Synergistic AML Cell Death Induction by Marine Cytotoxin (+)-1(R), 6(S), 1’(R), 6’(S), 11(R), 17(S)-Fistularin-3 and Bcl-2 Inhibitor Venetoclax

 ,
,  and
and 
Abstract
:1. Introduction
2. Results
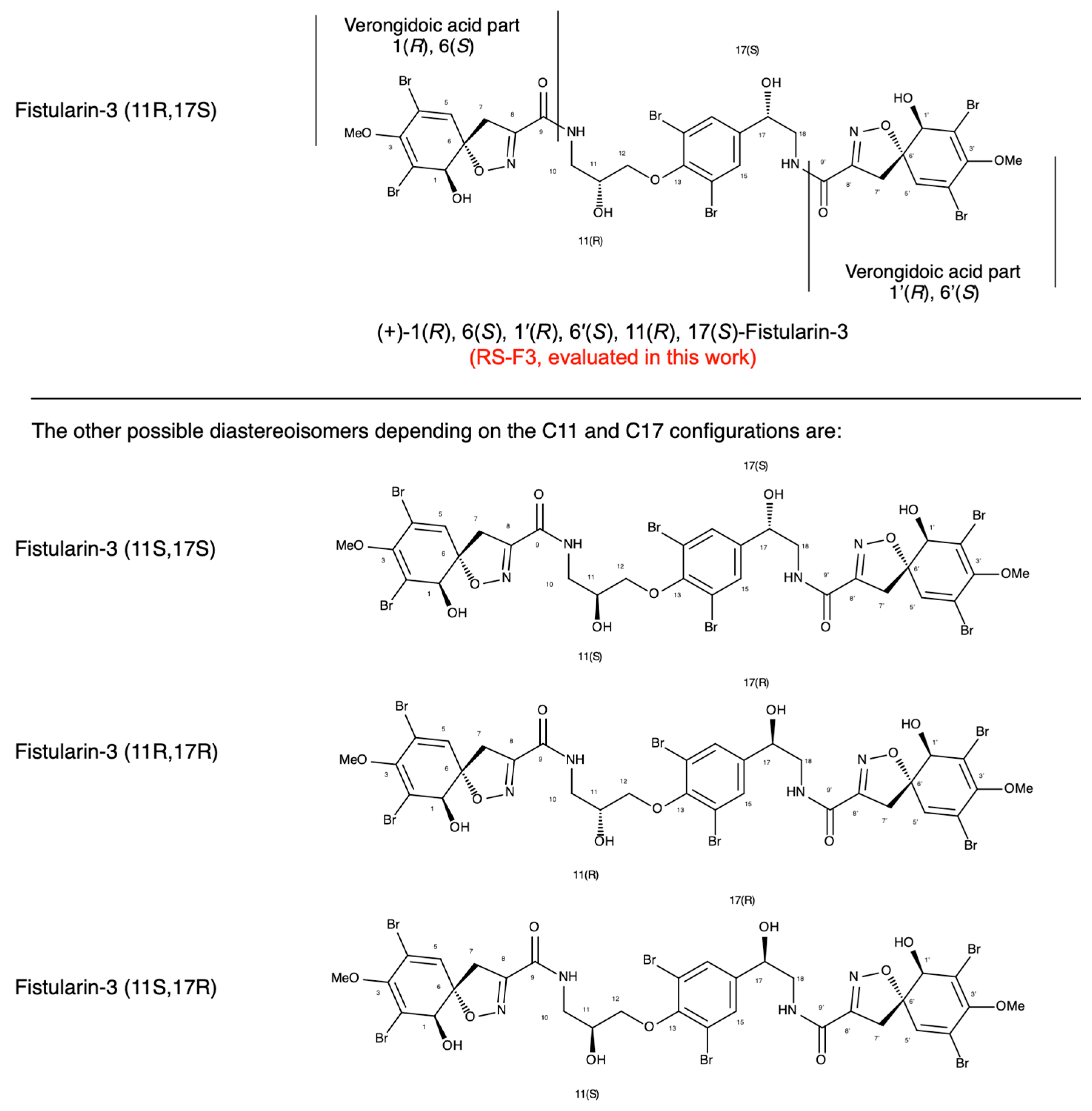
2.1. Extraction, Purification and Determination of RS-F3 Absolute Configuration
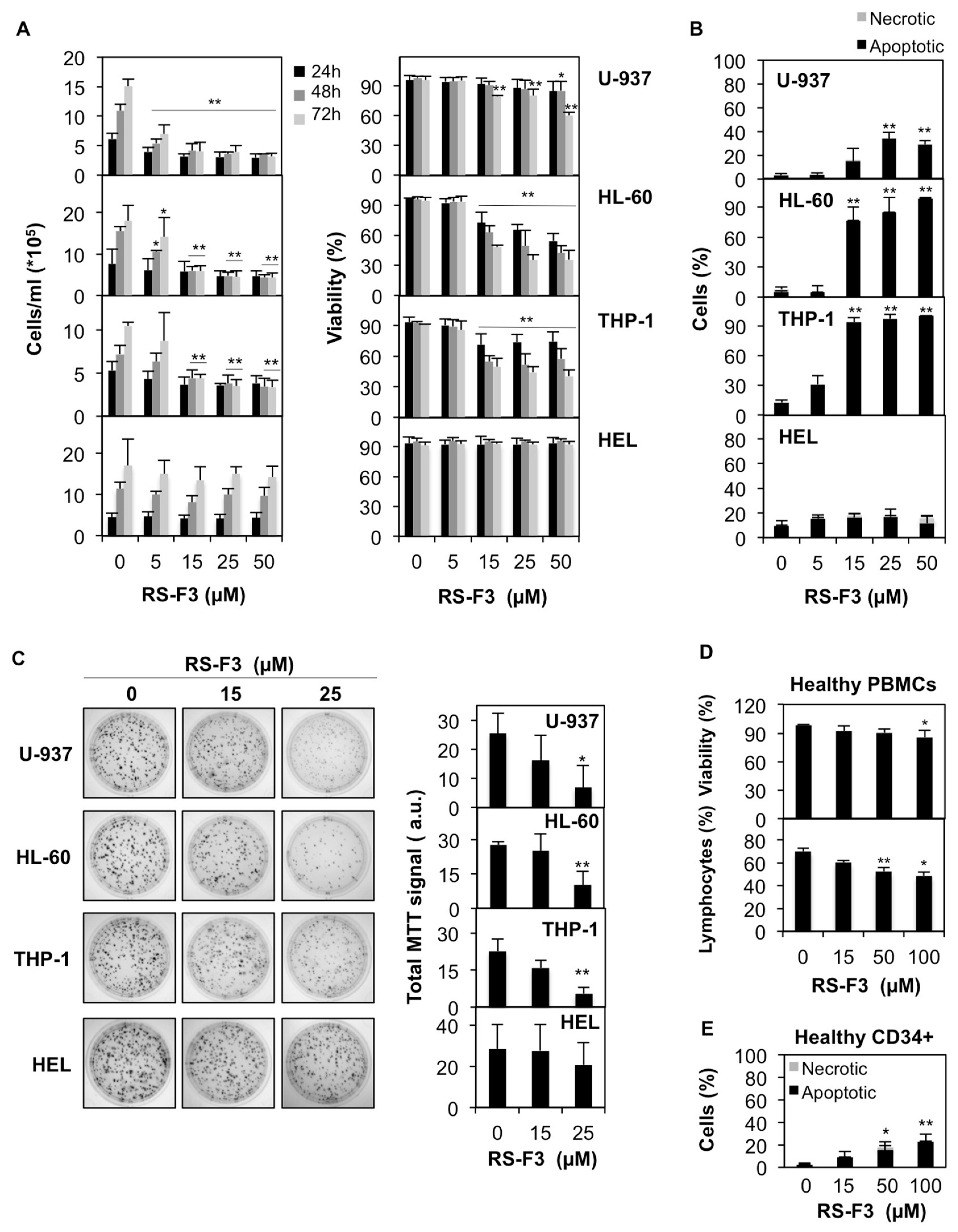
2.2. Differential Cytostatic and Cytotoxic Activities of RS-F3
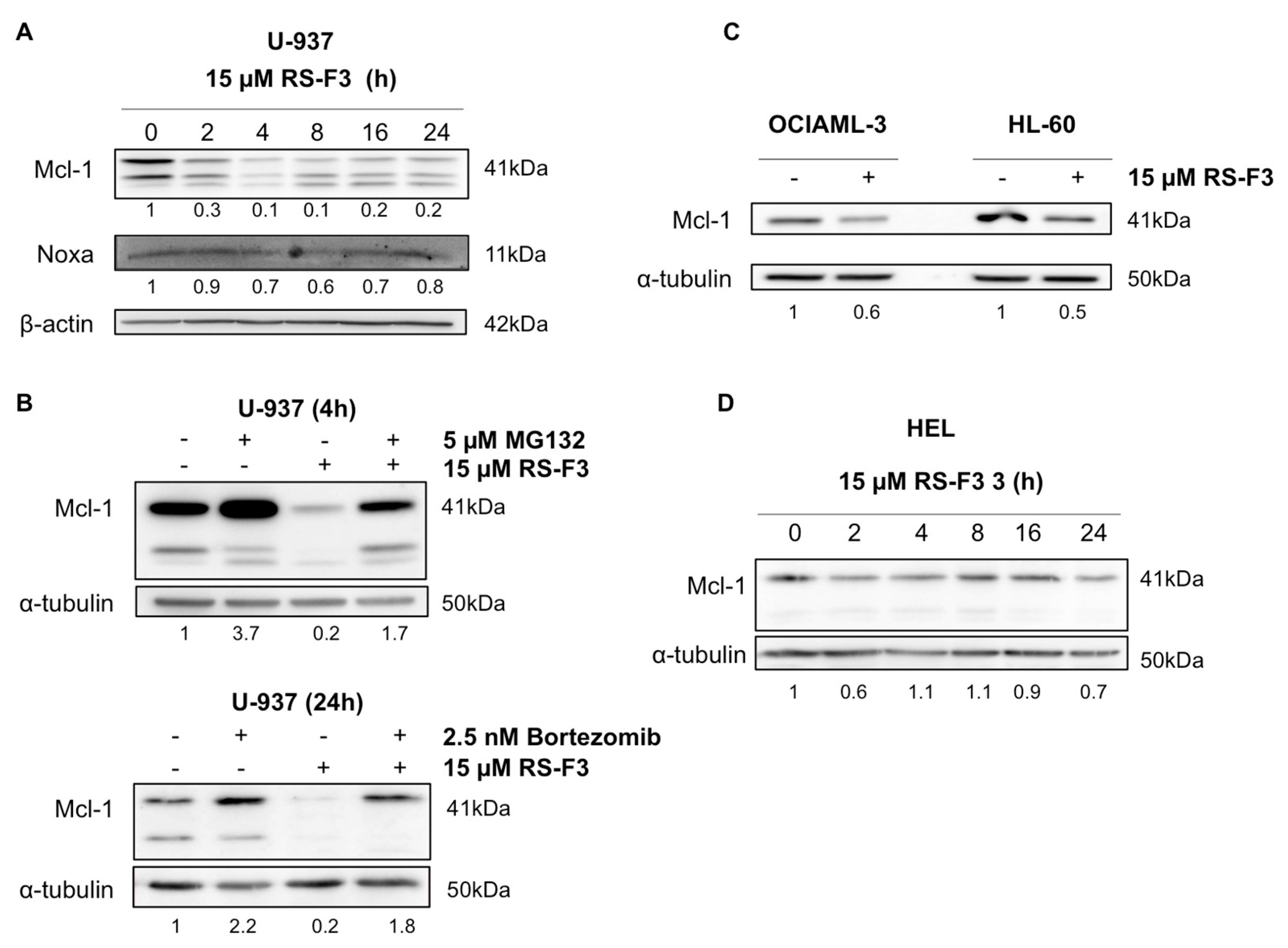
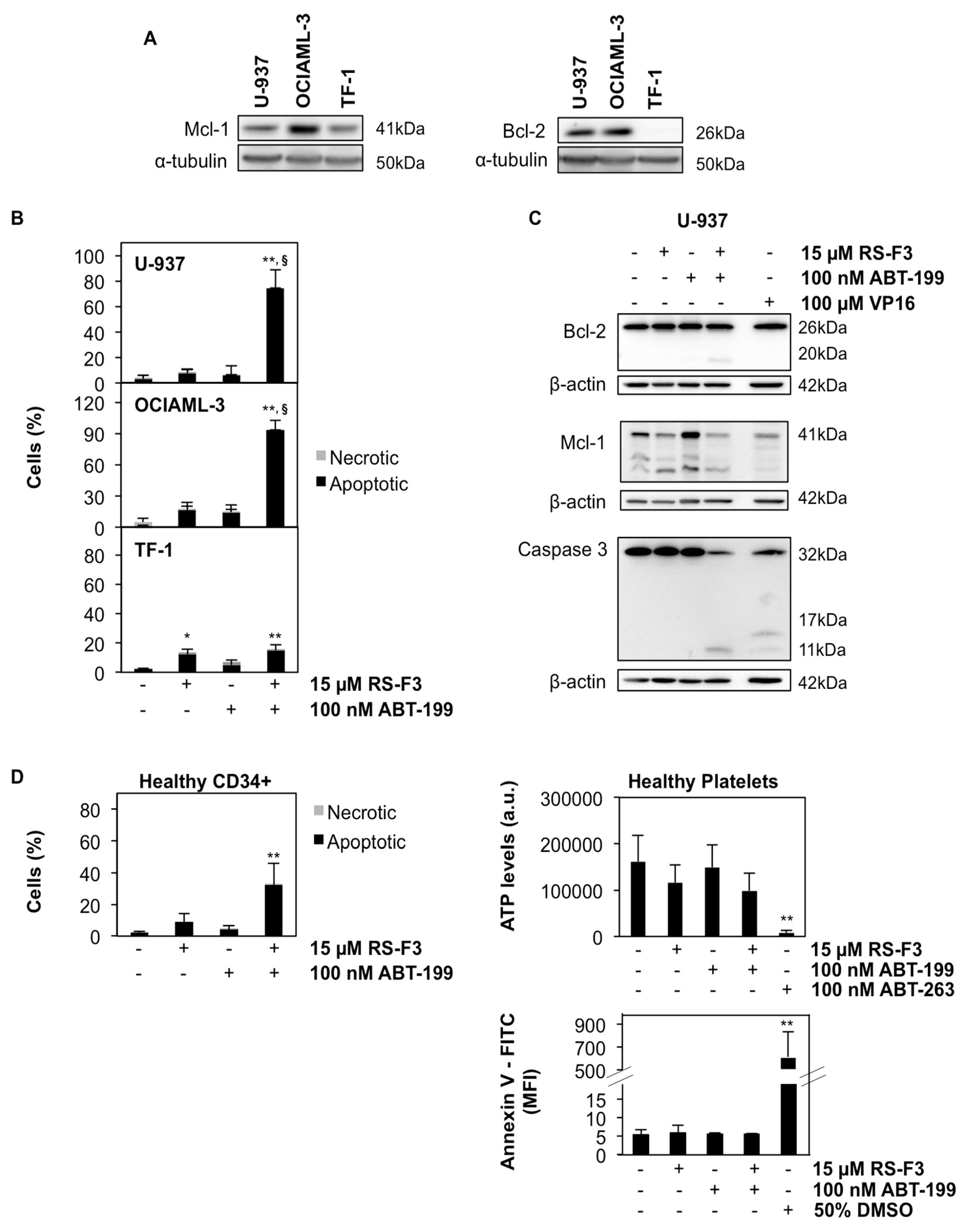
2.3. RS-F3 Strongly Down-Regulates the Anti-Apoptotic Protein Mcl-1
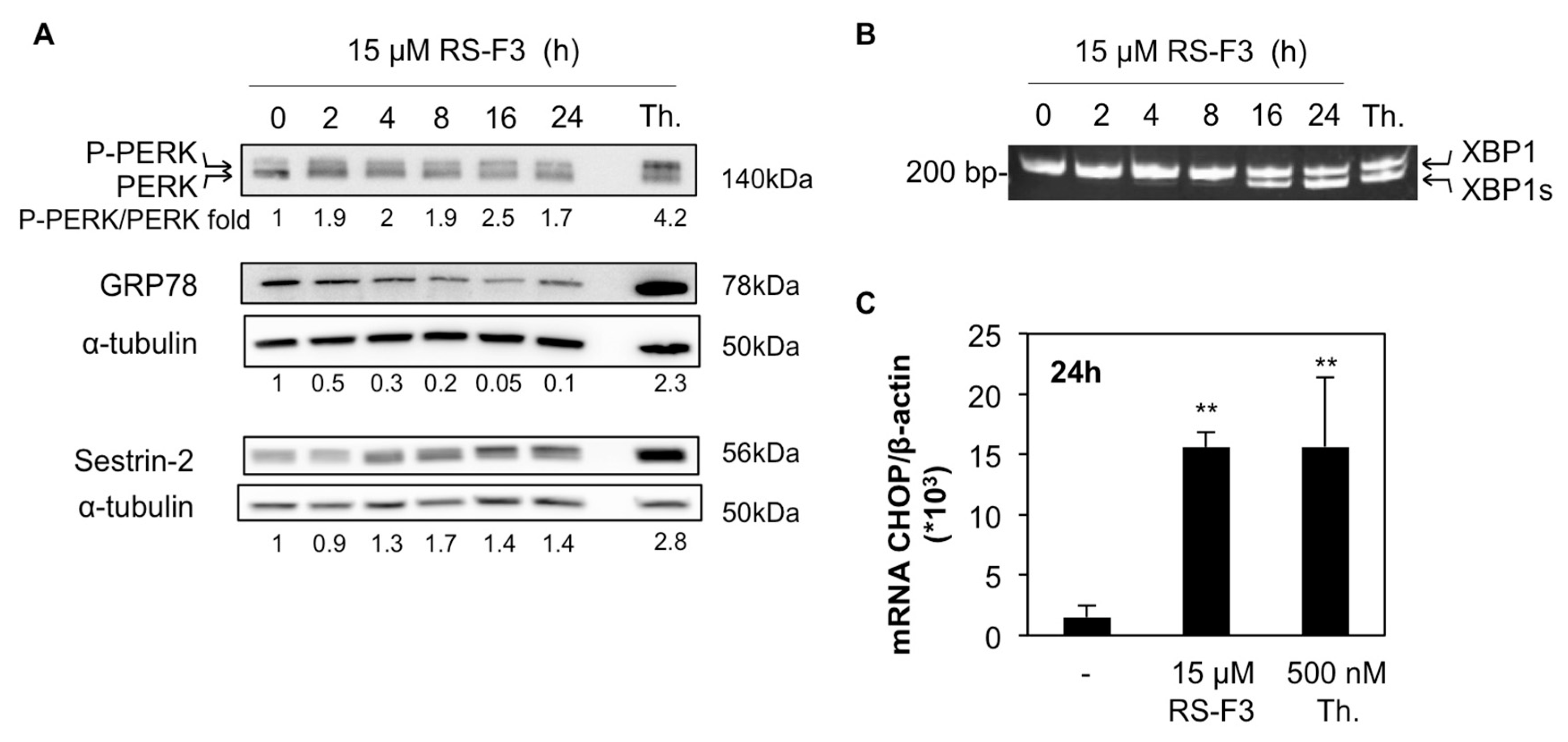
2.4. RS-F3 Triggers Endoplasmic Reticulum Stress in U-937 Cells
2.5. RS-F3 Sensitizes Bcl-2-Expressing AML Cell Lines to ABT-199
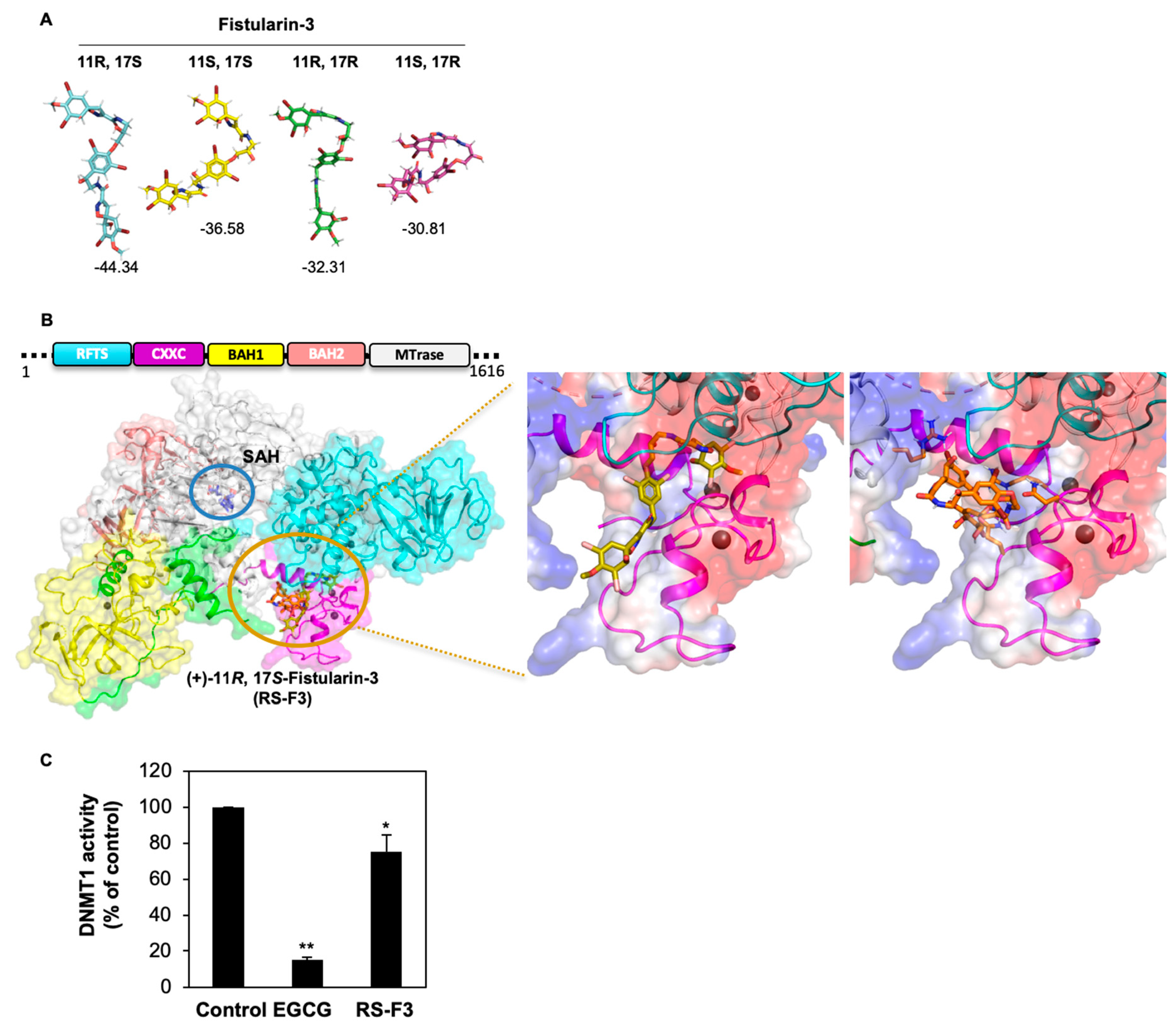
2.6. RS-F3 Docking Studies and In Vitro Activity Assay on DNMT1
3. Discussion
4. Materials and Methods
4.1. Compound Isolation and Structure Elucidation
4.1.1. Sponge Collection and Compound Isolation
4.1.2. Determination of (+)-11(R), 17(S)-fistularin-3 Absolute Configuration
4.1.3. Docking Studies
4.2. Biological Assays
4.2.1. Chemicals
4.2.2. Cell Proliferation and Viability Assays
4.2.3. Evaluation of Cell Death Type and Caspase-3/7 Activity Assay
4.2.4. Protein Extraction and Western Blotting
4.2.5. Gene Expression and XBP1 Splicing Analyses
4.2.6. In Vitro DNMT1 Activity Assay
4.2.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Briot, T.; Roger, E.; Thepot, S.; Lagarce, F. Advances in treatment formulations for acute myeloid leukemia. Drug Discov. Today 2018, 23, 1936–1949. [Google Scholar] [CrossRef] [PubMed]
- Muller, F.; Cerella, C.; Radogna, F.; Dicato, M.; Diederich, M. Effects of Natural Products on Mcl-1 Expression and Function. Curr. Med. Chem. 2015, 22, 3447–3461. [Google Scholar] [CrossRef] [PubMed]
- Gentile, M.; Petrungaro, A.; Uccello, G.; Vigna, E.; Recchia, A.G.; Caruso, N.; Bossio, S.; De Stefano, L.; Palummo, A.; Storino, F.; et al. Venetoclax for the treatment of chronic lymphocytic leukemia. Expert Opin. Investig. Drugs 2017, 26, 1307–1316. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Tiong, I.S. Midostaurin, enasidenib, CPX-351, gemtuzumab ozogamicin, and venetoclax bring new hope to AML. Blood 2017, 130, 2469–2474. [Google Scholar] [CrossRef] [PubMed]
- Grant, S. Rational combination strategies to enhance venetoclax activity and overcome resistance in hematologic malignancies. Leuk. Lymphoma 2018, 59, 1292–1299. [Google Scholar] [CrossRef]
- Cerella, C.; Gaigneaux, A.; Mazumder, A.; Lee, J.Y.; Saland, E.; Radogna, F.; Farge, T.; Vergez, F.; Recher, C.; Sarry, J.E.; et al. Bcl-2 protein family expression pattern determines synergistic pro-apoptotic effects of BH3 mimetics with hemisynthetic cardiac glycoside UNBS1450 in acute myeloid leukemia. Leukemia 2017, 31, 755–759. [Google Scholar] [CrossRef]
- Teh, T.C.; Nguyen, N.Y.; Moujalled, D.M.; Segal, D.; Pomilio, G.; Rijal, S.; Jabbour, A.; Cummins, K.; Lackovic, K.; Blombery, P.; et al. Enhancing venetoclax activity in acute myeloid leukemia by co-targeting MCL1. Leukemia 2018, 32, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, M.; Kelkel, M.; Dicato, M.; Diederich, M. Gold from the sea: Marine compounds as inhibitors of the hallmarks of cancer. Biotechnol. Adv. 2011, 29, 531–547. [Google Scholar] [CrossRef]
- Sawadogo, W.R.; Schumacher, M.; Teiten, M.H.; Cerella, C.; Dicato, M.; Diederich, M. A survey of marine natural compounds and their derivatives with anti-cancer activity reported in 2011. Molecules 2013, 18, 3641–3673. [Google Scholar] [CrossRef]
- Schumacher, M.; Kelkel, M.; Dicato, M.; Diederich, M. A survey of marine natural compounds and their derivatives with anti-cancer activity reported in 2010. Molecules 2011, 16, 5629–5646. [Google Scholar] [CrossRef]
- Folmer, F.; Schumacher, M.; Diederich, M.; Jaspars, M. Finding NEMO (inhibitors) the search for marine pharmacophores targeting the nuclear factor-kappa B. Chim. Oggi. 2008, 26, 40–46. [Google Scholar]
- Folmer, F.; Jaspars, M.; Schumacher, M.; Dicato, M.; Diederich, M. Marine natural products targeting phospholipases A2. Biochem. Pharmacol. 2010, 80, 1793–1800. [Google Scholar] [CrossRef] [PubMed]
- Folmer, F.; Jaspars, M.; Dicato, M.; Diederich, M. Photosynthetic marine organisms as a source of anticancer compounds. Phytochem. Rev. 2010, 9, 557–579. [Google Scholar] [CrossRef]
- Folmer, F.; Jaspars, M.; Dicato, M.; Diederich, M. Marine cytotoxins: Callers for the various dances of death. Gastroenterol. Hepatol. Bed Bench 2009, 2, S34–S50. [Google Scholar]
- Folmer, F.; Jaspars, M.; Dicato, M.; Diederich, M. Marine natural products as targeted modulators of the transcription factor NF-kappaB. Biochem. Pharmacol. 2008, 75, 603–617. [Google Scholar] [CrossRef] [PubMed]
- Kopf, S.; Viola, K.; Atanasov, A.G.; Jarukamjorn, K.; Rarova, L.; Kretschy, N.; Teichmann, M.; Vonach, C.; Saiko, P.; Giessrigl, B.; et al. In vitro characterisation of the anti-intravasative properties of the marine product heteronemin. Arch. Toxicol. 2013, 87, 1851–1861. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, M.; Cerella, C.; Eifes, S.; Chateauvieux, S.; Morceau, F.; Jaspars, M.; Dicato, M.; Diederich, M. Heteronemin, a spongean sesterterpene, inhibits TNF alpha-induced NF-kappa B activation through proteasome inhibition and induces apoptotic cell death. Biochem. Pharmacol. 2010, 79, 610–622. [Google Scholar] [CrossRef]
- Harms, H.; Orlikova, B.; Ji, S.; Nesaei-Mosaferan, D.; Konig, G.M.; Diederich, M. Epipolythiodiketopiperazines from the Marine Derived Fungus Dichotomomyces cejpii with NF-kappaB Inhibitory Potential. Mar. Drugs 2015, 13, 4949–4966. [Google Scholar] [CrossRef]
- Cardoso-Martínez, F.; de la Rosa, J.M.; Díaz-Marrero, A.R.; Darias, J.; D’Croz, L.; Cerella, C.; Diederich, M.; Cueto, M. Oximoaspergillimide, a Fungal Derivative from a Marine Isolate ofAspergillussp. Eur. J. Org. Chem. 2015, 2015, 2256–2261. [Google Scholar] [CrossRef]
- Cardoso-Martinez, F.; de la Rosa, J.M.; Diaz-Marrero, A.R.; Darias, J.; Cerella, C.; Diederich, M.; Cueto, M. Tanzawaic acids isolated from a marine-derived fungus of the genus Penicillium with cytotoxic activities. Org. Biomol. Chem. 2015, 13, 7248–7256. [Google Scholar] [CrossRef] [PubMed]
- Rateb, M.E.; Houssen, W.E.; Schumacher, M.; Harrison, W.T.; Diederich, M.; Ebel, R.; Jaspars, M. Bioactive diterpene derivatives from the marine sponge Spongionella sp. J. Nat. Prod. 2009, 72, 1471–1476. [Google Scholar] [CrossRef]
- Florean, C.; Schnekenburger, M.; Lee, J.Y.; Kim, K.R.; Mazumder, A.; Song, S.; Kim, J.M.; Grandjenette, C.; Kim, J.G.; Yoon, A.Y.; et al. Discovery and characterization of Isofistularin-3, a marine brominated alkaloid, as a new DNA demethylating agent inducing cell cycle arrest and sensitization to TRAIL in cancer cells. Oncotarget 2016, 7, 24027–24049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Compagnone, R.S.; Avila, R.; Suarez, A.I.; Abrams, O.V.; Rangel, H.R.; Arvelo, F.; Pina, I.C.; Merentes, E. 11-Deoxyfistularin-3, a new cytotoxic metabolite from the caribbean sponge Aplysina fistularis insularis. J. Nat. Prod. 1999, 62, 1443–1444. [Google Scholar] [CrossRef] [PubMed]
- Gunasekera, S.P.; Cross, S.S. Fistularin 3 and 11-ketofistularin 3. Feline leukemia virus active bromotyrosine metabolites from the marine sponge Aplysina archeri. J. Nat. Prod. 1992, 55, 509–512. [Google Scholar] [CrossRef] [PubMed]
- Rogers, E.W.; de Oliveira, M.F.; Berlinck, R.G.; Konig, G.M.; Molinski, T.F. Stereochemical heterogeneity in Verongid sponge metabolites. Absolute stereochemistry of (+)-fistularin-3 and (+)-11-epi-fistularin-3 by microscale LCMS-Marfey’s analysis. J. Nat. Prod. 2005, 68, 891–896. [Google Scholar] [CrossRef] [PubMed]
- Teeyapant, R.; Woerdenbag, H.J.; Kreis, P.; Hacker, J.; Wray, V.; Witte, L.; Proksch, P. Antibiotic and cytotoxic activity of brominated compounds from the marine sponge Verongia aerophoba. Z. Naturforsch. C 1993, 48, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Mijares, M.R.; Ochoa, M.; Barroeta, A.; Martinez, G.P.; Suarez, A.I.; Compagnone, R.S.; Chirinos, P.; Avila, R.; De Sanctis, J.B. Cytotoxic effects of Fisturalin-3 and 11-Deoxyfisturalin-3 on Jurkat and U937 cell lines. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech. Repub. 2013, 157, 222–226. [Google Scholar] [CrossRef] [Green Version]
- Nicacio, K.J.; Ioca, L.P.; Froes, A.M.; Leomil, L.; Appolinario, L.R.; Thompson, C.C.; Thompson, F.L.; Ferreira, A.G.; Williams, D.E.; Andersen, R.J.; et al. Cultures of the Marine Bacterium Pseudovibrio denitrificans Ab134 Produce Bromotyrosine-Derived Alkaloids Previously Only Isolated from Marine Sponges. J. Nat. Prod. 2017, 80, 235–240. [Google Scholar] [CrossRef]
- Bose, P.; Grant, S. Mcl-1 as a Therapeutic Target in Acute Myelogenous Leukemia (AML). Leukemia Res. Rep. 2013, 2, 12–14. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, S.H.; Karp, J.E.; Svingen, P.A.; Krajewski, S.; Burke, P.J.; Gore, S.D.; Reed, J.C. Elevated expression of the apoptotic regulator Mcl-1 at the time of leukemic relapse. Blood 1998, 91, 991–1000. [Google Scholar]
- Cerella, C.; Muller, F.; Gaigneaux, A.; Radogna, F.; Viry, E.; Chateauvieux, S.; Dicato, M.; Diederich, M. Early downregulation of Mcl-1 regulates apoptosis triggered by cardiac glycoside UNBS1450. Cell Death Dis. 2015, 6, e1782. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, R.M.; Schneider, G.; Saur, D.; Scheibel, M.; Schmid, R.M. Translational repression of MCL-1 couples stress-induced eIF2 alpha phosphorylation to mitochondrial apoptosis initiation. J. Biol. Chem. 2007, 282, 22551–22562. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, G.J.; DeSalvo, J.; Du, J.; Gao, N.; Leclerc, G.M.; Lehrman, M.A.; Lampidis, T.J.; Barredo, J.C. Mcl-1 downregulation leads to the heightened sensitivity exhibited by BCR-ABL positive ALL to induction of energy and ER-stress. Leuk. Res. 2015. [Google Scholar] [CrossRef]
- Sarosiek, K.A.; Letai, A. Directly targeting the mitochondrial pathway of apoptosis for cancer therapy using BH3 mimetics—recent successes, current challenges and future promise. FEBS J. 2016, 283, 3523–3533. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.; Hogdal, L.J.; Benito, J.M.; Bucci, D.; Han, L.; Borthakur, G.; Cortes, J.; DeAngelo, D.J.; Debose, L.; Mu, H.; et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov. 2014, 4, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Mashiach, E.; Schneidman-Duhovny, D.; Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: A web server for fast interaction refinement in molecular docking. Nucleic Acids Res. 2008, 36, W229–W232. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef]
- Zhang, Z.M.; Liu, S.; Lin, K.; Luo, Y.; Perry, J.J.; Wang, Y.; Song, J. Crystal Structure of Human DNA Methyltransferase 1. J. Mol. Biol. 2015, 427, 2520–2531. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Long, H.K.; Blackledge, N.P.; Klose, R.J. ZF-CxxC domain-containing proteins, CpG islands and the chromatin connection. Biochem. Soc. Trans. 2013, 41, 727–740. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Rechkoblit, O.; Bestor, T.H.; Patel, D.J. Structure of DNMT1-DNA complex reveals a role for autoinhibition in maintenance DNA methylation. Science 2011, 331, 1036–1040. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, M.; Esteve, P.O.; Chin, H.G.; Samaranayke, M.; Kim, G.D.; Pradhan, S. CXXC domain of human DNMT1 is essential for enzymatic activity. Biochemistry 2008, 47, 10000–10009. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Caballero, M.; Martinez-Poveda, B.; Medina, M.A.; Quesada, A.R. The Natural Antiangiogenic Compound AD0157 Induces Caspase-Dependent Apoptosis in Human Myeloid Leukemia Cells. Front. Pharmacol. 2017, 8, 802. [Google Scholar] [CrossRef] [PubMed]
- Saikia, M.; Retnakumari, A.P.; Anwar, S.; Anto, N.P.; Mittal, R.; Shah, S.; Pillai, K.S.; Balachandran, V.S.; Peter, V.; Thomas, R.; et al. Heteronemin, a marine natural product, sensitizes acute myeloid leukemia cells towards cytarabine chemotherapy by regulating farnesylation of Ras. Oncotarget 2018, 9, 18115–18127. [Google Scholar] [CrossRef]
- Mathieu, V.; Wauthoz, N.; Lefranc, F.; Niemann, H.; Amighi, K.; Kiss, R.; Proksch, P. Cyclic versus hemi-bastadins. pleiotropic anti-cancer effects: From apoptosis to anti-angiogenic and anti-migratory effects. Molecules 2013, 18, 3543–3561. [Google Scholar] [CrossRef] [PubMed]
- Schnekenburger, M.; Dicato, M.; Diederich, M. Epigenetic modulators from “The Big Blue”: A treasure to fight against cancer. Cancer Lett. 2014, 351, 182–197. [Google Scholar] [CrossRef]
- Zhang, Y.; Fang, L.; Zang, Y.; Ren, J.; Xu, Z. CIP2A Promotes Proliferation, Invasion and Chemoresistance to Cisplatin in Renal Cell Carcinoma. J. Cancer 2018, 9, 4029–4038. [Google Scholar] [CrossRef]
- Viet, C.T.; Dang, D.; Achdjian, S.; Ye, Y.; Katz, S.G.; Schmidt, B.L. Decitabine rescues cisplatin resistance in head and neck squamous cell carcinoma. PLoS ONE 2014, 9, e112880. [Google Scholar] [CrossRef]
- Shieh, J.M.; Shen, C.J.; Chang, W.C.; Cheng, H.C.; Chan, Y.Y.; Huang, W.C.; Chang, W.C.; Chen, B.K. An increase in reactive oxygen species by deregulation of ARNT enhances chemotherapeutic drug-induced cancer cell death. PLoS ONE 2014, 9, e99242. [Google Scholar] [CrossRef]
- Liston, D.R.; Davis, M. Clinically Relevant Concentrations of Anticancer Drugs: A Guide for Nonclinical Studies. Clin. Cancer Res. 2017, 23, 3489–3498. [Google Scholar] [CrossRef]
- Bayat Mokhtari, R.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef]
- Lee, J.Y.; Talhi, O.; Jang, D.; Cerella, C.; Gaigneaux, A.; Kim, K.W.; Lee, J.W.; Dicato, M.; Bachari, K.; Han, B.W.; et al. Cytostatic hydroxycoumarin OT52 induces ER/Golgi stress and STAT3 inhibition triggering non-canonical cell death and synergy with BH3 mimetics in lung cancer. Cancer Lett. 2018, 416, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, M.; Davis, E.M.; Crabtree, T.R.; Habibi, J.R.; Nguyen, T.K.; Dent, P.; Grant, S. The kinase inhibitor sorafenib induces cell death through a process involving induction of endoplasmic reticulum stress. Mol. Cell. Biol. 2007, 27, 5499–5513. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.J.; Hendershot, L.M. UPR activation alters chemosensitivity of tumor cells. Cancer Biol. Ther. 2006, 5, 736–740. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Fabritius, M.; Ip, C. Chemotherapeutic sensitization by endoplasmic reticulum stress: Increasing the efficacy of taxane against prostate cancer. Cancer Biol. Ther. 2009, 8, 146–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraus, M.; Malenke, E.; Gogel, J.; Muller, H.; Ruckrich, T.; Overkleeft, H.; Ovaa, H.; Koscielniak, E.; Hartmann, J.T.; Driessen, C. Ritonavir induces endoplasmic reticulum stress and sensitizes sarcoma cells toward bortezomib-induced apoptosis. Mol. Cancer Ther. 2008, 7, 1940–1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, M.D.; Mann, M.; Nitiss, J.L.; Hendershot, L.M. Activation of the unfolded protein response is necessary and sufficient for reducing topoisomerase IIalpha protein levels and decreasing sensitivity to topoisomerase-targeted drugs. Mol. Pharmacol. 2005, 68, 1699–1707. [Google Scholar] [PubMed]
- Ishizawa, J.; Kojima, K.; Chachad, D.; Ruvolo, P.; Ruvolo, V.; Jacamo, R.O.; Borthakur, G.; Mu, H.; Zeng, Z.; Tabe, Y.; et al. ATF4 induction through an atypical integrated stress response to ONC201 triggers p53-independent apoptosis in hematological malignancies. Sci. Signal. 2016, 9, ra17. [Google Scholar] [CrossRef]
- Galeano, E.; Thomas, O.P.; Robledo, S.; Munoz, D.; Martinez, A. Antiparasitic bromotyrosine derivatives from the marine sponge Verongula rigida. Mar. Drugs 2011, 9, 1902–1913. [Google Scholar] [CrossRef]
- Mallipeddi, P.L.; Kumar, G.; White, S.W.; Webb, T.R. Recent advances in computer-aided drug design as applied to anti-influenza drug discovery. Curr. Top. Med. Chem. 2014, 14, 1875–1889. [Google Scholar] [CrossRef]
- Schrodinger, L.L.C. The PyMOL Molecular Graphics System, Version 1.8. 2015. Available online: https://sourceforge.net/p/pymol/mailman/message/34691722/ (accessed on 29 November 2018).
- Schnekenburger, M.; Grandjenette, C.; Ghelfi, J.; Karius, T.; Foliguet, B.; Dicato, M.; Diederich, M. Sustained exposure to the DNA demethylating agent, 2’-deoxy-5-azacytidine, leads to apoptotic cell death in chronic myeloid leukemia by promoting differentiation, senescence, and autophagy. Biochem. Pharmacol. 2011, 81, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Schnekenburger, M.; Mathieu, V.; Lefranc, F.; Jang, J.Y.; Masi, M.; Kijjoa, A.; Evidente, A.; Kim, H.J.; Kiss, R.; Dicato, M.; et al. The Fungal Metabolite Eurochevalierine, a Sequiterpene Alkaloid, Displays Anti-Cancer Properties through Selective Sirtuin 1/2 Inhibition. Molecules 2018, 23, 333. [Google Scholar] [CrossRef] [PubMed]
- Seidel, C.; Schnekenburger, M.; Dicato, M.; Diederich, M. Antiproliferative and proapoptotic activities of 4-hydroxybenzoic acid-based inhibitors of histone deacetylases. Cancer Lett. 2014, 343, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Grandjenette, C.; Schnekenburger, M.; Karius, T.; Ghelfi, J.; Gaigneaux, A.; Henry, E.; Dicato, M.; Diederich, M. 5-aza-2’-deoxycytidine-mediated c-myc Down-regulation triggers telomere-dependent senescence by regulating human telomerase reverse transcriptase in chronic myeloid leukemia. Neoplasia 2014, 16, 511–528. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fistularin 3 (500 MHz) | ||
|---|---|---|
| Position | δC, Type | δH Mult. (J in Hz) |
| 1 | 75.2, CH | 4.18, d (8.0) |
| 2 | 122.1, C | |
| 3 | 148.8, C | |
| 4 | 113.8, C | |
| 5 | 132.3, CH | 6.52, s |
| 6 | 91.8, C | |
| 7 | 40.0, CH2 | 3.85, d (18.0) |
| 3.19, d (18.0) | ||
| 8 | 155.1, C | |
| 9 | 160.5, C | |
| 10 | 43.6, CH2 | 3.80, m |
| 3.54, m | ||
| 11 | 69.9, CH | 4.25, m |
| 12 | 75.9, CH2 | 4.06, dd (9.1, 5.5) |
| 4.02, dd (9.1, 5.5) | ||
| 13 | 152.7, C | |
| 14, 14’ | 118.4, C | |
| 15, 15’ | 131.5, CH | 7.66, s |
| 16 | 143.3, C | |
| 17 | 71.0, CH | 4.90, ddd (7.7, 5.5, 4.3) |
| 18 | 47.7, CH2 | 3.63, m |
| 3.49, m | ||
| 1’ | 75.3, CH | 4.19, d (8.0) |
| 2’ | 122.1, C | |
| 3’ | 148.8, C | |
| 4’ | 113.9, C | |
| 5’ | 132.4, CH | 6.53, s |
| 6’ | 91.8, C | |
| 7’ | 40.0, CH2 | 3.82, d (18.0) |
| 3.16, d (18.0) | ||
| 8’ | 155.2, C | |
| 9’ | 160.5, C | |
| OMe | 60.2, CH3 | 3.73, s |
| OMe’ | 60.2, CH3 | 3.73, s |
| NH | 7.62, bt (6.0) | |
| NH’ | 7.66, bt (6.0) | |
| OH 1-1’ | 5.41, d (8.0) | |
| OH 11 | 4.44, d (5.3) | |
| OH 17 | 5.00, d (4.3) | |
| Cell Line | GI50 (µM) | IC50 (µM) |
|---|---|---|
| U-937 | 3.2 | >50 |
| HL-60 | 10.86 | 12.1 |
| THP-1 | 15.17 | 10.36 |
| HEL | >50 | >50 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Florean, C.; Kim, K.R.; Schnekenburger, M.; Kim, H.-J.; Moriou, C.; Debitus, C.; Dicato, M.; Al-Mourabit, A.; Han, B.W.; Diederich, M. Synergistic AML Cell Death Induction by Marine Cytotoxin (+)-1(R), 6(S), 1’(R), 6’(S), 11(R), 17(S)-Fistularin-3 and Bcl-2 Inhibitor Venetoclax. Mar. Drugs 2018, 16, 518. https://doi.org/10.3390/md16120518
Florean C, Kim KR, Schnekenburger M, Kim H-J, Moriou C, Debitus C, Dicato M, Al-Mourabit A, Han BW, Diederich M. Synergistic AML Cell Death Induction by Marine Cytotoxin (+)-1(R), 6(S), 1’(R), 6’(S), 11(R), 17(S)-Fistularin-3 and Bcl-2 Inhibitor Venetoclax. Marine Drugs. 2018; 16(12):518. https://doi.org/10.3390/md16120518
Chicago/Turabian StyleFlorean, Cristina, Kyung Rok Kim, Michael Schnekenburger, Hyun-Jung Kim, Céline Moriou, Cécile Debitus, Mario Dicato, Ali Al-Mourabit, Byung Woo Han, and Marc Diederich. 2018. "Synergistic AML Cell Death Induction by Marine Cytotoxin (+)-1(R), 6(S), 1’(R), 6’(S), 11(R), 17(S)-Fistularin-3 and Bcl-2 Inhibitor Venetoclax" Marine Drugs 16, no. 12: 518. https://doi.org/10.3390/md16120518
APA StyleFlorean, C., Kim, K. R., Schnekenburger, M., Kim, H. -J., Moriou, C., Debitus, C., Dicato, M., Al-Mourabit, A., Han, B. W., & Diederich, M. (2018). Synergistic AML Cell Death Induction by Marine Cytotoxin (+)-1(R), 6(S), 1’(R), 6’(S), 11(R), 17(S)-Fistularin-3 and Bcl-2 Inhibitor Venetoclax. Marine Drugs, 16(12), 518. https://doi.org/10.3390/md16120518