inflammation: A cause or consequence of obesity-related insulin resistance? Clin. Sci. 2016, 130, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Moran-Salvador, E.; Lopez-Parra, M.; Garcia-Alonso, V.; Titos, E.; Martinez-Clemente, M.; Gonzalez-Periz, A.; Lopez-Vicario, C.; Barak, Y.; Arroyo, V.; Claria, J. Role for ppargamma in obesity-induced hepatic steatosis as determined by hepatocyte- and macrophage-specific conditional knockouts. FASEB J. 2011, 25, 2538–2550. [Google Scholar] [CrossRef] [PubMed]
- Jones, D. Potential remains for PPAR-targeted drugs. Nat. Rev. Drug Discov. 2010, 9, 668–669. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, J.M.; Moore, L.B.; Smith-Oliver, T.A.; Wilkison, W.O.; Willson, T.M.; Kliewer, S.A. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor γ (PPARγ). J. Biol. Chem. 1995, 270, 12953–12956. [Google Scholar] [CrossRef] [PubMed]
- Schoonjans, K.; Watanabe, M.; Suzuki, H.; Mahfoudi, A.; Krey, G.; Wahli, W.; Grimaldi, P.; Staels, B.; Yamamoto, T.; Auwerx, J. Induction of the Acyl-Coenzyme A Synthetase Gene by Fibrates and Fatty Acids is Mediated by a Peroxisome Proliferator Response Element in the C Promoter. J. Biol. Chem. 1995, 270, 19269–19276. [Google Scholar] [PubMed]
- Nissen, S.E.; Wolski, K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N. Engl. J. Med. 2007, 356, 2457–2471. [Google Scholar] [CrossRef] [PubMed]
- Jonas, D.; Van Scoyoc, E.; Gerrald, K.; Wines, R.; Amick, H.; Triplette, M.; Runge, T. Drug Class Review: Newer Diabetes Medications, TZDs, and Combinations: Final Original Report; Oregon Health & Science University: Portland, OR, USA, 2011. [Google Scholar]
- Amato, A.A.; Rajagopalan, S.; Lin, J.Z.; Carvalho, B.M.; Figueira, A.C.; Lu, J.; Ayers, S.D.; Mottin, M.; Silveira, R.L.; Souza, P.C.; et al. GQ-16, a novel peroxisome proliferator-activated receptor γ (PPARγ) ligand, promotes insulin sensitization without weight gain. J. Biol. Chem. 2012, 287, 28169–28179. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Banks, A.S.; Kamenecka, T.M.; Busby, S.A.; Chalmers, M.J.; Kumar, N.; Kuruvilla, D.S.; Shin, Y.; He, Y.; Bruning, J.B.; et al. Antidiabetic actions of a non-agonist PPARγ ligand blocking CDK5-mediated phosphorylation. Nature 2011, 477, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.K.; Zhuang, Y.; Wahli, W. Synthetic and natural peroxisome proliferator-activated receptor (PPAR) agonists as candidates for the therapy of the metabolic syndrome. Expert Opin. Ther. Targets 2017, 21, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Fievet, C.; Fruchart, J.C.; Staels, B. PPARα and PPARγ dual agonists for the treatment of type 2 diabetes and the metabolic syndrome. Curr. Opin. Pharmacol. 2006, 6, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Hasle, G.R.; Syvertsen, E.E. Marine diatoms. In Identifying Marine Phytoplankton; Academic Press Ltd.: Cambridge, MA, USA, 1997; pp. 5–385. [Google Scholar]
- Toyoda, K.; Nagasaki, K.; Williams, D.M.; Tomaru, Y. PCR-RFLP analysis for species-level distinction of the genus chaetoceros Ehrenberg (Bacillariophyceae). Hiyoshi Rev. Nat. Sci. 2011, 50, 21–29. [Google Scholar]
- Huseby, S.; Degerlund, M.; Eriksen, G.K.; Ingebrigtsen, R.A.; Eilertsen, H.C.; Hansen, E. Chemical diversity as a function of temperature in six northern diatom species. Mar. Drugs 2013, 11, 4232–4245. [Google Scholar] [CrossRef] [PubMed]
- Ingebrigtsen, R.; Hansen, E.; Andersen, J.; Eilertsen, H. Light and temperature effects on bioactivity in diatoms. J. Appl. Phycol. 2015, 28, 939–950. [Google Scholar] [CrossRef] [PubMed]
- D’Ippolito, G.; Cutignano, A.; Briante, R.; Febbraio, F.; Cimino, G.; Fontana, A. New C16 fatty-acid-based oxylipin pathway in the marine diatom Thalassiosira rotula. Org. Biomol. Chem. 2005, 3, 4065–4070. [Google Scholar] [CrossRef] [PubMed]
- Dang, H.T.; Lee, H.J.; Yoo, E.S.; Shinde, P.B.; Lee, Y.M.; Hong, J.; Kim, D.K.; Jung, J.H. Anti-inflammatory constituents of the red alga gracilaria verrucosa and their synthetic analogues. J. Nat. Prod. 2008, 71, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, H.E.; Kotlyar, V.; Nudelman, A. NMR chemical shifts of common laboratory solvents as trace impurities. J. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef] [PubMed]
- Kenner, G.W.; Stenhagen, E. Location of double bonds by mass spectrometry. Acta Chem. Scand. 1964, 18, 1551–1552. [Google Scholar] [CrossRef]
- Jain, M.R.; Giri, S.R.; Trivedi, C.; Bhoi, B.; Rath, A.; Vanage, G.; Vyas, P.; Ranvir, R.; Patel, P.R. Saroglitazar, a novel PPARα/γagonist with predominant PPARα activity, shows lipid-lowering and insulin-sensitizing effects in preclinical models. Pharmacol. Res. Perspect. 2015, 3, e00136. [Google Scholar] [CrossRef] [PubMed]
- Yore, M.M.; Syed, I.; Moraes-Vieira, P.M.; Zhang, T.; Herman, M.A.; Homan, E.A.; Patel, R.T.; Lee, J.; Chen, S.; Peroni, O.D.; et al. Discovery of a class of endogenous mammalian lipids with anti-diabetic and anti-inflammatory effects. Cell 2014, 159, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Svenson, J. Mabcent: Arctic marine bioprospecting in Norway. Phytochem. Rev. 2013, 12, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Weedon-Fekjaer, M.S.; Dalen, K.T.; Solaas, K.; Staff, A.C.; Duttaroy, A.K.; Nebb, H.I. Activation of LXR increases acyl-CoA synthetase activity through direct regulation of ACSL3 in human placental trophoblast cells. J. Lipid Res. 2010, 51, 1886–1896. [Google Scholar] [CrossRef] [PubMed]
- Napal, L.; Marrero, P.F.; Haro, D. An intronic peroxisome proliferator-activated receptor-binding sequence mediates fatty acid induction of the human carnitine palmitoyltransferase 1A. J. Mol. Biol. 2005, 354, 751–759. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 a | 2 a | ||
|---|---|---|---|---|
| 13C | 1H | 13C | 1H | |
| 1 | 175.0, t | - | 175.2, t | - |
| 2 | 34.2, t | 2.27, t, (7.4) | 34.3, t | 2.27, t, (7.5) |
| 3 | 25.6, t | 1.57, m | 25.6, t | 1.57, m |
| 4 | 29.5, t | 1.34, m | 29.8, t | 1.31, m, overlapped |
| 5 | 28.7, t | 1.48, m | 29.8, t | 1.31, m, overlapped |
| 6 | 33.0, t | 2.22, ddt, (7.5, 7.0, 1.5) | 29.8, t | 1.31, m, overlapped |
| 7 | 148.2, d | 6.84, dt, (15.9, 7.0) | 25.1, t | 1.55, m |
| 8 | 131.1, d | 131.1, dt, (15.9, 1.5) | 40.4, t | 2.54, t, (7.4) |
| 9 | 201.4, s | - | 201.4, s | - |
| 10 | 40.4, t | 2.54, t, (7.4) | 131.2, d | 6.08, dt, (15.9, 1.5) |
| 11 | 25.1, t | 1.55, m | 148.1, d | 6.85, dt, (15.9, 7.0) |
| 12 | 29.8, t | 1.30, m | 33.0, t | 2.22, ddt. (7.5, 7.0, 1.5) |
| 13 | 30.4, t | 1.29, m | 28.6, t | 1.48, tt, (7.5, 7.5) |
| 14 | 32.5, t | 1.30, m | 32.2, t | 1.33, m |
| 15 | 23.3, t | 1.32, m | 23.3, t | 1.34, m |
| 16 | 14.4, q | 0.90, t, (7.0) | 14.4, q | 0.91, t, (7.0) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moldes-Anaya, A.; Sæther, T.; Uhlig, S.; Nebb, H.I.; Larsen, T.; Eilertsen, H.C.; Paulsen, S.M. Two Isomeric C16 Oxo-Fatty Acids from the Diatom Chaetoceros karianus Show Dual Agonist Activity towards Human Peroxisome Proliferator-Activated Receptors (PPARs) α/γ. Mar. Drugs 2017, 15, 148. https://doi.org/10.3390/md15060148
Moldes-Anaya A, Sæther T, Uhlig S, Nebb HI, Larsen T, Eilertsen HC, Paulsen SM. Two Isomeric C16 Oxo-Fatty Acids from the Diatom Chaetoceros karianus Show Dual Agonist Activity towards Human Peroxisome Proliferator-Activated Receptors (PPARs) α/γ. Marine Drugs. 2017; 15(6):148. https://doi.org/10.3390/md15060148
Chicago/Turabian StyleMoldes-Anaya, Angel, Thomas Sæther, Silvio Uhlig, Hilde I. Nebb, Terje Larsen, Hans C. Eilertsen, and Steinar M. Paulsen. 2017. "Two Isomeric C16 Oxo-Fatty Acids from the Diatom Chaetoceros karianus Show Dual Agonist Activity towards Human Peroxisome Proliferator-Activated Receptors (PPARs) α/γ" Marine Drugs 15, no. 6: 148. https://doi.org/10.3390/md15060148
APA StyleMoldes-Anaya, A., Sæther, T., Uhlig, S., Nebb, H. I., Larsen, T., Eilertsen, H. C., & Paulsen, S. M. (2017). Two Isomeric C16 Oxo-Fatty Acids from the Diatom Chaetoceros karianus Show Dual Agonist Activity towards Human Peroxisome Proliferator-Activated Receptors (PPARs) α/γ. Marine Drugs, 15(6), 148. https://doi.org/10.3390/md15060148