2. Results
The zoanthid
P. tuberculosa was collected from the northern seashore of Taiwan. The animal materials were extracted by ethanol three times and partitioned between EtOAc and H
2O. The H
2O layer was further partitioned between BuOH and H
2O to give a relatively low polarity layer. Repeated column chromatography of the BuOH portion yielded three new compounds, tuberazines A–C (
1–
3), and six known compounds: palythazine (
4) [
6], 3,4,5,6-tetrahydro-7
-hydroxy-5,6-dimethyl-1
H-azepino[5,4,3-
cd]indole (
5) [
7], tryptamine (
6) [
8], tyramine (
7) [
9],
N-methylserotonin (
8) [
10], and phenethylamine (
9) [
11]. The EtOAc extract was also further partitioned between hexanes and 75% MeOH(aq) layers. Five known compounds [isobutylamine (
10) [
12], isoamylamine (
11) [
13], (2
S)-2-hydroxy-3-[[(6
Z,9
Z,12
Z)-1-oxo-6,9,12-octadecatrien-1-yl]oxy]propyl-β-
d-galactopyranoside (
12) [
14], 20-hydroxyecdysone-3-acetate (
13) [
15], and 20-hydroxyecdysone-2-acetate (
14) [
15] were isolated from the 75% MeOH(aq) layer. Compounds
5–
12 were obtained from the genus
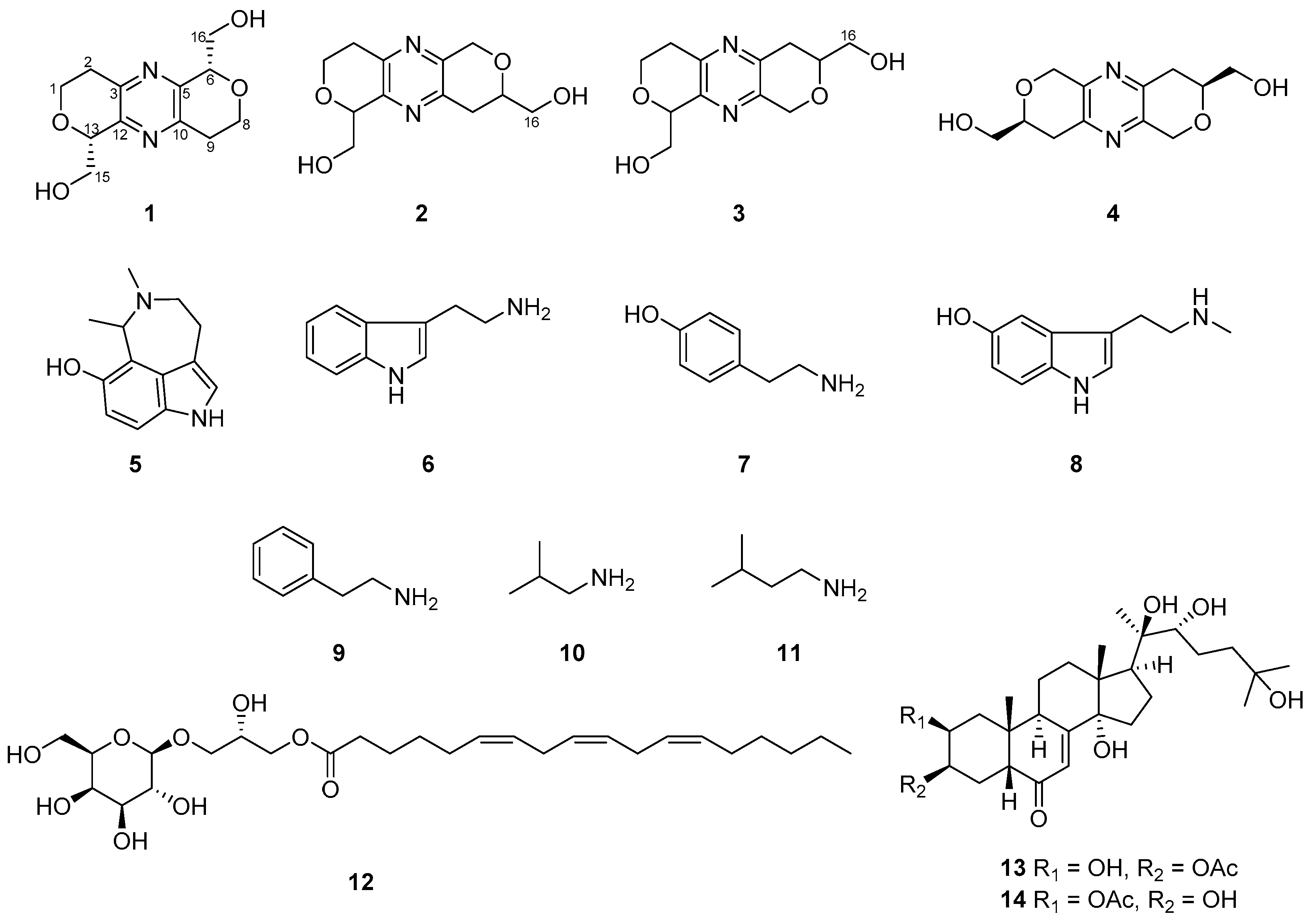
Palythoa for the first time. The structures of all the isolated compounds (
1−
14) are illustrated in
Figure 1.
Tuberazine A (
1),
+96 (
c 0.05, MeOH), was isolated as white amorphous powder. The molecular formula of C
12H
16N
2O
4 and six degrees of unsaturation were inferred from its high-resolution electrospray ionisation mass spectrometry (HRESIMS) data (
m/
z 275.10028 [M + Na]
+). In the infrared radiation (IR) spectrum of
1, absorption at 3389 cm
−1 revealed the existence of hydroxy functionality. The ultraviolet (UV) maximum absorption at 289 nm was similar to palythazine (
4), suggesting these two compounds had the pyrazine framework. The
1H NMR data (
Table 1) revealed the presences of one methylene (δ
H 2.84 and 3.15), two oxygen-bearing methylenes (δ
H 3.91 and 4.30; δ
H 3.98 and 4.06), and one oxymethine (δ
H 4.74). In the
13C NMR and distortionless enhancement by polarization transfer (DEPT) spectra of
1 (
Table 2), six carbon signals can be classified into two aromatic nonprotonated carbons (δ
C 149.5 and 149.9), one aliphatic methylene (δ
C 32.4), two oxygen-bearing methylenes (δ
C 64.5 and 64.7), and one oxymethine (δ
C 79.8). Only of the half carbon and proton NMR signals was detected in comparison with the molecular formula of
1, implying that
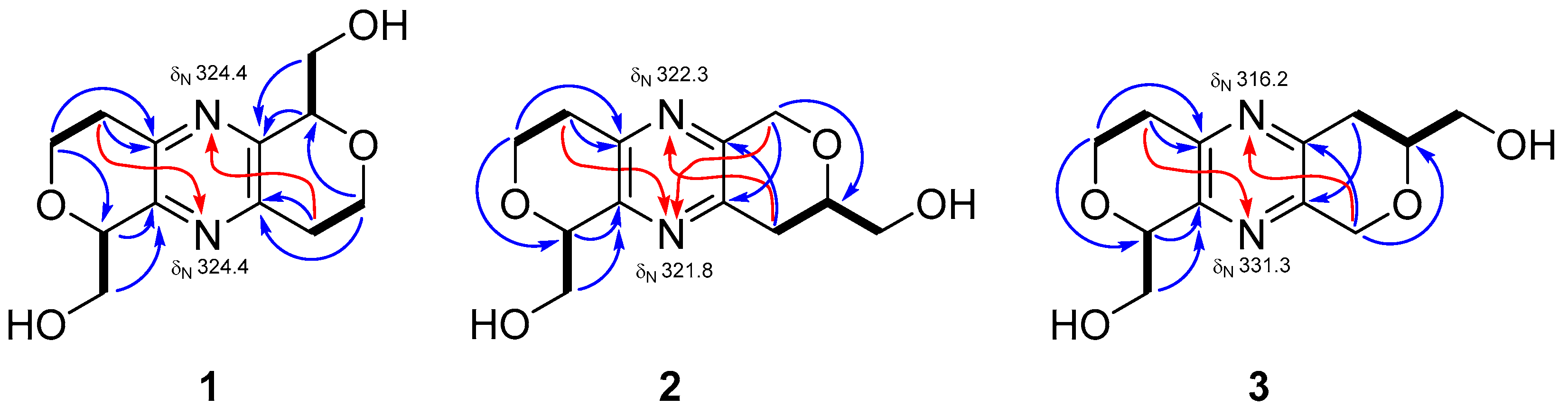
1 is a symmetric compound. In the COSY spectrum (
Figure 2), correlations of H
2-1 (δ
H 3.91 and 4.30)/H
2-2 (δ
H 2.84 and 3.15) and H-13 (δ
H 4.74)/H
2-15 (δ
H 3.98 and 4.06) were found. These two proton sequences were linked by virtue of the HMBC correlation (
Figure 2) of H
2-1/C-13 (δ
C 79.8). The deshielded chemical shifts of C-1 (δ
C 64.5) and C-13 (δ
C 79.8) suggested that an oxygen atom should be located between them. The HMBC correlations from H-13 and H-15 to C-12 (δc 149.9) indicated that C-13 and C-12 were connected. In addition, HMBC correlations from H
2-1 and H
2-2 to C-3 (δ
C 149.5) suggested the connection of C-2/C-3. Due to the lacks of direct linkages among nonprotonated carbons and protons in the
1H-
13C HMBC spectrum, the existence of a pyrazine moiety (C-3, N-4, C-5, C-10, N-11, and C-12) was assured by comparing of the characteristic UV and NMR data with the congener, palythazine (
4). However, two possible
1H and
13C symmetric structures (
1 and
1a,
Figure S22) matched the aforementioned 1D and 2D NMR data. In order to differentiate those structures, the
1H-
15N HMBC experiment was executed using a high sensitivity CryoProbe NMR. In the
1H-
15N HMBC spectrum, correlations from H
2-2/H
2-9 to N-4/N-11 (δ
N 324.4) were detected (
Figure 2). These correlations are consistent with the
4J1H-
15N correlations reported for 3,5-dialkylpyridines [
16]. The single nitrogen chemical shift revealed
1 was a nitrogen symmetric compound, which excluded the possibility of structure
1a. Therefore, the planar structure of
1 was established.
The absolute stereochemistry of
1 was determined by its optical rotation and comparing the experimental electronic circular dichroism (ECD) spectrum with the computer generated ECD spectra. The positive optical rotation value of
1 suggested compound
1 was optically active and was not a
meso compound (6
R13
S-
1 or 6
S13
R-
1). Thus, the ECD spectra of two stereoisomers 6
S13
S-
1 and 6
R13
R-
1 were calculated. The experimental ECD spectrum of
1 demonstrated positive Cotton effect at 232 nm and negative Cotton effect at 212 nm, which was consistent with the trend of the calculated ECD spectrum of 6
R13
R-
1 (
Figure 3). From all of those spectroscopic data, structure
1 was unambiguously assigned to tuberazine A.
Tuberazine B (
2) was obtained as white amorphous powder. The molecular formula C
12H
16N
2O
4 and six indices of hydrogen deficiency of
2 were determined by the sodiated ion peak at
m/
z 275.10025 in the HRESIMS. The IR absorption at 3387 cm
−1 revealed the presence of hydroxy functionality. The pyrazine skeleton as
1 was deduced by the UV maximum absorptions at 287 and 212 nm. The UV, IR,
1H, and
13C NMR data of
2 (
Table 1 and
Table 2) were similar to those of
1, which revealed that these two compounds were close related. The major difference between
2 and
1 was twelve carbon peaks were found in the
13C NMR spectrum of
2, suggesting that
2 was an asymmetric compound. In the COSY spectrum, three proton sequences of H
2-1 (δ
H 4.29 and 3.93)/H
2-2 (δ
H 3.12 and 2.84), H-13 (δ
H 4.73)/H
2-15 (δ
H 4.08 and 4.01), and H
2-9 (δ
H 2.89)/H-8 (δ
H 3.93)/H
2-16 (δ
H 3.74 and 3.69) were observed. The deshielded chemical shift of C-1 (δ
C 64.4) and C-13 (δ
C 79.7) together with the HMBC correlations from H
2-1 to C-13 suggested the former two proton sequences were connected by an ether bridge. In addition, HMBC correlations from H
2-2 to C-3 (δ
C 149.2) and from H-13 to C-12 (δ
C 150.3) revealed C-2 and C-13 were connected to the pyrazine moiety. The HMBC correlations from H
2-6 (δ
H 4.84 and 4.76) to C-8 (δ
C 77.2), C-10 (δ
C 149.4) and from H
2-9 to C-5 (δ
C 148.4) denoted C-5, C-6, O-7, C-8, C-9, and C-10 formed a hexacyclic ring. Considering the structures of congeners, palythazine and isopalythazine [
17], one oxygen atom might locate on the position of 1 or 14 and the other on the position of 7 or 8. Therefore, four possible structures
2,
2a,
2b, and
3 were proposed (
Figure S23). The structure of
2 was also confirmed by the
1H-
15N HMBC experiment. As a result, H
2-2 and H
2-6 showed
4J correlations to N-11 (δ
N 321.8), and H
2-9 correlated to N-4 (δ
N 322.3). The same as
1, the computer generated ECD spectra of
2 were proposed for determining its absolute stereochemistry. The calculated ECD spectra for isomers weren’t significantly different, so only the planar structure without stereochemistry is illustrated.
The molecular formula of tuberazine C (
3), C
12H
16N
2O
4, was deduced from the pseudo-molecular ion peak at
m/
z 275.10023 [M + Na]
+. The IR, UV, Mass, and NMR spectrometric data of
3 indicated it was a close analogue of
2. In the COSY spectrum, cross-peaks of H
2-1 (δ
H 4.30 and 3.93)/H
2-2 (δ
H 3.15 and 2.83) and H-13 (δ
H 4.71)/H
2-15 (δ
H 4.06 and 3.99) were found (
Figure 2). These two proton sequences and the HMBC correlations of H
2-1/C-13 (δ
C 79.7), H
2-2/C-3 (δ
C 149.6), and H-13/C-12 (δ
C 149.9) were used to establish the dihydro-2
H-pyran moiety connecting a pyrazine. On the other hand, the COSY correlations of H
2-6 (δ
H 2.86)/H-7 (δ
H 3.91)/H
2-16 (δ
H 3.73 and 3.69) together with the HMBC correlations of H
2-9 (δ
H 4.85 and 4.76)/C-5 (δ
C 149.5), C-7 (δ
C 77.1) and H
2-6/C-10 (δ
C 148.4) supported another dihydro-2
H-pyran moiety attaching at the pyrazine. Therefore, compound
3 had the same possible structures (
Figure S23) as those of
2. The difference between
3 and
2 could be the oxygen atom locating in different position. This assumption was confirmed by the
1H-
15N HMBC correlations from H
2-2 (δ
H 3.15 and 2.83) to N-11 (δ
N 331.3) and from H
2-9 (δ
H 4.85 and 4.76) to N-4 (δ
N 316.2). Hence, two oxygen atoms were placed in positions 8 and 14, and the structure of
3 was determined as shown.
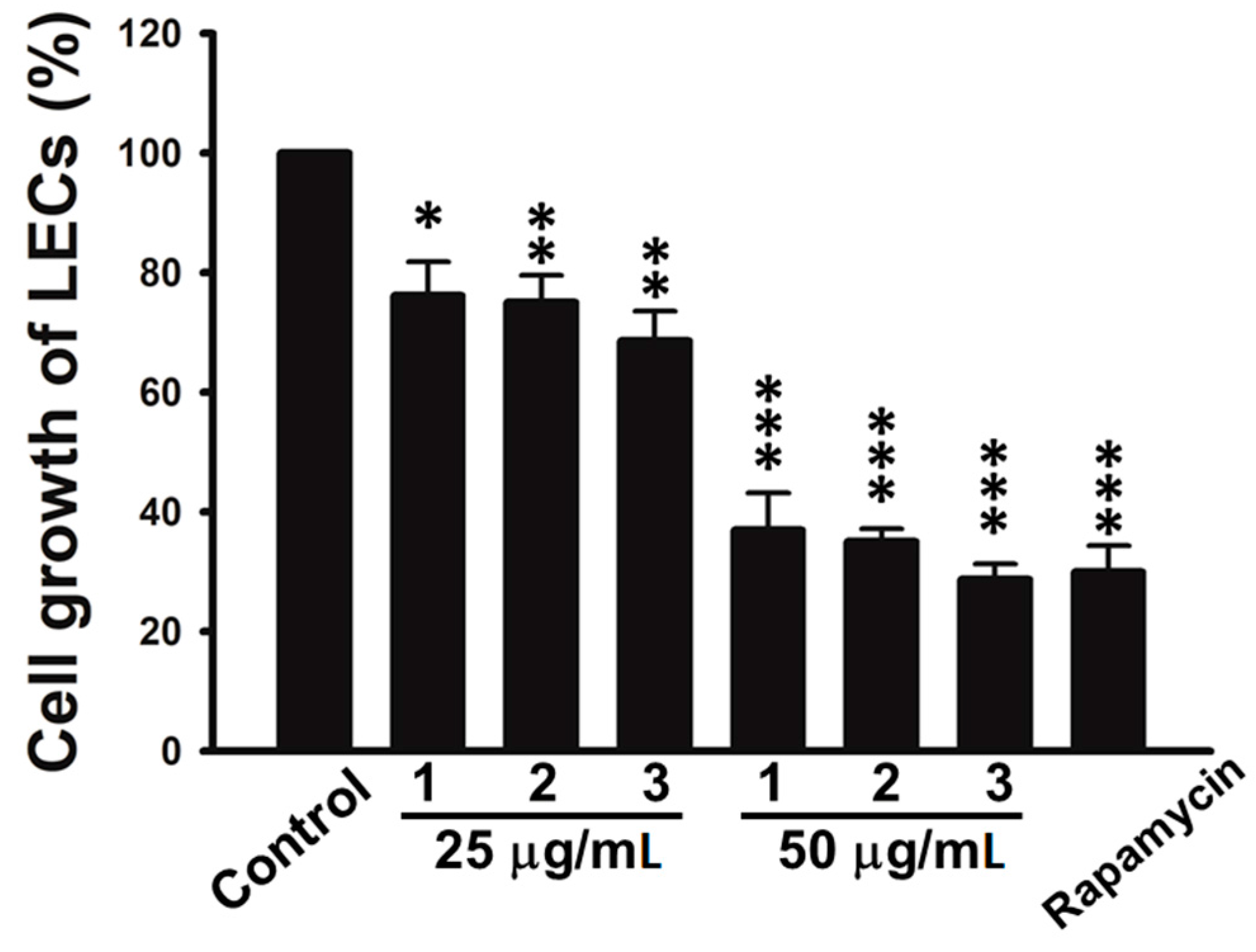
In this study, all isolated compounds were evaluated for their anti-cancer activities. The results showed that the isolates (
1–
14) exhibited no significant activities against three human cancer cell lines (A549, HepG2, and MDA-MB231). However, we found that tuberazines A–C (
1–
3) exerted promising anti-lymphangiogeneic activities in human lymphatic endothelial cells (LECs). Lymphangiogenesis has been shown to provoke tumor progression and lymphatic metastasis. Vascular endothelial growth factor-C (VEGF-C) is the most dominant lymphangiogenic factor, acting through VEGF receptor-3 (VEGFR-3) that is specifically expressed by LECs [
18]. The activation of VEGF-C/VERFR-3 axis is responsible for LECs growth, migration and tube formation during lymphangiogenic process [
2]. As shown in
Figure 4, tuberazines A–C (
1–
3) inhibited cell growth of LECs in a concentration dependent manner. Tuberazines C (
3) exhibited the most potent anti-lymphangiogeneic activity by acting the inhibitory effect on LECs growth (IC
50 = 33 ± 1 μg/mL). Rapamycin, a well-known lymphangiogenesis inhibitor, was used as a positive control for in vitro anti-lymphangiogenesis assay (the IC
50 value of compounds
1–
3 and rapamycin are shown in
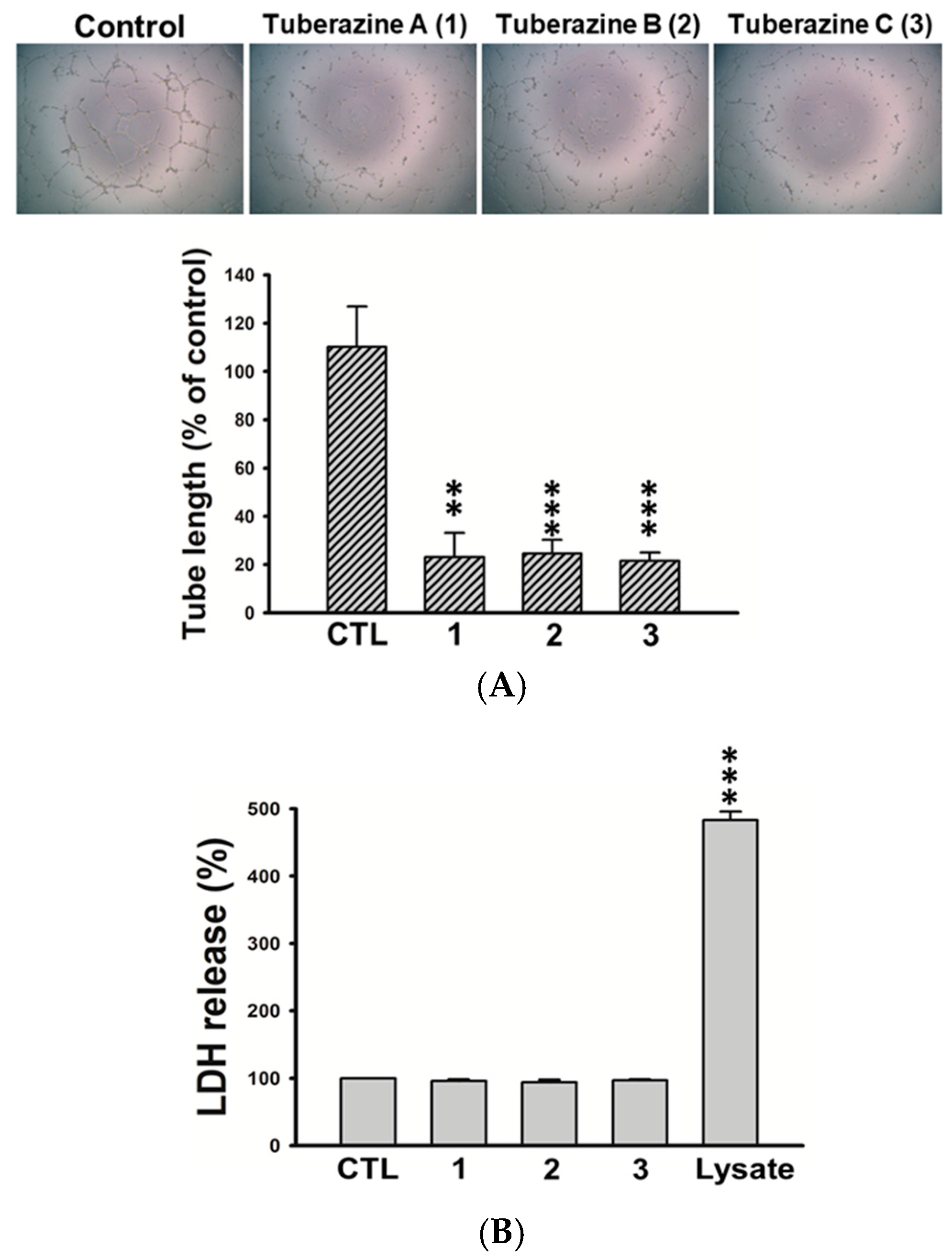
Table S1). Capillary-like tubules are regarded as representative stage during lymphangiogenesis, we next performed tube formation assay to validate the anti-lymphangiogeneic effect of three new compounds (
1–
3) in human LECs. The results showed that tuberazines A–C (
1–
3) manifestly suppressed tube formation of LECs (
Figure 5A). In addition, we found that tuberazines A–C (
1–
3) did not induce the significant lactate dehydrogenase (LDH) release in LECs (
Figure 5B). Based upon these findings, we suggest that inhibitory activities of these compounds on lymphangiogenesis are not due to their cytotoxicity. Therefore, tuberazines A–C (
1–
3) are the promising marine products worthy of further development for impeding tumor lymphangiogenesis and metastasis.
4. Materials and Methods
4.1. General Experimental Procedures
Perkin Elmer (Waltham, MA, USA) system 2000 FT-IR spectrophotometer was used for IR spectrum measurement. JASCO (Tokyo, Japan) P-1020 digital polarimeter was utilized for optical rotation measurement. UV and ECD spectra were recorded on JASCO V-530 UV/VIS spectrophotometer and JASCO J-815 CD spectrometer, respectively. Electrospray ionization (ESI) mass data were obtained from Waters (Milford, MA, USA) 2695 separations module and Bruker (Billerica, MA, USA) APEX II spectrometer (high resolution). NMR spectra were obtained by Bruker AVIII HD 700 MHz FT-NMR and AVAN CEIII 600 MHz FT-NMR. Merck (Darmstadt, Germany) silica gel 60 and GE Healthcare (Chicago, IL, USA) Sephadex LH-20 were used for column chromatography. The instrumentation for HPLC was composed of a Shimadzu (Kyoto, Japan) LC-20AD pump and a Shimadzu SPD-M20A PDA detector.
4.2. Animal Material
Specimens of Palythoa tuberculosa were collected in Taitung County, Taiwan, in April 2016. The research samples were identified by Dr. Yuan-Bin Cheng. A voucher specimen (no. KMU-PT1) was deposited in the Graduate Institute of Natural Products, College of Pharmacy, Kaohsiung Medical University.
4.3. Extraction and Isolation
The animal materials were lyophilized and the dry materials were extracted by ethanol three times. The ethanolic extract was partitioned between ethyl acetate and water (1:1) to afford two different polarity layers. The water layer was further partitioned by n-butanol and water (1:1) to give an n-butanol soluble extract. This extract (3.2 g) was chromatographed by a Sephadex LH-20 column eluted with methanol to give five fractions (A–E). Fraction D (958.0 mg) was separated by a Si gel open column stepwise eluted with dichloromethane and methanol (30:1 to 0:1) to furnish seven fractions (D1–D7). Fraction D1 (46.6 mg) was isolated by RP-HPLC (phenyl-hexyl, 15% methanol, isocratic elution) to afford compound 1 (1.5 mg). Fraction D-3 (40.6 mg) was purified by RP-HPLC (phenyl-hexyl, 20% methanol, isocratic elution) to yield compounds 2 (2.3 mg), 3 (2.3 mg) and 5 (12.4 mg). Fraction D6 (30.1 mg) was separated by RP-HPLC (C18, 5% to 100% methanol, gradient elution) to give compounds 4 (1.5 mg) and 9 (5.8 mg). Fraction D7 (230.4 mg) was chromatographed by a Si gel column stepwise eluted with dichloromethane and methanol (10:1 to 0:1) to furnish nine fractions (D7A–D7I). Fraction D7H (87.3 mg) was isolated by RP-HPLC (C18, 5% to 95% acetonitrile, gradient elution) to give compound 6 (8.4 mg). Fraction D7I (36.2 mg) was purified by RP-HPLC (C18, 1% methanol, isocratic elution) to yield compounds 7 (1.9 mg) and 8 (2.1 mg). On the other hand, the ethyl acetate was subsequently partitioned between n-hexane and 75% methanol. The 75% methanolic layer (4.1 g) was subjected to a Sephadex LH-20 column eluted with methanol to afford fractions F–I. Fraction G (4.1 g) was isolated by a Si gel open column stepwise eluted with dichloromethane and methanol (34:1 to 5:1) to give fractions G1–G6. Fraction G2 (255.8 mg) was purified by another Si gel column isocratic eluted with ethyl acetate and methanol (6:1) to give subfractions G2A–G2C. Subfraction G2B (16.7 mg) was isolated by RP-HPLC (phenyl-hexyl, 25% to 55% acetonitrile, gradient elution) to give compounds 13 (0.5 mg) and 14 (0.6 mg). Subfraction G2C (42.6 mg) was purified by RP-HPLC (phenyl-hexyl, 50% acetonitrile, isocratic elution) to give compound 12 (6.3 mg). Fraction G3 (357.9 mg) was separated by Si gel column isocratic eluted with dichloromethane and methanol (9:1) to afford subfractions G3A–G3E. Subfraction G3D (58.6 mg) was further isolated by RP-HPLC (phenyl-hexyl, 20% acetonitrile, isocratic elution) to yield compounds 10 (2.7 mg) and 11 (12.0 mg).
Tuberazine A (
1): colorless amorphous powder;
+96 (
c 0.05, MeOH); UV (MeOH)
λmax (log ε) 289 (4.04), 212 (4.23) nm; ECD (MeOH)
λmax (Δε): 212 (−4.65), 232 (+7.64) nm; IR (ATR)
νmax: 3389, 2972, 2920, 1685, 1520, 1086, 882 cm
−1;
1H NMR and
13C NMR data, see
Table 1 and
Table 2; HRESIMS
m/
z 275.10028 [M + Na]
+ (calcd. for C
12H
16N
2O
4Na, 275.10023).
Tuberazine B (
2): colorless amorphous powder;
+22 (
c 0.05, MeOH); UV (MeOH)
λmax (log ε) 287 (4.08), 212 (4.12) nm; ECD (MeOH)
λmax (Δε): 215 (−1.27), 319 (+0.89) nm; IR (ATR)
νmax: 3387, 2974, 2916, 1657, 1231, 1091, 1085, 884 cm
−1;
1H NMR and
13C NMR data, see
Table 1 and
Table 2; HRESIMS
m/
z 275.10025 [M + Na]
+ (calcd. for C
12H
16N
2O
4Na, 275.10023).
Tuberazine C (
3): colorless amorphous powder;
+45 (
c 0.05, MeOH); UV (MeOH)
λmax (log ε) 288 (4.04), 213 (4.23) nm; ECD (MeOH)
λmax (Δε): 214 (−0.95), 291 (+1.11) nm; IR (ATR)
νmax: 3387, 2922, 1648, 1407, 1082, 1059, 876 cm
−1;
1H NMR and
13C NMR data, see
Table 1 and
Table 2; HRESIMS
m/
z 275.10014 [M + Na]
+ (calcd. for C
12H
16N
2O
4Na, 275.10023).
4.4. Ecd Calculations
The lowest energies of 6S13S-1 and 6R13R-1 were calculated and the data were performed by the Gaussian 09 software (Gaussian Inc., Wallingford, CT, USA). The density functional theory (DFT) at the B3LYP/6-31G(d) level in the gas phase were used to obtain the restricted conformation. The minima energies of 20 conformers were computed by the time-dependent density functional theory (TDDFT) methodology at the B3LYP/6-311++G(d,p) level. The final ECD files were generated by GaussSum 2.2.5 software with a bandwidth σ of 0.5 eV. The calculated ECD and experimental ECD curves were drawn by Excel.
4.5. Cell Culture of Human LECs
The human telomerase-immortalized human dermal lymphatic endothelial cells (hTERT-HDLECs), an immortalized human LEC line, was purchased from Lonza (Walkersville, MD, USA). The cultivation and maintenance of human LECs were performed as described previously [
25]. Briefly, LECs were grown in EGM-2MV BulletKit medium consisting of EBM-2 basal medium plus SingleQuots kit (Lonza, Basel, Switzerland). Cells were seeded onto 1% gelatin-coated plastic ware and cultured at 37 °C with 5% CO
2 for further treatment. Experiments were conducted on LECs between passages 10 and 20.
4.6. Cell Growth Assay
LECs were seeded onto 96-well plates in at the density of 5 × 103 cells per well. After 24 h incubation, the culture medium was removed and cells were treated with EGM-2MV BulletKit medium in the absence or presence of tested compounds for 48 h. Then, cells were fixed with 50% TCA to terminate reaction, and 0.4% SRB (Sigma, St. Louis, MO, USA) in 1% acetic acid was added to each well. After a 15-min incubation, the plates were washed, and dye was dissolved by 10 mM Tris buffer. The 96-well plate was read by enzyme-linked immunosorbent assay (ELISA) reader (515 nm) to get the absorbance density values.
4.7. Capillary Tube Formation Assay
Matrigel (50 μL) was added to 96-well plates for determining the differentiation of LECs into a capillary tube-like structure. Matrigel-coated 96-well plates were incubated at 37 °C for 30 min to allow for polymerization. After gel formation, LECs were seeded per well at a density of 2 × 104/200 μL in EGM-2MV BulletKit medium with the indicated concentration of tested compounds. After 8 h incubation, LECs tube formation were taken with the inverted phase contrast microscope. The number of tube branches and total tube length were calculated using the MacBiophotonics Image J software.
4.8. Cytotoxicity Assay
LECs were seeded onto 96-well plates at the density of 5 × 103 cells per well. After 24 h incubation, cells were treated with the EGM-2MV BulletKit medium with the indicated concentration of tested compounds. Then, the percentage of LDH release in the collected medium was determined by non-radioactive cytotoxicity assay kit (Promega, Madison, WI, USA).
4.9. Anti-Cancer Assay
A549, HepG2, and MDA-MB-231 cells were cultured on 96-well plate at the density of 5 × 103 cells per well. Tested compound were afterwards added for 72 h. The cell viability was determined by the MTT assay. Doxorubicin and paclitaxel were used as positive controls. ELISA reader (Thermo Electron Cooperation, Waltham, MA, USA) was used for absorbance (550 nm) measurement.
4.10. Statistical Analysis
Data are presented as the mean ± SEM for the indicated number of separate experiment. Statistical analyses of data were performed with one-way ANOVA followed by Student’s t-test, and p-values less than 0.05 were considered significant.

 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}