2. Results and Discussion
The structures of known polyketides (
2–
9) were determined by the analysis of their NMR, MS, and specific rotation data, along with comparisons of these data with those of the previously-published values in the literature. They were identified as curvulone B (
2) [
10], curvularin (
3) [
11], (11
R,15
S)-11-hydroxycurvularin (
4) [
12], (11
S,15
S)-11-hydroxycurvularin (
5) [
12], (11
R,15
S)-11-methoxycurvularin (
6) [
13], (11
S,15
S)-11-methoxycurvularin (
7) [
13], (10
E,15
S)-10,11-dehydrocurvularin (
8) [
12], and (10
Z,15
S)-10,11-dehydrocurvularin (
9) [
14] (
Figure 1).
Among the curvularin-type fungal polyketides with a 12-membered lactone ring, the absolute configuration of the C-15 chiral center adjacent to the lactone oxygen is known to be variable depending on the reduction pathway by β-ketoacyl reductase in fungal macrolide biosynthesis. Until now, most of the C-15 chiral centers in this class have been described to have 15
S configuration with the exception of curvalarins isolated from two marine fungal strains belonging to the genus
Curvularia sp. [
10,
12]. On the basis of synthetic studies and analysis of chiroptical data of several curvularins, the absolute configuration 15
S has been correlated with the negative specific rotations [
12]. Therefore, the absolute configurations of the isolated metabolites (
2–
9) were considered to belong to the 15
S series of curvularin-type metabolites based on the observation of negative signs of their specific rotation values (
Supplementary Materials, Table S1). It is noteworthy that (10
Z,15
S*)-10,11-dehyrocurvularin that has the same planar structure as that of
9 was previously isolated from a hybrid strain derived from
Penicillium sp. with a positive specific rotation {
= +7.3 (c 0.78, EtOH)} [
14], whereas
9 has a negative specific rotation
= −19.9 (
c = 0.15, EtOH). Considering the relationship between the sign of the specific rotation and the absolute configuration at C-15, it was suggested that
9 is the first report of naturally-occurring (10
Z,15
S)-10,11-dehyrocurvularin, and the previously-reported (10
Z,15
S*)-10,11-dehyrocurvularin would have been a enantiomer of
9.
Compound
1 was isolated as a colorless oil with an optical rotation of +13.6 (
c = 0.22, EtOH). Its molecular formula was determined as C
16H
20O
6 based on the observation of a sodium adduct ion at
m/
z 331.1158 [M + Na]
+ and a protonated molecular ion at
m/
z 309.1336 [M + H]
+ in its positive ion mode of HRESIMS. In the
1H-NMR spectrum of
1 recorded in CDCl
3, the broad singlet signals for two aromatic protons and two oxymethine proton signals, and a signal for a methyl group that is coupled to the one of oxymethine protons were clearly observed. Analysis of the
13C-NMR and DEPT data of
1 suggested the presence of 16 carbons, including two sp
3 oxymethines, two sp
2 methine carbons, two carbonyl carbons, and four sp
2 quaternary carbons. The signals for the four remaining methylene carbons and a methyl carbon were also observed in the
13C-NMR spectrum of
1. Based on these structural features, including the molecular formula, scrutiny of the literature revealed that
13C-NMR data of
1 closely matched with the previously-published
13C-NMR chemical shift values for curvulone B [
10]. Although
1H-NMR data of
1 recorded in CDCl
3 contained some overlapped signals hampering direct comparison of the data with that of curvulone B, the
1H-NMR spectrum of
1 recorded in acetone-
d6 (
Table 1) pointed out that the chemical shifts of
1 were in close agreement with those of
2, except for missing the methoxy signal in curvulone B.
Therefore, compound
1 was suggested to be a new analogue of curvulone B, having the carboxylic acid functionality instead of the methyl ester group in curvulone B. The relative configuration of the tetrahydropyran ring in
1 was deduced from the NOESY spectrum (
Supplementary Materials, Figure S5). NOESY cross-peaks observed between H-11 and H-15 corroborated the
cis-orientation of the substituents at C-11 and C-15. Thus, the relative configuration at C-11 and C-15 was proposed to be the same as that in curvulone B, whose absolute configuration was reported as 11
R and 15
R [
10]. The specific rotation of
1 showed the opposite sign although the reported specific rotation (−15.24,
c = 0.18, EtOH) of curvulone B [
10] was in agreement with that of
1 (+13.6,
c = 0.22, EtOH) regarding its magnitude. Therefore, the absolute configuration of
1 was assigned as 11
S and 15
S. As a result,
1 was determined as a new analogue of curvularin-type macrolides, and named curvulone C.
All isolated compounds (
1–
9) were evaluated for their anti-inflammatory effects. In the treatment of inflammatory disorders, the pharmacological manipulation of Nitric oxide (NO) and prostaglandin E
2 (PGE
2) overproduction has been suggested [
15,
16,
17]. NO is a small molecule which is an intracellular mediator produced in various immune cells. It plays a pivotal role in the physiological and pathological condition of inflammatory symptoms [
17]. PGE
2 can also regulate the immune and inflammatory responses [
15]. In this study, RAW264.7 macrophages were pre-treated for 3 h in the medium containing non-toxic concentrations (
Table 2) of each compound (
1–
9), and then LPS (1 μg/mL) was treated for 24 h. According to the LPS stimulation of RAW264.7 macrophages, production of NO and PGE
2 was increased, and the effects of all compounds on the production level of NO and PGE
2 were evaluated by the Griess reaction and a PGE
2 kit, respectively. As a result, compounds
3–
9 dose-dependently inhibited the LPS-induced production of NO and PGE
2, and their IC
50 values are shown in
Table 2. Based on the comparison of IC
50 values for compounds
1–
9, it was evident that curvularin-type metabolites exhibit structure-dependent anti-inflammatory properties. Noticeably, the presence of a 12-membered macrolactone ring was essential for their anti-inflammatory activity as indicated by the observation that
1 and
2, which are supposed to be derived from the cleavage of the lactone bond in the macrolactone ring, appeared to lose the inhibitory activity. To evaluate the impact of phenol and resorcinol groups in the curvularin-type metabolites for the anti-inflammatory activity, 5-
O-methyl and 5,7,-di-
O-methyl derivatives of curvularin (
3a and
3b, respectively), as well as 5-
O-acetyl and 5,7,-di-
O-acetyl derivatives of curvularin (
3c and
3d, respectively), were prepared, and the structures of
3a–
3d were identified by comparison the
1H and
13C-NMR spectroscopic data with those of curvularin (
3), and confirmed by their HRESIMS. These compounds exhibited the similar chemical shifts, except for the additional appearances of the methoxy signals and acetyl signals. The position of the methoxy group in
3a was confirmed by HMBC correlation between methoxy signal at δ
H 3.77 (3H, s, 5-OCH
3) and δ
C 161.8 (C-5). The NOESY cross-peaks between acetyl proton (5-OCH
3) with H-2, H-4, and H-16 of
3c were used to identify the position of the acetyl group in
3c. According to the anti-inflammatory effects of
3a–
3d, as shown in
Table 2, the blocking of the phenol and resorcinol groups appeared to lead to the significant decrease in the anti-inflammatory activity, implying the crucial role of the intact phenols of curvularin-type metabolites for their anti-inflammatory activity. However, this observation is inconsistent with the previous study reporting that
3d showed higher inhibitory effect on the cytokine-mediated induction of the iNOS promotor than that of (
S)-curvularin (
3) in the assay of human iNOS promotor activity in human alveolar epithelial A549/8 cells [
11]. Different cell types employed in both studies may have resulted in these inconsistent biological effects. In addition, the present study showed that the modification of the aliphatic part around the macrolactone ring could influence the anti-inflammatory activity of curvularin-type metabolites as shown that unsaturated (
S)-curvularin-derivatives (
8 and
9) and 11-methoxy and 11-hydroxy derivatives (
4–
7) possess stronger anti-inflammatory activities than that of
3. Further systematic investigations are needed to clarify the structure activity relationship associated with the modification of the aliphatic part of the molecule.
Among the curvularin-type metabolites encountered in this study, compound
8 was identified as the most active anti-inflammatory metabolite based on its IC
50 values (
Table 2). Therefore, we further tested whether inhibitory effects of
8 against NO and PGE
2 productions are correlated with the protein expression of pro-inflammatory enzymes (i.e., iNOS and COX-2, respectively), which are known to catalyze the production of NO and PGEs in LPS-stimulated cells. When RAW264.7 macrophages were pretreated with the indicated concentrations of
8 for 3 h before stimulation with LPS for 24 h, the presence of
8 led to the attenuation of the excessive protein expression of iNOS and COX-2 in a dose-dependent manner (
Figure 2a). Upon the stimulation by LPS, macrophages can also trigger the production of pro-inflammatory cytokines such as TNF-α, and ILs [
18]. The overproduction of these cytokines contributes to the pathogenesis of inflammatory diseases [
19,
20,
21]. Thus, we further evaluated the effects of
8 on the mRNA expression of pro-inflammatory cytokines in the LPS-induced cells. The cells were pre-treated with indicated concentration for 3 h, followed by LPS stimulation (1 μg/mL) for 6 h. The mRNA expression of pro-inflammatory cytokines was determined by RT-qPCR. As demonstrated in
Figure 2b–d, compound
8 markedly suppressed the mRNA expression of IL-1β, IL-6, and TNF-α in a dose-dependent manner. These results indicated that
8 attenuated the gene expression of pro-inflammatory cytokines at the transcriptional level.
It has been reported that many cellular signaling pathways and transcription factors are related to the expression of pro-inflammatory genes and enzymes in immune cells [
18]. Nuclear factor-κB (NF-κB) is one of the important transcriptional factors involved in inflammation-related disorders. It is known to modulate inflammatory genes and the expression of pro-inflammatory mediators, such as iNOS and COX-2 [
22]. In normal cells, NF-κB consists of inactive subunits of p50 and p65 bound to the inhibitor of NF-κB (IκB-α) [
23]. The NF-κB signaling pathway can be activated by LPS or other stimuli, which then phosphorylates IκB-α, leading to degradation and subsequent translocation of NF-κB into the nucleus. The data from our study indicated that pre-treatment of
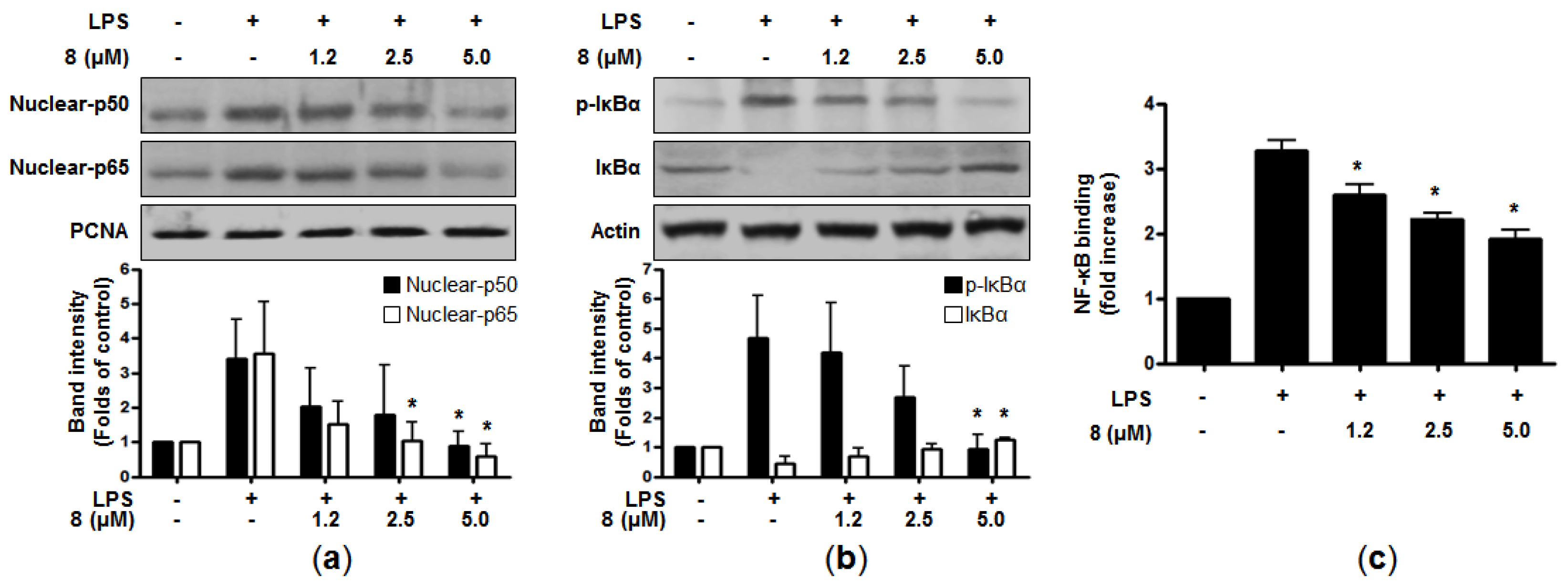
8 clearly suppressed the nuclear translocation of p50 and p65 in a dose-dependent manner (
Figure 3a). In addition, when the cells were treated with LPS alone, the phosphorylation level of IκB-α was increased, whereas
8 attenuated this phosphorylation of IκB-α. Compound
8 also blocked the degradation of IκB-α in a concentration-dependent manner (
Figure 3b). In line with these, LPS-induced DNA binding activity of NF-κB was declined in the nuclear extracts of the cells co-treated with
8 (
Figure 3c). Taken together, it was suggested that
8 could suppress the induction of pro-inflammatory mediators and cytokines through the downregulation of the NF-κB signaling pathway.
Mitogen-activated protein kinase (MAPK) pathways are also known to be involved in the expression of pro-inflammatory cytokines in macrophages [
24]. Thus, the effects of
8 on the LPS-induced phosphorylation of MAPK were examined. Although the treatment of LPS for 30 min with the cells caused the phosphorylation of p38, c-Jun N-terminal kinase (JNK), and extracellular signal-regulated kinase (ERK), our data indicated that
8 did not suppress these phosphorylations (
Figure 4a–c). Consequently, the anti-inflammatory effects of
8 did not seem to be mediated through MAPK signaling pathways, and further study is needed in order to elucidate the specific target of
8 involved in its anti-inflammatory activity.
Curvularin-type metabolites are macrocyclic lactones produced by several fungi of the genera
Curvularia,
Penicillium, and
Alternaria [
11,
25]. They have been reported to possess a variety of biological activities. For example, curvularin (
3) inhibited inducible transcription and synthesis of iNOS and COX-2 through blocking the activation of the transcription factor signal transducer and activator of transcription (STAT)-1α in human alveolar epithelial A549/8 cells [
11,
26]. The suppressive effect of
3 against the expression of various pro-inflammatory cytokines and chemokines was also observed in the mice model of chronic induced arthritis [
27]. Recently, cytotoxicity of compounds
6–
8 against a small panel of human tumor cell lines has been observed, and this was partially linked to their suppressive effect on the activation of the TNFα-induced NF-κB pathway [
28]. It was also reported that
8 acts as a covalent inhibitor of p97, interfering with its ATPase activity [
29]. Furthermore, the antibacterial activity inhibiting the growth of
E. coli of 11-hydroxylcurvularin (
4–
5) was also demonstrated [
30]. In the present study, nine curvularin-type metabolites, including a new curvularin derivative (
1), were isolated from a marine-derived fungal strain
Penicillium sp. SF-5859. From the evaluation of anti-inflammatory effects of these metabolites in LPS-stimulated RAW 264.7 cells that has not been reported previously, it was suggested that structural variation of curvularin-type metabolites could lead to the development of a promising agent for the treatment of inflammation-related diseases. In addition, this study demonstrated that the most active metabolite, (10
E,15
S)-10,11-dehydrocurvularin (
8) can significantly suppress the induction of pro-inflammatory mediators and cytokines via inhibition of the NF-κB signaling pathway, and could play a role as a lead compound in the search for anti-inflammatory drugs.
3. Materials and Methods
3.1. General Experimental Procedures
Optical rotations were recorded using a Jasco P-2000 digital polarimeter (Jasco, Easton, PA, USA). NMR spectra (1D and 2D) were recorded in a JEOL JNM ECP-400 spectrometer (400 MHz for 1H and 100 MHz for 13C, JEOL Ltd., Akishima, Japan), and chemical shifts were referenced relative to the corresponding residual solvents signals (δH 2.05/δC 29.8 for acetone-d6, δH 7.26/δC 77.2 for CDCl3, and δH 3.30/δC 49.0 for CD3OD). HMQC and HMBC experiments were optimized for 1JCH = 140 Hz and nJCH = 8 Hz, respectively. HRESIMS data were obtained using an ESI Q-TOF MS/MS system (AB SCIEX Triple, SCIEX, Framingham, MA, USA). Flash column chromatography was performed on silica gel (Kieselgel 60, 70–230 mesh and 230–400 mesh, Merck, Kenilworth, NJ, USA) and YMC octadecyl-functionalized silica gel (C18, YMC CO., Kyoto, Japan). YMC semiprep-C18 column (20 × 150 mm; 4 μm particle size; 80 Å pore size, 5 mL/min, YMC CO., Kyoto, Japan) and Shodex Ohpak SB 802.5 (8 × 300 mm; 6 μm particle size; 80 Å pore size, 0.6 mL/min, Showa Denko K.K., Tokyo, Japan) were used for HPLC (YoungLin, Anyang, Korea) separations. TLC was performed on Kieselgel 60 F254 (Merck, Kenilworth, NJ, USA) or reversed-phase (RP)-18 F254s (Merck, Kenilworth, NJ, USA) plates. Spots were visualized by spraying with 10% aqueous H2SO4 solution, followed by heating. All compounds were detected by UV absorption at 210 and 254 nm.
RPMI1640, fetal bovine serum (FBS), and other tissue culture reagents were purchased from Gibco BRL Co. (Grand Island, NY, USA). All other chemicals were obtained from Sigma-Aldrich Co. (St. Louis, MO, USA). Primary antibodies (COX-2: sc-1745; iNOS: sc-650; IκB-α: sc-371; p-IκB-α: sc-8404; p50: sc-7178; p65: sc-8008, Santa Cruz Biotechnology, Dallas, TX, USA, p-ERK: #9101; ERK: #9102; p-JNK: #9251; JNK: #9252S; p-p38: #9211; p38: 9212S, Cell Signaling Technology, Danvers, MA, USA) and secondary antibodies (mouse: ap124p; goat: ap106p; rabbit: ap132p, Millipore, Billerica MA, USA). Enzyme-linked immunosorbent assay (ELISA) kits for PGE2 were purchased from R and D Systems, Inc. (Minneapolis, MN, USA).
3.2. Fungal Material and Fermentation
Penicillium sp. SF-5859 was isolated from an unidentified sponge that was collected using a dredge in the Ross Sea (76 06.25635 S 169 12.6752 E) on 8 February 2011. The surface of the sponge was sterilized, and one gram of the sample was ground with a mortar and pestle, followed by mixing with sterile seawater (10 mL). A portion (0.1 mL) of the sample was processed utilizing the spread plate method in potato dextrose agar (PDA) medium containing sterile seawater collected in the Busan area. The plate was incubated at 25 °C for 14 days. After subculturing the isolates several times, the final pure cultures were selected and preserved at −70 °C. The fungal strain SF-5859 was identified based upon the analysis of their ribosomal RNA (rRNA) sequences. A GenBank search with the 28S rRNA gene of SF-5859 (GenBank accession number KF745792) indicated Penicillium chrysogenum (FJ890400), P. steckii (HM469415), P. paxilli (FJ890408), and P. citrinum (JN938950), as the closest match showing sequence identities of 99.48%, 98.69%, 98.69%, and 98.43%, respectively. Therefore, the marine-derived fungal strain SF-5859 was characterized as Penicillium sp., but could not be definitively identified to a specific species.
3.3. Extraction and Isolation
The fungal strain Penicillium sp. SF-5859 was cultured on ten Fernbach-style flasks each containing 100 g of semi-solid vermiculite and 400 mL of PDB with 3% (w/v) NaCl. The flasks were individually inoculated with 2 mL seed cultures of the fungal strain and incubated at 25 °C for 14 days then extracted with EtOAc (4 L per one flask). The combined extract solutions were filtered through filter paper and evaporated to dryness resulting in a crude extract SF5859 (2.2 g). The crude extract was fractionated on reversed phase (RP) C18 flash column chromatography (5 × 30 cm), eluting with a stepwise gradient of 20, 40, 60, 80, and 100% (v/v) MeOH in H2O (500 mL each) to give six fractions, SF5859-1 to SF5859-6, consecutively. The fraction SF5859-3 was applied to a chromatographic column packed with silica gel (2 × 30 cm). The column was subsequently eluted with gradients of CH2Cl2 in EtOAc (8/1 v/v, 200 mL) and (4/1 v/v, 150 mL), to yield 8 (30.0 mg) and seven other fractions, SF5859-31 to SF5859-38, which were pooled based on TLC analysis. The fourth fraction, SF5859-34, was further purified by semi-preparative reverse-phase HPLC eluting with a gradient of MeOH (60% to 80% in 20 min) in water (0.1% HCOOH) to afford 9 (1.5 mg, tR = 13.5 min) and 2 (0.5 mg, tR = 19 min). Similarly, the sixth fraction SF5859-36 was subjected to semi-preparative RP HPLC column (50–80% MeOH in H2O (0.1% HCOOH) over 30 min), giving two sub-fractions, SF5859-361 and SF5859-362, and 6 (3.5 mg, tR = 46 min) was isolated from the sub-fraction SF5859-362 by performing on Shodex Ohpak SB 802.5 HPLC column (30–75% MeOH in H2O over 50 min). The seventh fraction SF5859-37 was separated into 5 (1.5 mg, tR = 28 min) and two other sub-fractions by using semi-preparative RP HPLC column (30–60% MeOH in H2O (0.1% HCOOH) in 30 min). Among these sub-fractions, SF5859-373 was further separated on semi-preparative RP HPLC column (40–65% MeOH in H2O (0.1% HCOOH) in 25 min), giving compound 4 (2.5 mg, tR = 20.5 min). The eighth fraction SF5859-38 was separated firstly by C18 chromatographic column (1.5 × 20 cm), eluting with MeOH in H2O (1/3 v/v), and the sub-fraction SF5859-383 was further purified by semi-preparative RP HPLC column (30–55% CH3CN in H2O (0.1% HCOOH) over 25 min), yielding 1 (1.3 mg, tR = 22 min). The fraction SF5859-4 was chromatographed on silica gel column (3 × 30 cm), eluting with CH2Cl2 in EtOAc (7/1 v/v). From this, the major metabolite 3 (450.0 mg) was obtained, along with four another fractions. The fifth fraction SF5859-45 was subjected to a final purification on semi-preparative RP HPLC column (60–75% MeOH in H2O (0.1% HCOOH) over 15 min) to afford 7 (2 mg, tR = 13 min).
Curvulone C (
1): colorless oil;
= +13.6 (c 0.22, EtOH);
1H-NMR data (CDCl
3, acetone-
d6, 400 MHz) and
13C-NMR data (CDCl
3, 100 MHz), see
Table 1; HRESIMS
m/
z 309.1336 [M + H]
+ (calcd. for C
16H
21O
6, 309.1338), 331.1158 [M + Na]
+ (calcd. for C
16H
20O
6Na, 331.1158).
3.4. Preparation of Compounds 3a and 3b
N,N-diisopropylethylamine (50 μL) was added to a solution of curvularin (3, 15 mg) in 1 mL of MeOH, followed by the addition of TMSCHN2 (110 μL, 2.2 M in n-hexane). The reaction mixture was stirred for 15 h at room temperature. The solution was then concentrated in vacuo and extracted with EtOAc and H2O prior to the evaporation of organic phase. Subsequently, the residual material was subjected to semi-preparative RP HPLC eluting with the gradient of methanol in water (0.1% HCOOH) from 70% to 86% over 18 min to afford methylated products 3a (4 mg, tR = 14 min) and 3b (6 mg, tR = 16 min).
5-
O-methylcurvularin (
3a): white amorphous powder;
1H-NMR (CD
3OD
, 400 MHz) and
13C-NMR data (CD
3OD, 100 MHz),
Supplementary Materials Figures S7 and S8; HRESIMS
m/
z 309.1667 [M + H]
+ (calcd. for C
17H
21D
2O
5 due to deuterium exchange, 309.1671).
5,7-Di-
O-methylcurvularin (
3b): white amorphous powder;
1H-NMR (CD
3OD, 400 MHz) and
13C-NMR data (CD
3OD, 100 MHz),
Supplementary Materials, Figures S13 and S14; HRESIMS
m/
z 321.1706 [M + H]
+ (calcd. for C
18H
25O
5, 321.1702).
3.5. Preparation of Compounds 3c and 3d
Curvularin (3, 10 mg) was dissolved in 600 μL acetone, followed by the addition of acetic anhydride (600 μL). The reaction was started with adding a catalytic amount of N,N-dimethylpyridin-4-amine. The reaction mixture was stirred for 3 h at room temperature. The resulting solution was dried in vacuo then partitioned with EtOAc and H2O prior to the evaporation of the organic phase. Thereafter, the residual material was subjected to semi-preparative RP HPLC eluting with the gradient of methanol in water (0.1% HCOOH) from 62% to 80% over 19 min to afford the acetylated products 3c (2 mg, tR = 14 min) and 3d (3.5 mg, tR = 16 min).
5-
O-acetylcurvularin (
3c): white amorphous powder;
1H-NMR (acetone-
d6, 400 MHz),
Supplementary Materials, Figure S16; HRESIMS
m/
z 357.1336 [M + Na]
+ (calcd. for C
18H
22NaO
6, 357.1314).
5,7-Di-
O-acetylcurvularin (
3d): white amorphous powder;
1H-NMR (acetone-
d6, 400 MHz),
Supplementary Materials, Figure S19; HRESIMS
m/
z 399.1441 [M + Na]
+ (calcd. for C
20H
24NaO
7, 399.1420).
3.6. Cell Culture and Cytotoxic Assay
RAW264.7 macrophages were maintained at a density of 5 × 105 cells/mL in RPMI1640 medium supplemented with 10% heat-inactivated FBS, penicillin G (100 units/mL), streptomycin (100 mg/mL), and L-glutamine (2 mM), and were incubated at 37 °C in a humidified atmosphere containing 5% CO2. For determination of cell viability, cells (1 × 105 cells/well in 96-well plates) were incubated with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) at a final concentration of 0.5 mg/mL for 3 h, and the formazan formed was dissolved in acidic 2-propanol. The optical density was measured at 540 nm with a microplate reader (BioRad, Hercules, CA, USA). The optical density of the formazan formed in control (untreated) cells was considered to represent 100% viability.
3.7. Determination of Nitrite and PGE2
As an indicator of NO production in RAW264.7 macrophages, production of nitrite, a stable end-product of NO oxidation, was estimated. Briefly, the concentration of nitrite in the conditioned media was determined by a method based on the Griess reaction [
31], and the details of the assay were described previously [
32].
3.8. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Total RNA was isolated from RAW264.7 macrophages using Trizol (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol and quantified spectrophotometrically at 260 nm. Total RNA (1 μg) was reverse-transcribed using the High Capacity RNA-to-cDNA kit from Applied Biosystems (Carlsbad, CA, USA). The cDNA was amplified using the SYBR Premix Ex Taq kit from TaKaRa Bio Inc. (Shiga, Japan) and a StepOnePlus Real-Time PCR system from Applied Biosystems. qRT-PCR was performed in a 20 μL total volume of 0.8 μM of each primer, 2.5 μL of cDNA sample, diethyl pyrocarbonate (DEPC)-treated water, and 10 μL SYBR Green PCR Master Mix. The primer sequences were designed using Primer Quest software from Integrated DNA Technologies (Cambridge, MA, USA). The primer sequences used in this study were provided previously [
32]. The optimal conditions for PCR amplification of cDNA were established using the manufacturer’s instructions. In addition, the data were analyzed using StepOne software from Applied Biosystems (Carlsbad, CA, USA). The cycle number at the linear amplification threshold (Ct) values for the endogenous control Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and the target gene were recorded. Relative gene expression (target gene expression normalized to the expression of the endogenous control gene) was calculated using the comparative Ct method (2
−ΔΔCt).
3.9. Western Blot Analysis
The proteins of iNOS, COX-2, NF-κB, and MAPKs were measured by Western blot analysis. The details of procedures for Western blot analysis were described previously [
32].
3.10. Preparation of Nuclear and Cytosolic Fractions
The proteins of nuclear NF-κB (p50 and p65) and cytosolic IκBα (p-IκBα and IκBα) were obtained by using cytosolic and nuclear fractions. The details of this procedure have been described previously [
32].
3.11. DNA Binding Activity of NF-κB
TransAM kit (Active Motif, Carlsbad, CA, USA) was used to estimate the DNA-binding activity of NF-κB in the nuclear extract according to the manufacturer’s instructions as described previously [
32].
3.12. Statistical Analysis
Data were presented as the mean ± standard deviation (S.D.) of at least three independent experiments. One-way analysis of variance (ANOVA), followed by Tukey’s multiple comparison tests, was used to compare three or more groups. Statistical analysis was performed using GraphPad Prism software, version 4.00 (GraphPad Software Inc., San Diego, CA, USA).






{kind=link}
{kind=link}
{kind=link}
{kind=link}