Fucosterol from an Edible Brown Alga Ecklonia stolonifera Prevents Soluble Amyloid Beta-Induced Cognitive Dysfunction in Aging Rats


Abstract
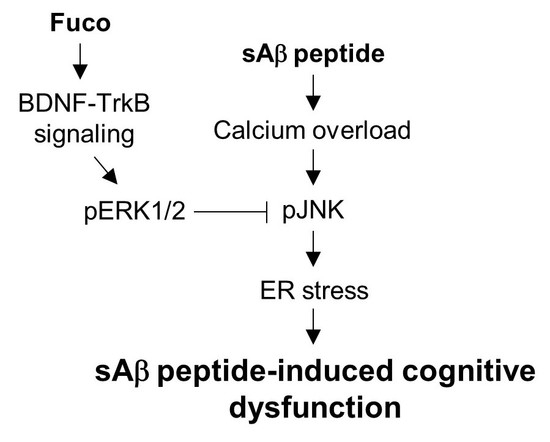
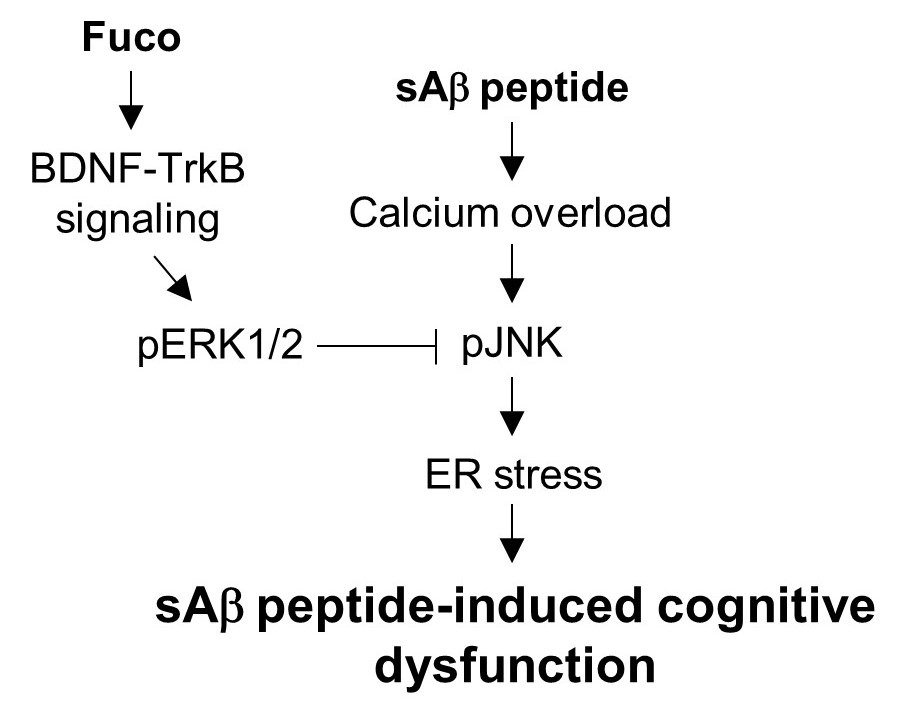
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Fucosterol Attenuated sAβ1-42-Induced Decrease in the Viability of Hippocampal Neurons
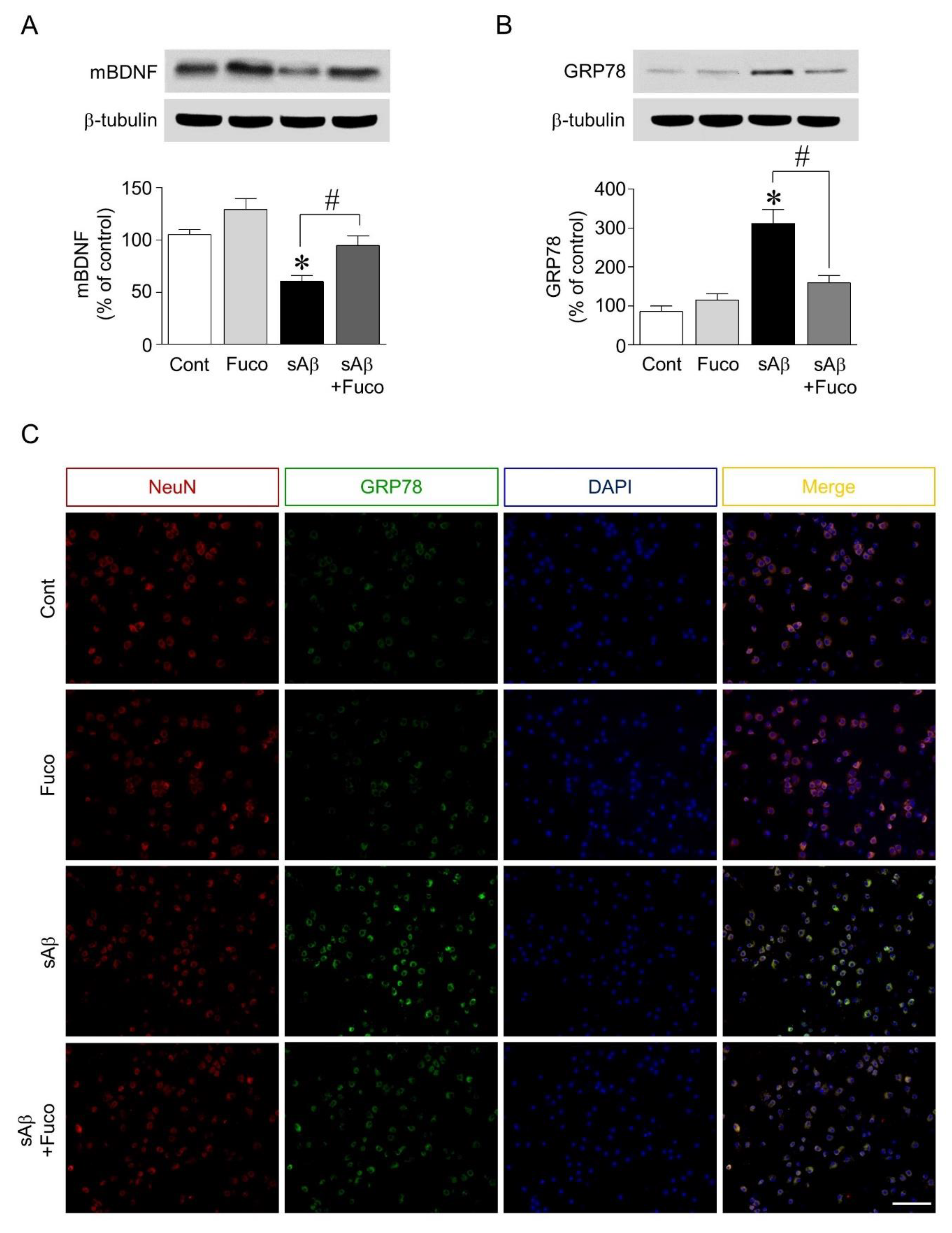
2.2. Fucosterol Pretreatment Downregulated sAβ1-42-Induced Decrease in Mature BDNF Expression and Increase in GRP78 Expression in Hippocampal Neurons
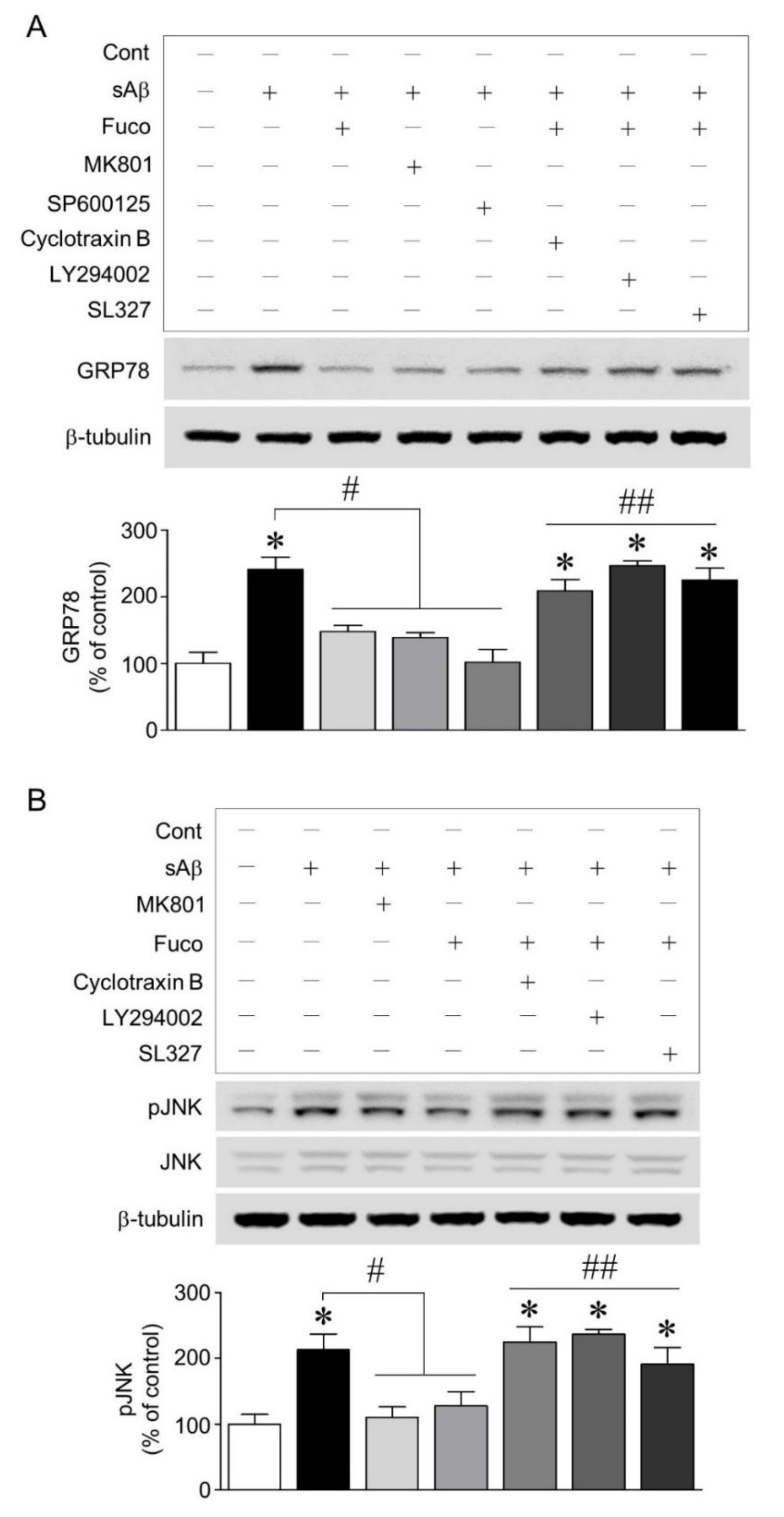
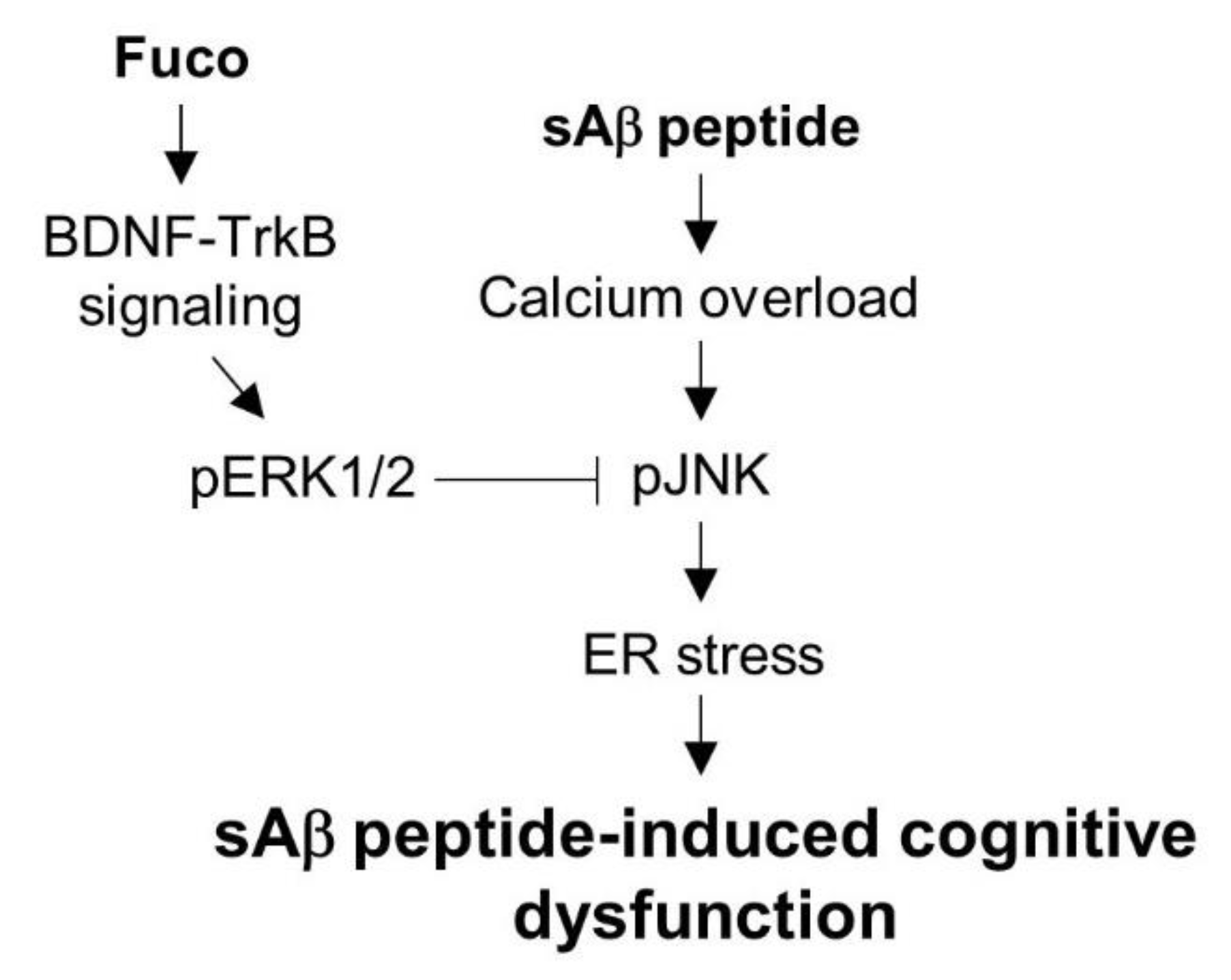
2.3. Fucosterol Reduced sAβ1-42-Induced Increase in GRP78 Expression and JNK Phosphorylation Coupled to N-Methyl d-aspartate (NMDA) Receptor via Activation of TrkB-Mediated ERK1/2 Signaling
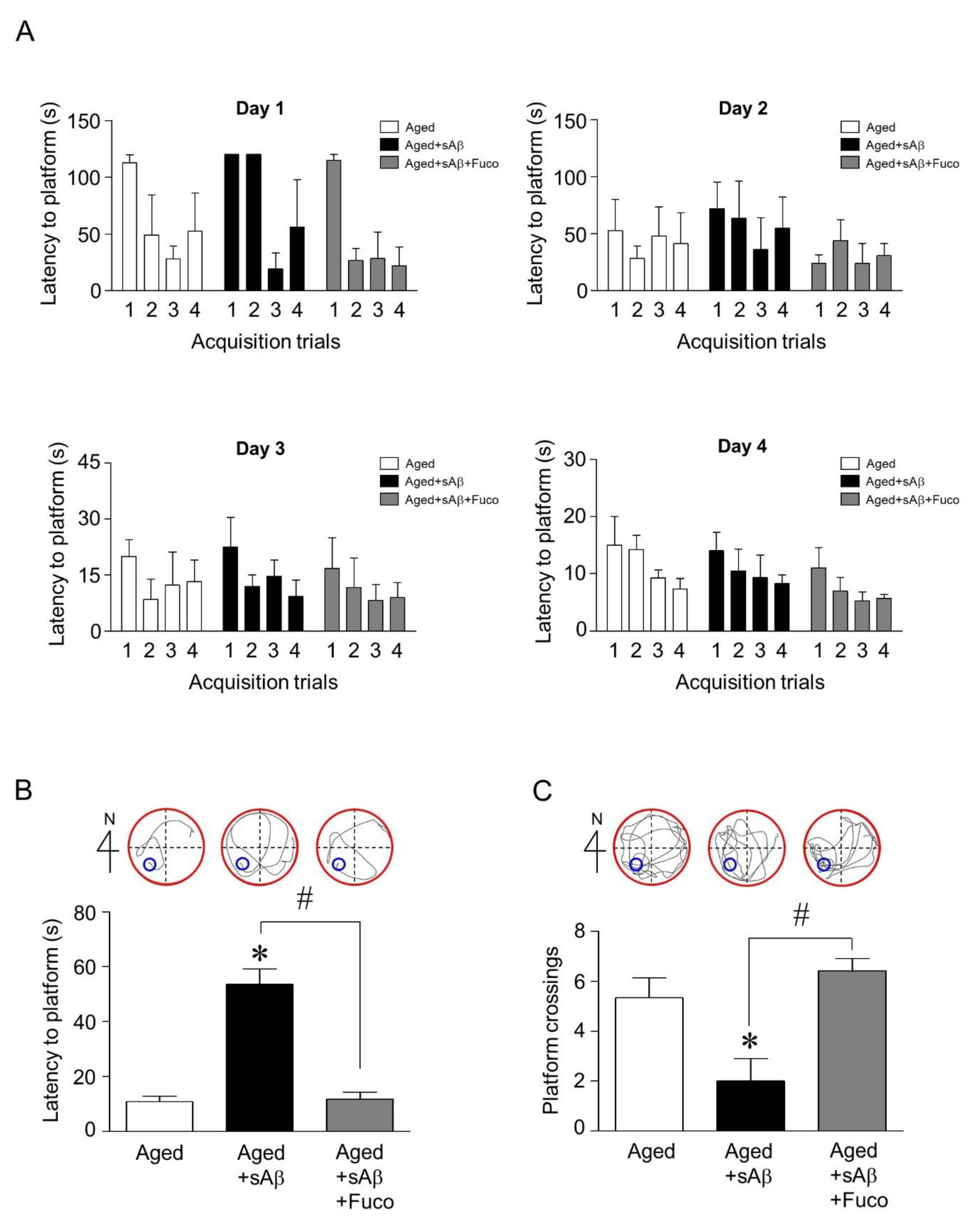
2.4. Fucosterol Co-Infusion Attenuated sAβ1-42-Induced Cognitive Impairment in the Dentate Gyrus of the Dorsal Hippocampus of Aging Rats
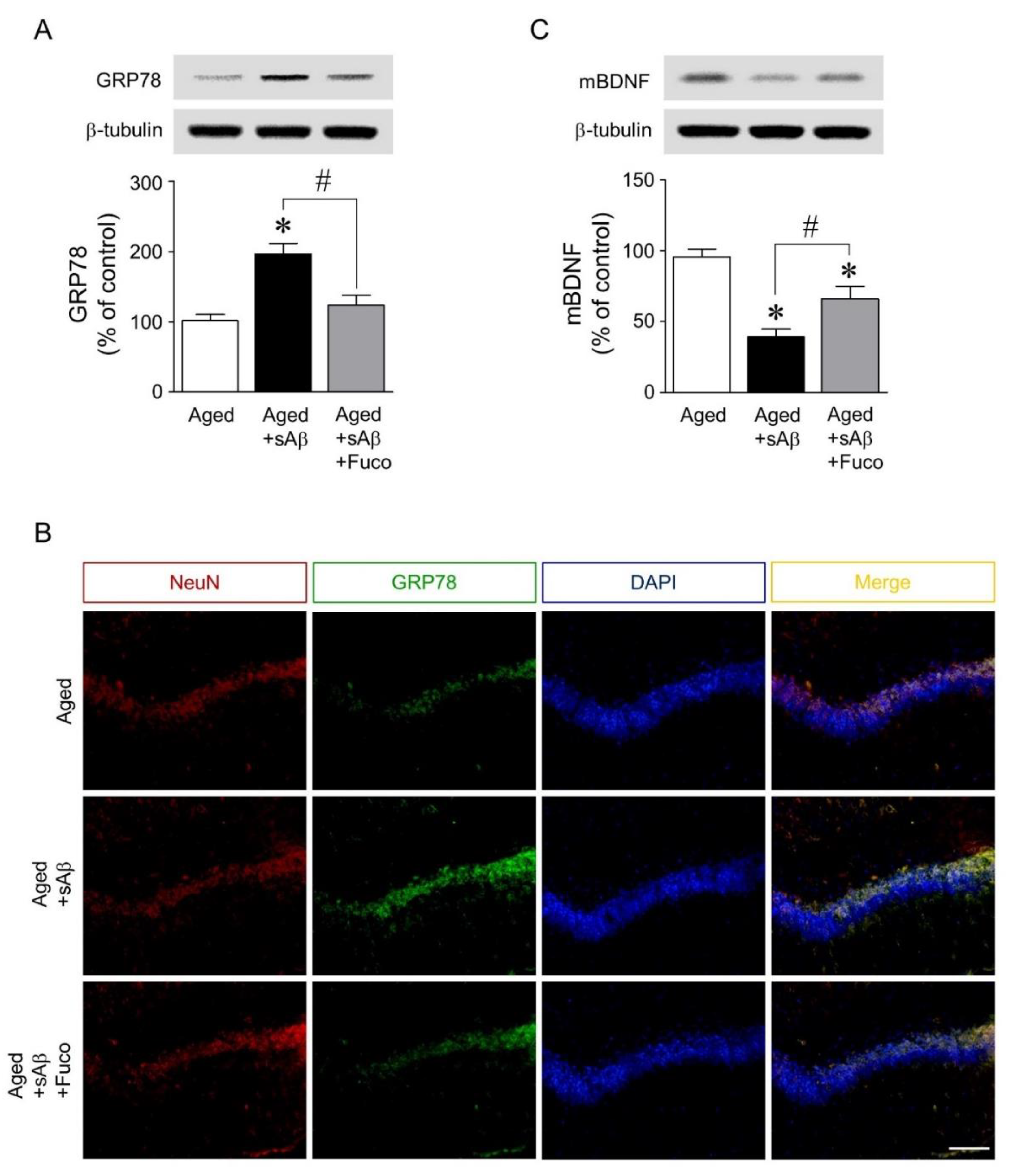
2.5. Fucosterol Infusion Downregulated sAβ1-42-Induced Increase in GRP78 Expression and Upregulated Mature BDNF Expression in the Dorsal Hippocampus of Aging Rats
3. Discussion
4. Material and Methods
4.1. Fucosterol Extraction from E. stolonifera
4.2. Primary Hippocampal Neuronal Culture
4.3. Cell Viability Assay
4.4. Intracellular Calcium Level
4.5. Immunoblotting
4.6. Double-Immunofluorescence Staining
4.7. Implantation of An Osmotic Pump for Sustained Fucosterol Delivery in vivo
4.8. Morris Water Maze Test for Spatial Learning and Memory
4.9. Statistics
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| BDNF | Brain-derived neurotrophic factor |
| DPBS | Dulbecco’s phosphate-buffered saline |
| ER | Endoplasmic reticulum |
| JNK | Jun N-terminal kinase |
| NeuN | Neuronal nuclear antigen |
| NMDA | N-methyl D-aspartate |
| sAβ1-42 | soluble amyloid beta peptide (1–42) |
| GRP78 | glucose-regulated protein 78 |
| TrkB | tyrosine receptor kinase B |
References
- Hetz, C.; Mollereau, B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 2014, 15, 233–249. [Google Scholar] [CrossRef] [PubMed]
- Martínez, G.; Duran-Aniotz, C.; Cabral-Miranda, F.; Vivar, J.P.; Hetz, C. Endoplasmic reticulum proteostasis impairment in aging. Aging Cell 2017, 16, 615–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darling, N.J.; Cook, S.J. The role of MAPK signalling pathways in the response to endoplasmic reticulum stress. Biochim. Biophys. Acta. 2014, 1843, 2150–2163. [Google Scholar] [CrossRef] [PubMed]
- Gavilán, M.P.; Pintado, C.; Gavilán, E.; Jiménez, S.; Ríos, R.M.; Vitorica, J.; Castaño, A.; Ruano, D. Dysfunction of the unfolded protein response increases neurodegeneration in aged rat hippocampus following proteasome inhibition. Aging Cell 2009, 8, 654–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeeshan, H.M.; Lee, G.H.; Kim, H.R.; Chae, H.J. Endoplasmic reticulum stress and associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef] [PubMed]
- Gardner, B.M.; Pincus, D.; Gotthardt, K.; Gallagher, C.M.; Walter, P. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb. Perspect. Biol. 2013, 5, a013169. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.T.; Klein, W.L. The Aβ oligomer hypothesis for synapse failure and memory loss in Alzheimer’s disease. Neurobiol. Learn. Mem. 2011, 96, 529–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonseca, A.C.; Oliveira, C.R.; Pereira, C.F.; Cardoso, S.M. Loss of proteostasis induced by amyloid beta peptide in brain endothelial cells. Biochim. Biophys. Acta. 2014, 1843, 1150–1161. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, K.C.; Lacor, P.N.; Pitt, L.; Klein, W.L. Aβ oligomer-induced synapse degeneration in Alzheimer’s disease. Cell Mol. Neurobiol. 2011, 31, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. Proteostasis and aging. Nat. Med. 2015, 21, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Scheper, W.; Hoozemans, J.J. The unfolded protein response in neurodegenerative diseases: A neuropathological perspective. Acta. Neuropathol. 2015, 130, 315–331. [Google Scholar] [CrossRef] [PubMed]
- Bekinschtein, P.; Cammarota, M.; Igaz, L.M.; Bevilaqua, L.R.; Izquierdo, I.; Medina, J.H. Persistence of long-term memory storage requires a late protein synthesis and BDNF-dependent phase in the hippocampus. Neuron 2007, 53, 261–277. [Google Scholar] [CrossRef] [PubMed]
- Takei, N.; Kawamura, M.; Hara, K.; Yonezawa, K.; Nawa, H. Brain-derived neurotrophic factor enhances neuronal translation by activating multiple initiation processes: Comparison with the effects of insulin. J. Biol. Chem. 2001, 276, 42818–42825. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, J.; Horiike, Y.; Matsuzaki, M.; Miyazaki, T.; Ellis-Davies, G.C.; Kasai, H. Protein synthesis and neurotrophin-dependent structural plasticity of single dendritic spines. Science 2008, 319, 1683–1687. [Google Scholar] [CrossRef] [PubMed]
- Qiu, B.; Hu, S.; Liu, L.; Chen, M.; Wang, L.; Zeng, X.; Zhu, S. CART attenuates endoplasmic reticulum stress response induced by cerebral ischemia and reperfusion through upregulating BDNF synthesis and secretion. Biochem. Biophys. Res. Commun. 2013, 436, 655–659. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.J.; Xu, J.H.; Li, M.H.; Tang, J.P.; Zou, W.; Zhang, P.; Wang, L.; Wang, C.Y.; Tang, X.Q. Hydrogen sulfide inhibits homocysteine-induced endoplasmic reticulum stress and neuronal apoptosis in rat hippocampus via upregulation of the BDNF-TrkB pathway. Acta. Pharmacol. Sin. 2014, 35, 707–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Autry, A.E.; Monteggia, L.M. Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharmacol. Rev. 2012, 64, 238–258. [Google Scholar] [CrossRef] [PubMed]
- Boulle, F.; van den Hove, D.L.; Jakob, S.B.; Rutten, B.P.; Hamon, M.; van Os, J.; Lesch, K.P.; Lanfumey, L.; Steinbusch, H.W.; Kenis, G. Epigenetic regulation of the BDNF gene: Implications for psychiatric disorders. Mol. Psychiatry. 2012, 17, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Erickson, K.I.; Miller, D.L.; Roecklein, K.A. The aging hippocampus: Interactions between exercise, depression, and BDNF. Neuroscientist 2012, 18, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Petzold, A.; Psotta, L.; Brigadski, T.; Endres, T.; Lessmann, V. Chronic BDNF deficiency leads to an age-dependent impairment in spatial learning. Neurobiol. Learn. Mem. 2015, 120, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.A.; Jin, S.E.; Ahn, B.R.; Lee, C.M.; Choi, J.S. Anti-inflammatory activity of edible brown alga Eisenia bicyclis and its constituents fucosterol and phlorotannins in LPS-stimulated RAW264.7 macrophages. Food Chem. Toxicol. 2013, 59, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.A.; Jung, H.J.; Jeong, H.Y.; Kwon, H.J.; Kim, M.S.; Choi, J.S. Anti-adipogenic activity of the edible brown alga Ecklonia stolonifera and its constituent fucosterol in 3T3-L1 adipocytes. Arch. Pharm. Res. 2014, 37, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Hwang, E.; Park, S.Y.; Sun, Z.W.; Shin, H.S.; Lee, D.G.; Yi, T.H. The protective effects of fucosterol against skin damage in UVB-irradiated human dermal fibroblasts. Mar. Biotechnol. 2014, 16, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.N.; Chung, H.Y.; Kim, H.R.; Choi, J.S. Acetyl- and butyryl-cholinesterase inhibitory activities of sterols and phlorotannins from Ecklonia stolonifera. Fish Sci. 2008, 74, 200–207. [Google Scholar] [CrossRef]
- Jung, H.A.; Ali, M.Y.; Choi, R.J.; Jeong, H.O.; Chung, H.Y.; Choi, J.S. Kinetics and molecular docking studies of fucosterol and fucoxanthin, BACE1 inhibitors from brown algae Undaria pinnatifida and Ecklonia stolonifera. Food Chem. Toxicol. 2016, 89, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Magi, S.; Castaldo, P.; Macrì, M.L.; Maiolino, M.; Matteucci, A.; Bastioli, G.; Gratteri, S.; Amoroso, S.; Lariccia, V. Intracellular calcium dysregulation: Implications for Alzheimer’s Disease. Biomed. Res. Int. 2016, 2016, 6701324. [Google Scholar] [CrossRef] [PubMed]
- Thibault, O.; Hadley, R.; Landfield, P.W. Elevated postsynaptic [Ca2+]i and L-type calcium channel activity in aged hippocampal neurons: Relationship to impaired synaptic plasticity. J. Neurosci. 2001, 21, 9744–9756. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.J.; Reichardt, L.F. Trk receptors: Roles in neuronal signal transduction. Annu. Rev. Biochem. 2003, 72, 609–642. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.K.; You, Y.; Gupta, V.B.; Klistorner, A.; Graham, S.L. TrkB receptor signalling: Implications in neurodegenerative, psychiatric and proliferative disorders. Int. J. Mol. Sci. 2013, 14, 10122–10142. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.W.; Jang, J.M.; Lim, S.S. Isolation of fucosterol from Pelvetia siliquosa by high-speed countercurrent chromatography. Fish Aquat. Sci. 2012, 15, 191–195. [Google Scholar] [CrossRef]
- Lee, D.G.; Park, J.H.; Yoo, K.H.; Chung, I.S.; Lee, Y.H.; Lee, J.K.; Han, D.S.; Cho, S.M.; Baek, N.I. 24-Ethylcholesta-4,24(28)-dien-3,6-dione from Osmanthus fragrans var. aurantiacus flowers inhibits the growth of human colon cancer cell line, HCT-116. J. Korean Soc. Appl. Biol. Chem. 2011, 54, 206–210. [Google Scholar]
- Kaech, S.; Banker, G. Culturing hippocampal neurons. Nat. Protoc. 2006, 1, 2406–2415. [Google Scholar] [CrossRef] [PubMed]
- Mhillaj, E.; Morgese, M.G.; Tucci, P.; Furiano, A.; Luongo, L.; Bove, M.; Maione, S.; Cuomo, V.; Schiavone, S.; Trabace, L. Celecoxib prevents cognitive impairment and neuroinflammation in soluble amyloid β-treated rats. Neuroscience 2018, 372, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Sotthibundhu, A.; Sykes, A.M.; Fox, B.; Underwood, C.K.; Thangnipon, W.; Coulson, E.J. Beta-amyloid1–42 induces neuronal death through the p75 neurotrophin receptor. J. Neurosci. 2008, 28, 3941–3946. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.H.; Kim, E.Y.; Nam, T.J. Phycoerythrin-derived tryptic peptide of a red alga Pyropia yezoensis attenuates glutamate-induced ER stress and neuronal senescence in primary rat hippocampal neurons. Mol. Nutr. Food Res. 2018, 62, e1700469. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.K.; Franciosi, S.; Sattayaprasert, P.; Kim, S.U.; McLarnon, J.G. Minocycline inhibits neuronal death and glial activation induced by beta-amyloid peptide in rat hippocampus. Glia 2004, 48, 85–90. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, J.H.; Choi, J.S.; Nam, T.-J. Fucosterol from an Edible Brown Alga Ecklonia stolonifera Prevents Soluble Amyloid Beta-Induced Cognitive Dysfunction in Aging Rats. Mar. Drugs 2018, 16, 368. https://doi.org/10.3390/md16100368
Oh JH, Choi JS, Nam T-J. Fucosterol from an Edible Brown Alga Ecklonia stolonifera Prevents Soluble Amyloid Beta-Induced Cognitive Dysfunction in Aging Rats. Marine Drugs. 2018; 16(10):368. https://doi.org/10.3390/md16100368
Chicago/Turabian StyleOh, Jeong Hwan, Jae Sue Choi, and Taek-Jeong Nam. 2018. "Fucosterol from an Edible Brown Alga Ecklonia stolonifera Prevents Soluble Amyloid Beta-Induced Cognitive Dysfunction in Aging Rats" Marine Drugs 16, no. 10: 368. https://doi.org/10.3390/md16100368
APA StyleOh, J. H., Choi, J. S., & Nam, T. -J. (2018). Fucosterol from an Edible Brown Alga Ecklonia stolonifera Prevents Soluble Amyloid Beta-Induced Cognitive Dysfunction in Aging Rats. Marine Drugs, 16(10), 368. https://doi.org/10.3390/md16100368