ATG5 Promotes Death Signaling in Response to the Cyclic Depsipeptides Coibamide A and Apratoxin A

 ,
,  ,
, 

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
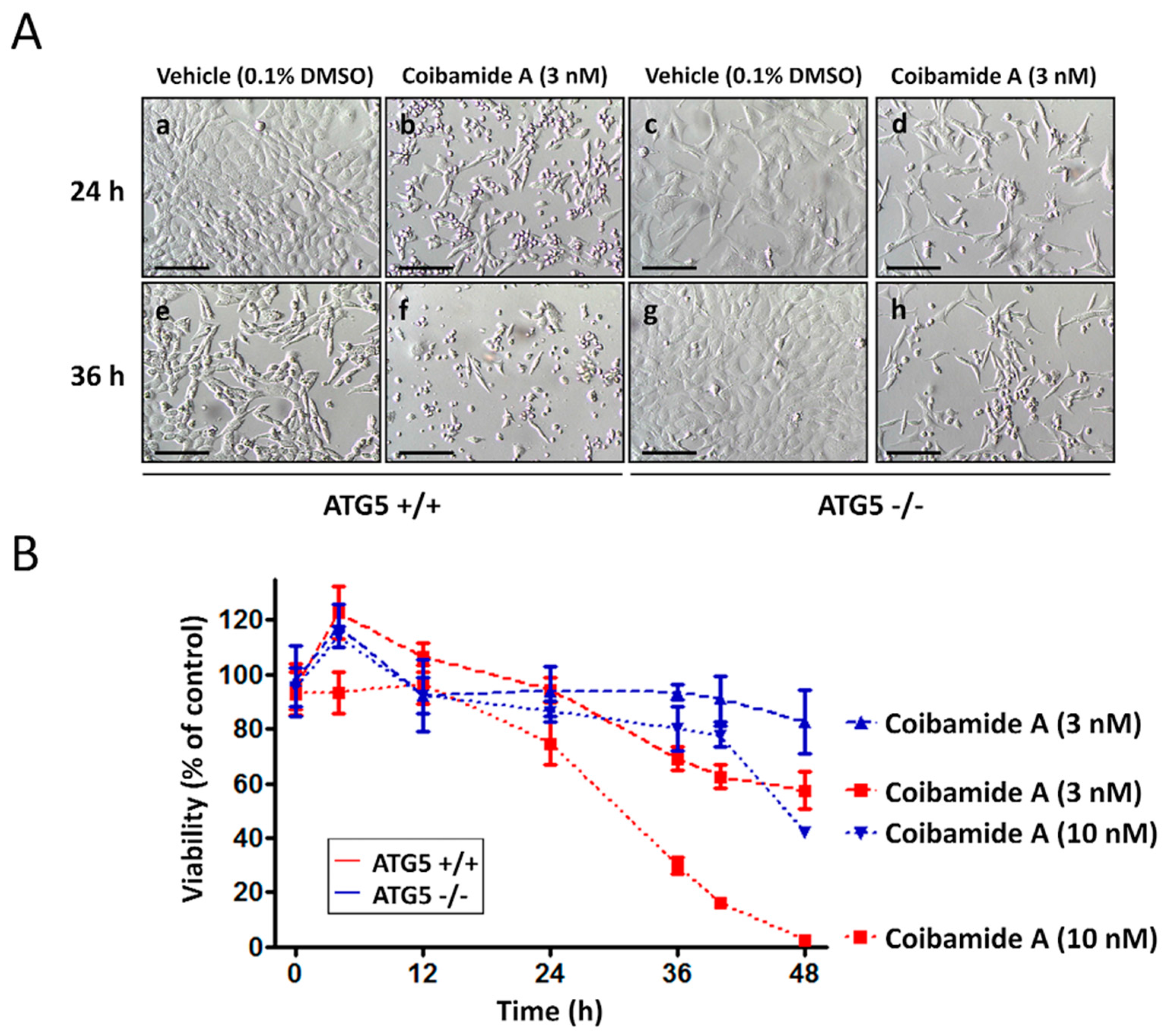
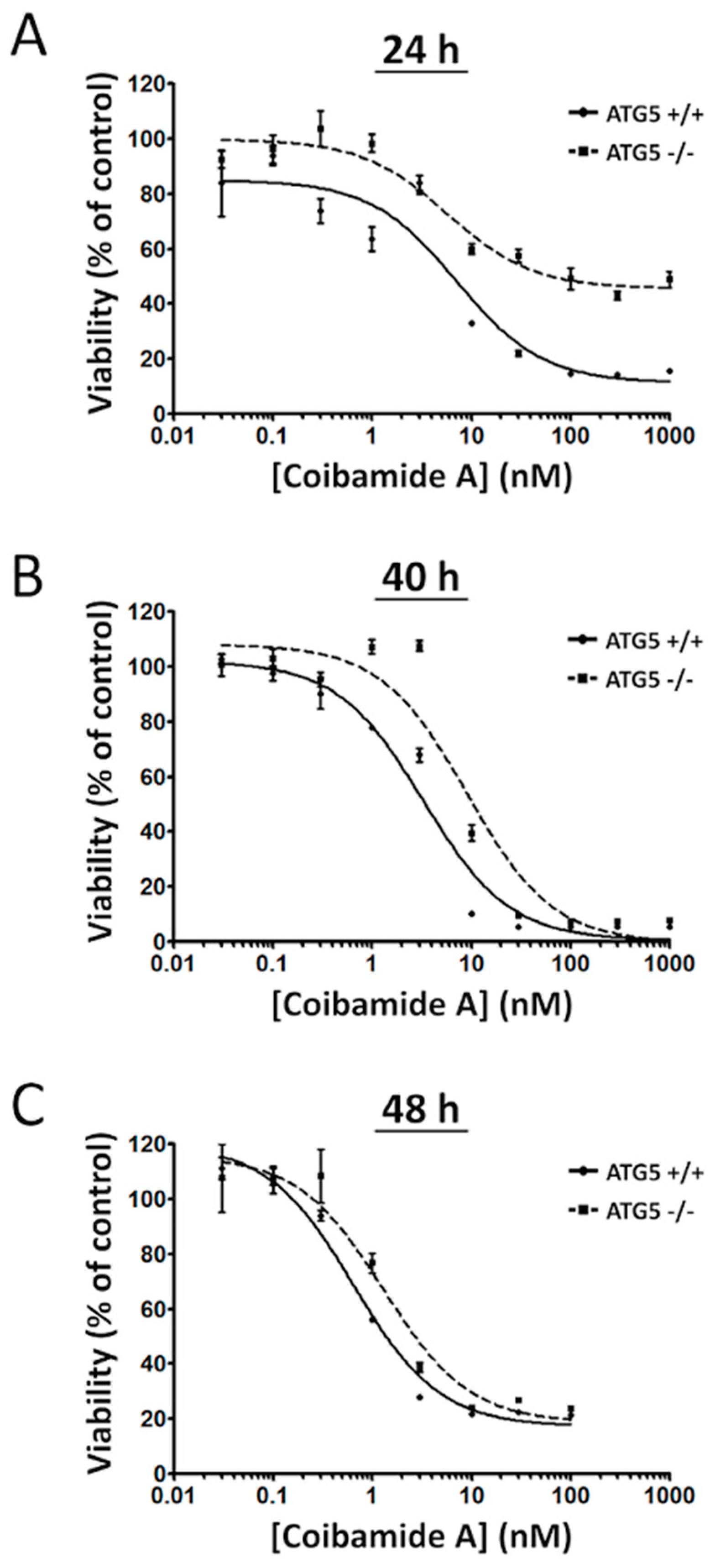
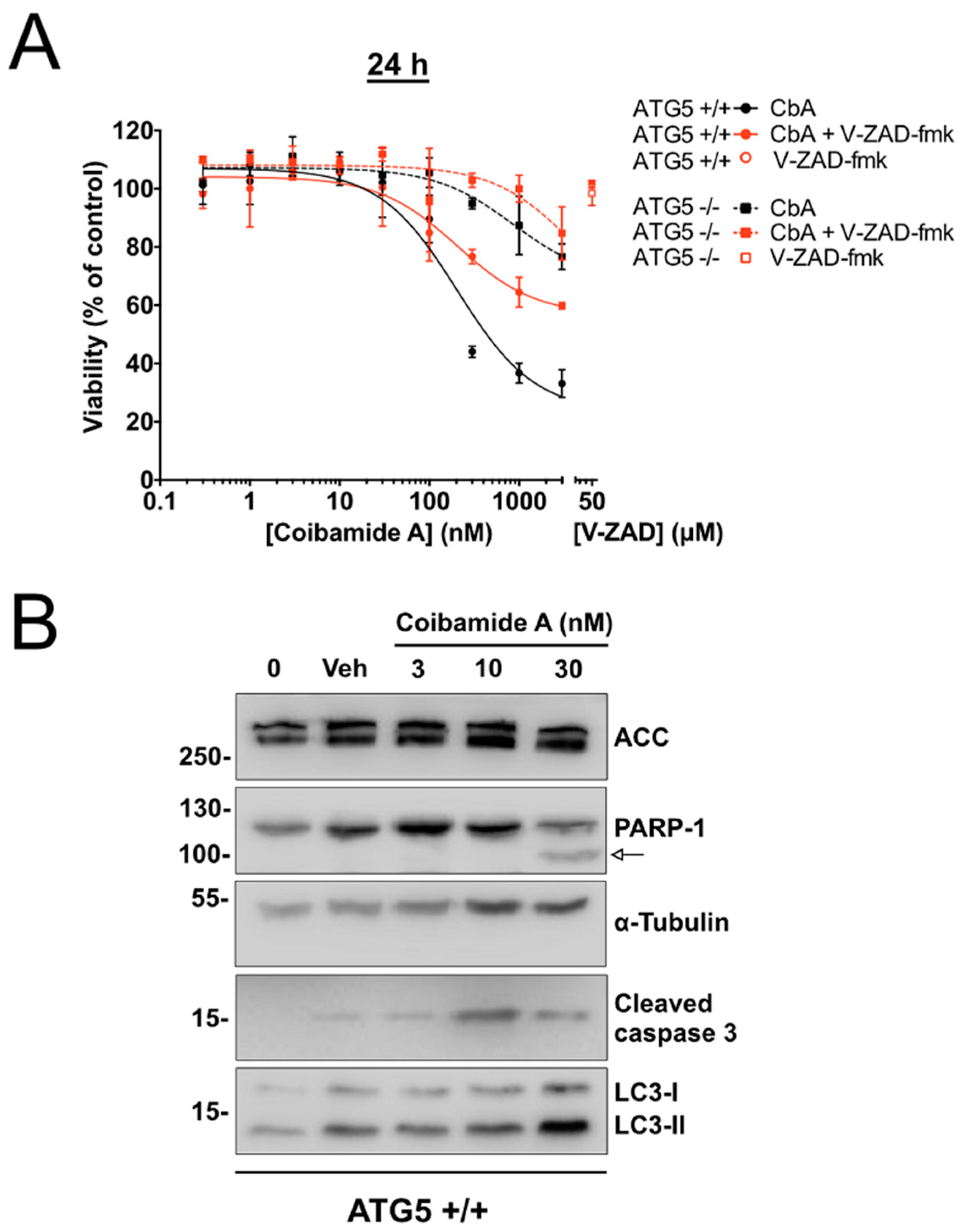
2.1. Wild-Type and ATG5-Null MEFs Show Differential Sensitivity to Coibamide A
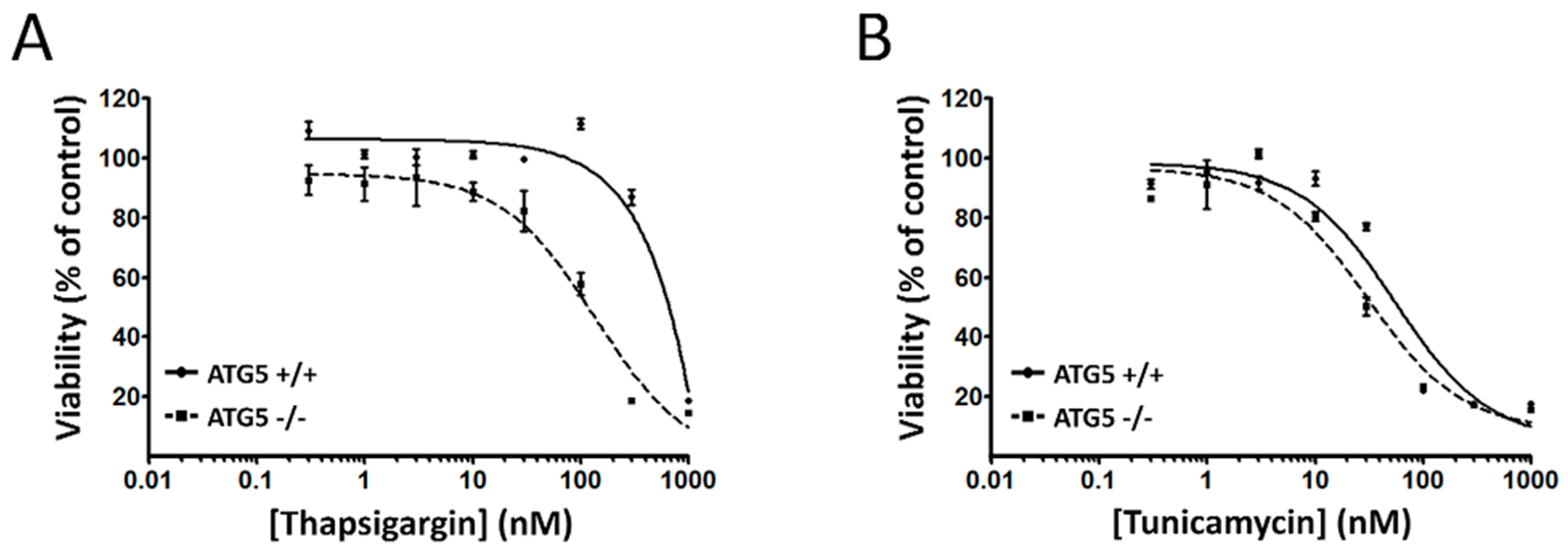
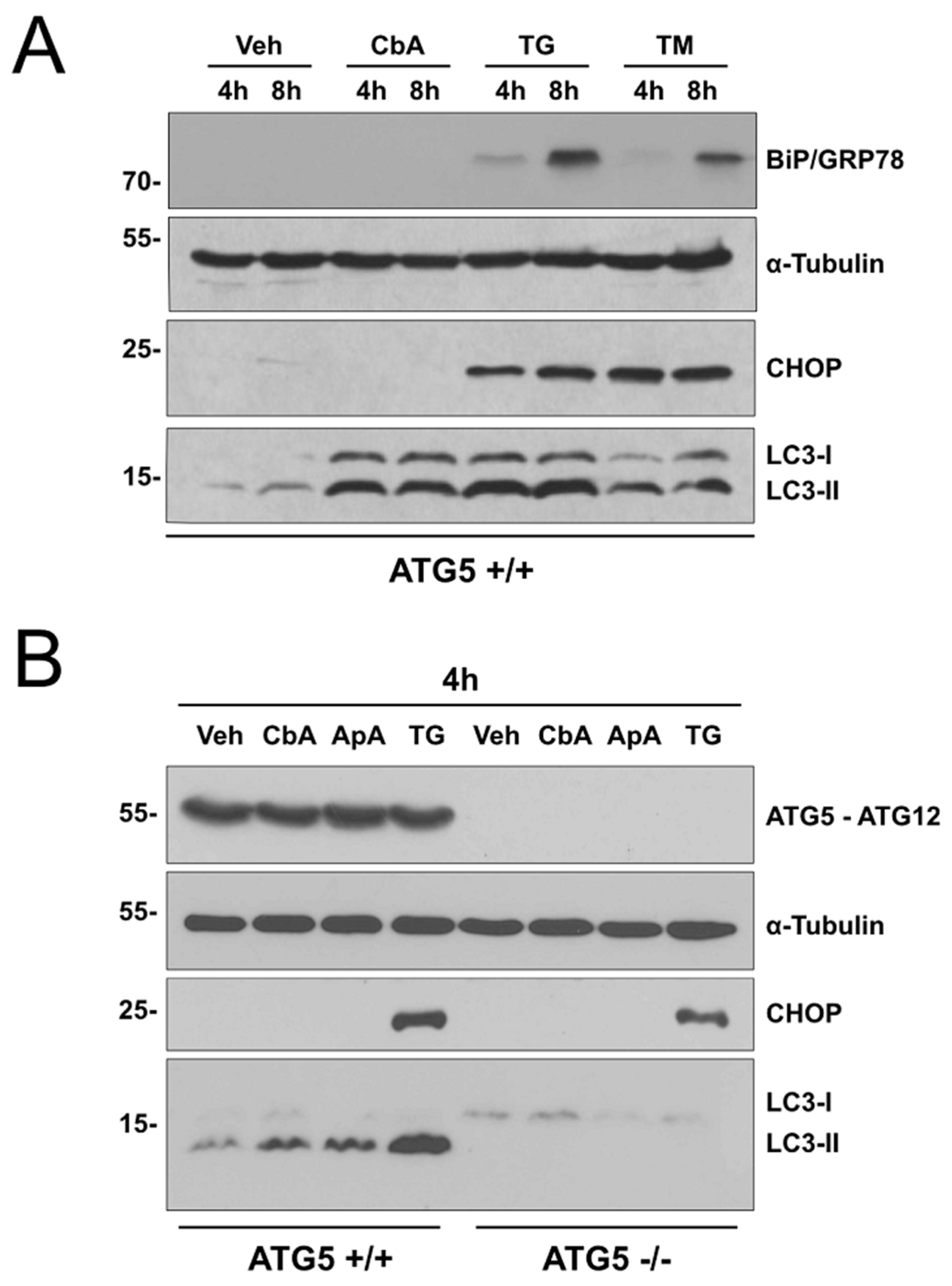
2.2. Coibamide A-Induced Autophagy Is Not Triggered by Acute ER Stress
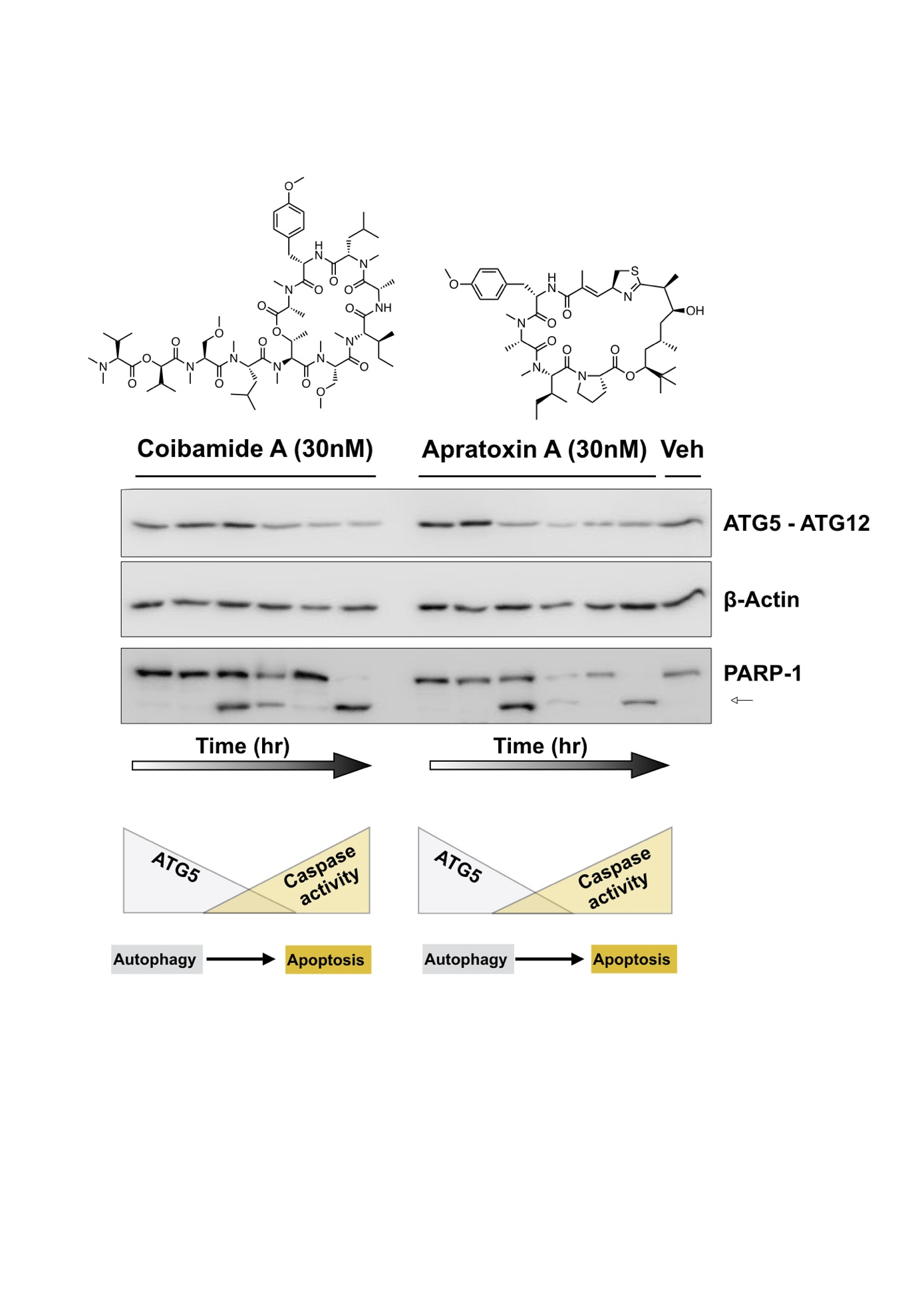
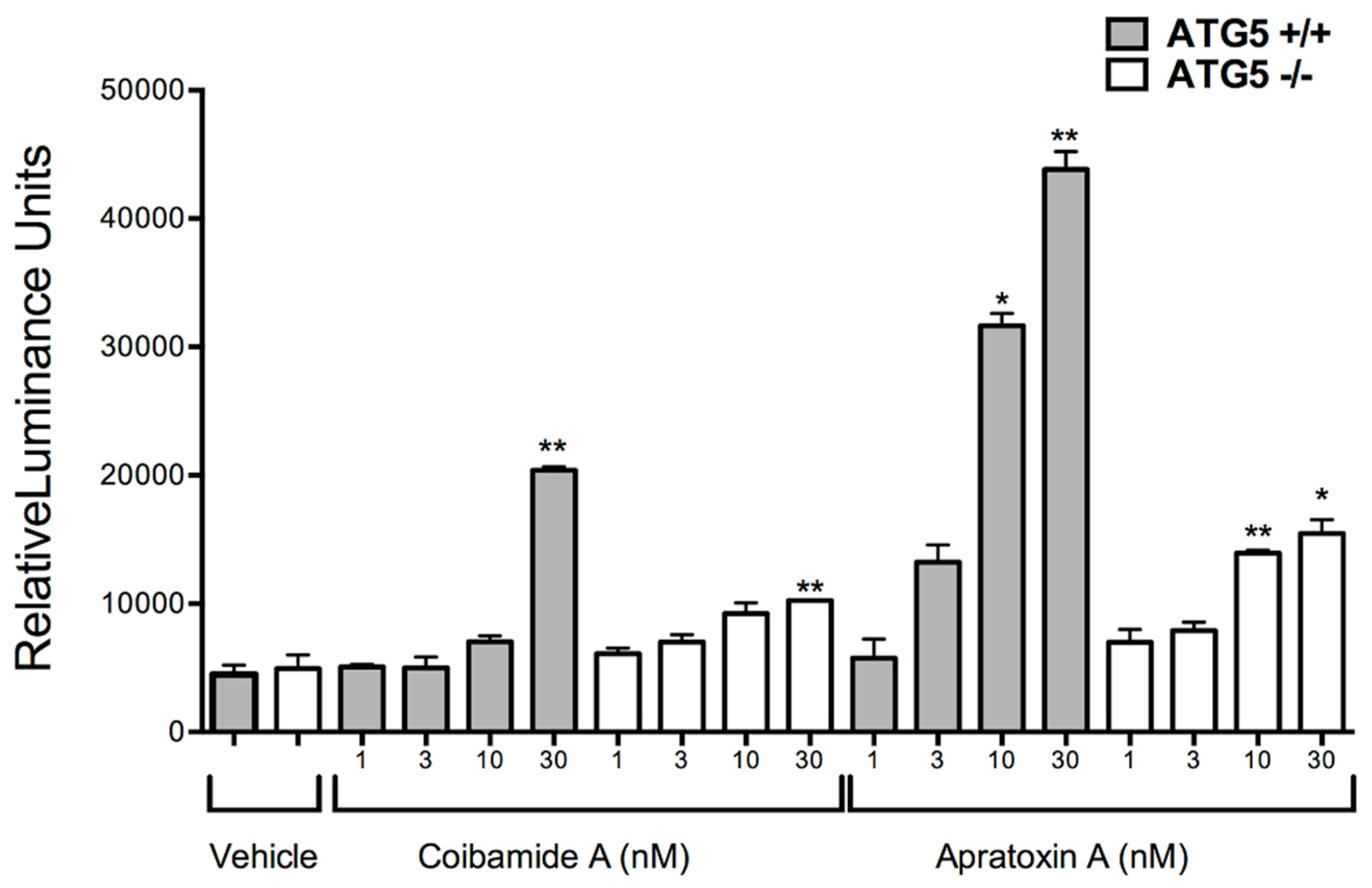
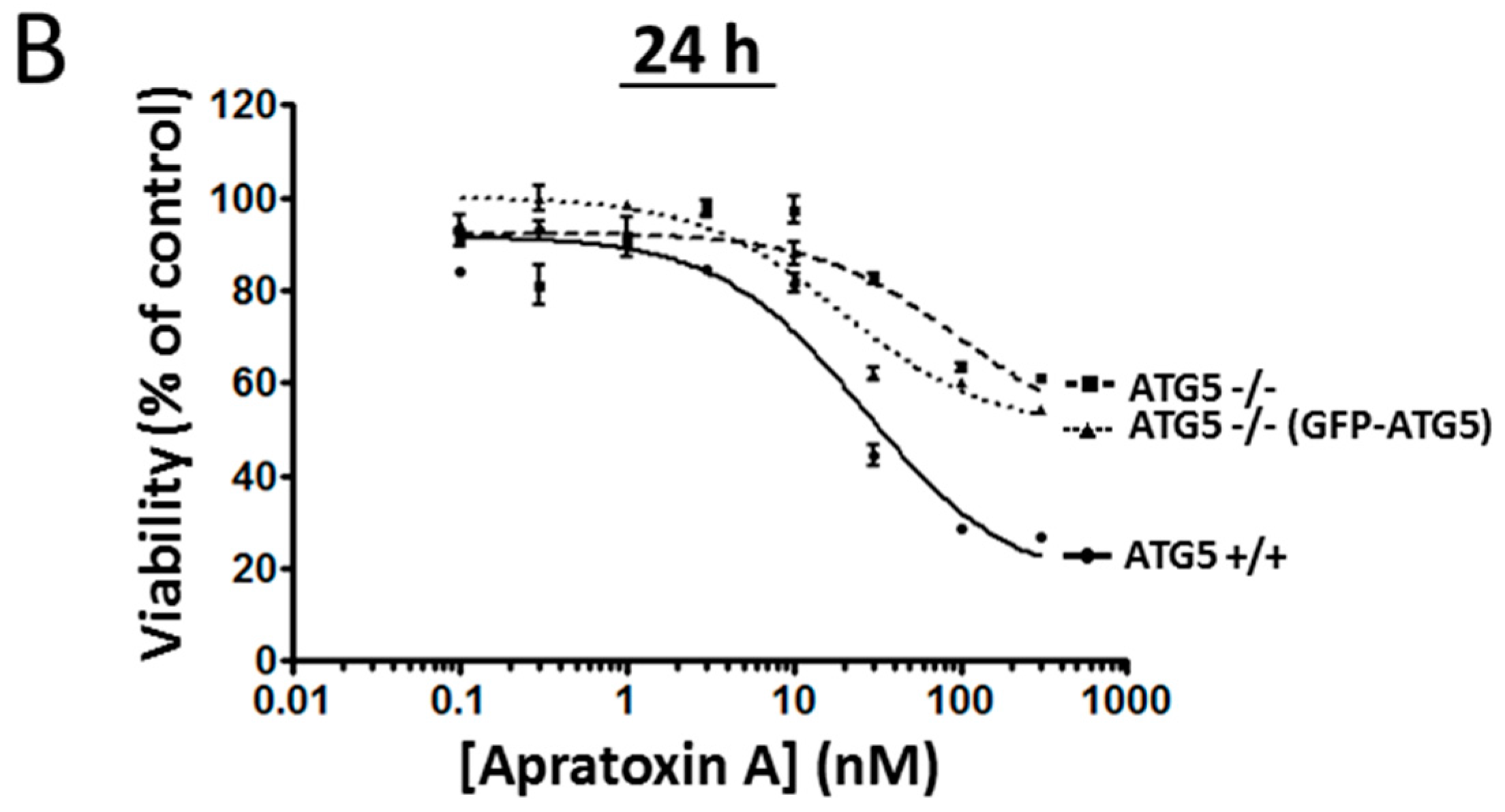
2.3. Comparison of Coibamide- and Apratoxin-Induced Cell Death
2.4. Partial Rescue of the Wild-Type Phenotype by Expression of ATG5 in ATG5-Null MEFs
3. Discussion
4. Materials and Methods
4.1. Chemicals, Reagents and Antibodies
4.2. Mammalian Cell Culture
4.3. Analysis of Cell Morphology, Viability and Caspase Activity
4.4. Detection of Annexin V-FITC and Propidium Iodide by FACS
4.5. Immunoblot Analysis
4.6. Data Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| PVDF | Polyvinylidene difluoride |
| SDS-PAGE | sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| SCUBA | self-contained underwater breathing apparatus |
| Z-VAD-fmk | N-Benzyloxycarbonyl-Val-Ala-Asp(O-Me) fluoromethyl ketone |
| FACS | fluorescence-activated cell sorting |
| DMSO | dimethylsulfoxide |
References
- Rubinstein, A.D.; Kimchi, A. Life in the balance—A mechanistic view of the crosstalk between autophagy and apoptosis. J. Cell Sci. 2012, 125 Pt 22, 5259–5268. [Google Scholar] [CrossRef] [PubMed]
- Marino, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Alnemri, E.S.; Altucci, L.; Andrews, D.; Annicchiarico-Petruzzelli, M.; et al. Essential versus accessory aspects of cell death: Recommendations of the NCCD 2015. Cell Death Differ. 2015, 22, 58–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denton, D.; Xu, T.; Kumar, S. Autophagy as a pro-death pathway. Immunol. Cell Biol. 2015, 93, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Klionsky, D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell. 2004, 6, 463–477. [Google Scholar] [CrossRef]
- Levine, B.; Packer, M.; Codogno, P. Development of autophagy inducers in clinical medicine. J. Clin. Investig. 2015, 125, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Mah, L.Y.; Ryan, K.M. Autophagy and cancer. Cold Spring Harb. Perspect. Biol. 2012, 4, a008821. [Google Scholar] [CrossRef] [PubMed]
- Driggers, E.M.; Hale, S.P.; Lee, J.; Terrett, N.K. The exploration of macrocycles for drug discovery—An underexploited structural class. Nat. Rev. Drug Discov. 2008, 7, 608–624. [Google Scholar] [CrossRef] [PubMed]
- Bockus, A.T.; McEwen, C.M.; Lokey, R.S. Form and function in cyclic peptide natural products: A pharmacokinetic perspective. Curr. Top. Med. Chem. 2013, 13, 821–836. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werner, G.; Hagenmaier, H.; Drautz, H.; Baumgartner, A.; Zahner, H. Metabolic products of microorganisms. 224. Bafilomycins, a new group of macrolide antibiotics. Production, isolation, chemical structure and biological activity. J. Antibiot. (Tokyo) 1984, 37, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Bowman, E.J.; Siebers, A.; Altendorf, K. Bafilomycins: A class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc. Natl. Acad. Sci. USA 1988, 85, 7972–7976. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Feng, Y.; Forgac, M. Proton conduction and bafilomycin binding by the V0 domain of the coated vesicle V-ATPase. J. Biol. Chem. 1994, 269, 23518–23523. [Google Scholar] [PubMed]
- Yoshimori, T.; Yamamoto, A.; Moriyama, Y.; Futai, M.; Tashiro, Y. Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J. Biol. Chem. 1991, 266, 17707–17712. [Google Scholar] [PubMed]
- Yamamoto, A.; Tagawa, Y.; Yoshimori, T.; Moriyama, Y.; Masaki, R.; Tashiro, Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct. Funct. 1998, 23, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Mauvezin, C.; Neufeld, T.P. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy 2015, 11, 1437–1438. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhu, L.; Sun, D.; Gan, F.; Gao, S.; Yin, Y.; Chen, L. Natural products as modulator of autophagy with potential clinical prospects. Apoptosis 2017, 22, 325–356. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, S.N.; Baker, H.; Vezina, C. Rapamycin (AY-22,989), a new antifungal antibiotic. II. Fermentation, isolation and characterization. J. Antibiot. (Tokyo) 1975, 28, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.J.; Albers, M.W.; Shin, T.B.; Ichikawa, K.; Keith, C.T.; Lane, W.S.; Schreiber, S.L. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 1994, 369, 756–758. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Ruegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Hau, A.M.; Greenwood, J.A.; Lohr, C.V.; Serrill, J.D.; Proteau, P.J.; Ganley, I.G.; McPhail, K.L.; Ishmael, J.E. Coibamide A induces mTOR-independent autophagy and cell death in human glioblastoma cells. PLoS ONE 2013, 8, e65250. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Noda, T.; Yoshimori, T.; Tanaka, Y.; Ishii, T.; George, M.D.; Klionsky, D.J.; Ohsumi, M.; Ohsumi, Y. A protein conjugation system essential for autophagy. Nature 1998, 395, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Nomenclature Committee on Cell, D. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009, 16, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Pyrko, P.; Schonthal, A.H.; Hofman, F.M.; Chen, T.C.; Lee, A.S. The unfolded protein response regulator GRP78/BiP as a novel target for increasing chemosensitivity in malignant gliomas. Cancer Res. 2007, 67, 9809–9816. [Google Scholar] [CrossRef] [PubMed]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J.; Corbett, T.H. Total structure determination of apratoxin, A.; a potent novel cytotoxin from the marine cyanobacterium Lyngbya majuscula. J. Am. Chem. Soc. 2001, 123, 5418–5423. [Google Scholar] [CrossRef] [PubMed]
- Serrill, J.D.; Wan, X.; Hau, A.M.; Jang, H.S.; Coleman, D.J.; Indra, A.K.; Alani, A.W.; McPhail, K.L.; Ishmael, J.E. Coibamide A, a natural lariat depsipeptide, inhibits VEGFA/VEGFR2 expression and suppresses tumor growth in glioblastoma xenografts. Investig. New Drugs 2016, 34, 24–40. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, S.; Perozzo, R.; Schmid, I.; Ziemiecki, A.; Schaffner, T.; Scapozza, L.; Brunner, T.; Simon, H.U. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat. Cell Biol. 2006, 8, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- Ganley, I.G.; Wong, P.M.; Gammoh, N.; Jiang, X. Distinct autophagosomal-lysosomal fusion mechanism revealed by thapsigargin-induced autophagy arrest. Mol. Cell 2011, 42, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Medina, R.A.; Goeger, D.E.; Hills, P.; Mooberry, S.L.; Huang, N.; Romero, L.; Ortega-Barria, E.; Gerwick, W.H.; McPhail, K.L. Coibamide, A, a Potent Antiproliferative Cyclic Depsipeptide from the Panamanian Marine Cyanobacterium Leptolyngbya sp. J. Am. Chem. Soc. 2008, 130, 6324–6325. [Google Scholar] [CrossRef] [PubMed]
- Luesch, H.; Chanda, S.K.; Raya, R.M.; DeJesus, P.D.; Orth, A.P.; Walker, J.R.; Izpisua Belmonte, J.C.; Schultz, P.G. A functional genomics approach to the mode of action of apratoxin, A. Nat. Chem. Biol. 2006, 2, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Law, B.K.; Luesch, H. Apratoxin a reversibly inhibits the secretory pathway by preventing cotranslational translocation. Mol. Pharmacol. 2009, 76, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Paatero, A.O.; Kellosalo, J.; Dunyak, B.M.; Almaliti, J.; Gestwicki, J.E.; Gerwick, W.H.; Taunton, J.; Paavilainen, V.O. Apratoxin Kills Cells by Direct Blockade of the Sec61 Protein Translocation Channel. Cell Chem. Biol. 2016, 23, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.C.; Chen, Z.; Jiang, Y.; Akare, S.; Kolber-Simonds, D.; Condon, K.; Agoulnik, S.; Tendyke, K.; Shen, Y.; Wu, K.M.; et al. Apratoxin A Shows Novel Pancreas-Targeting Activity through the Binding of Sec 61. Mol. Cancer Ther. 2016, 15, 1208–1216. [Google Scholar] [CrossRef] [PubMed]
- Ogata, M.; Hino, S.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K.; et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell. Biol. 2006, 26, 9220–9231. [Google Scholar] [CrossRef] [PubMed]
- Yorimitsu, T.; Nair, U.; Yang, Z.; Klionsky, D.J. Endoplasmic reticulum stress triggers autophagy. J. Biol. Chem. 2006, 281, 30299–30304. [Google Scholar] [CrossRef] [PubMed]
- Li, W.W.; Alexandre, S.; Cao, X.; Lee, A.S. Transactivation of the grp78 promoter by Ca2+ depletion. A comparative analysis with A23187 and the endoplasmic reticulum Ca(2+)-ATPase inhibitor thapsigargin. J. Biol. Chem. 1993, 268, 12003–12009. [Google Scholar] [PubMed]
- Wang, X.Z.; Lawson, B.; Brewer, J.W.; Zinszner, H.; Sanjay, A.; Mi, L.J.; Boorstein, R.; Kreibich, G.; Hendershot, L.M.; Ron, D. Signals from the stressed endoplasmic reticulum induce C/EBP-homologous protein (CHOP/GADD153). Mol. Cell. Biol. 1996, 16, 4273–4280. [Google Scholar] [CrossRef] [PubMed]
- Zinszner, H.; Kuroda, M.; Wang, X.; Batchvarova, N.; Lightfoot, R.T.; Remotti, H.; Stevens, J.L.; Ron, D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998, 12, 982–995. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.S.; Hill, K.; McKenna, M.; Ogbechi, J.; High, S.; Willis, A.E.; Simmonds, R.E. The pathogenic mechanism of the Mycobacterium ulcerans virulence factor, mycolactone, depends on blockade of protein translocation into the, E.R. PLoS Pathog. 2014, 10, e1004061. [Google Scholar] [CrossRef] [PubMed]
- Baron, L.; Paatero, A.O.; Morel, J.D.; Impens, F.; Guenin-Mace, L.; Saint-Auret, S.; Blanchard, N.; Dillmann, R.; Niang, F.; Pellegrini, S.; et al. Mycolactone subverts immunity by selectively blocking the Sec61 translocon. J. Exp. Med. 2016, 213, 2885–2896. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Sugita, H.; Yoshimori, T.; Ohsumi, Y. A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J. Biol. Chem. 1998, 273, 33889–33892. [Google Scholar] [CrossRef] [PubMed]
- Kuma, A.; Hatano, M.; Matsui, M.; Yamamoto, A.; Nakaya, H.; Yoshimori, T.; Ohsumi, Y.; Tokuhisa, T.; Mizushima, N. The role of autophagy during the early neonatal starvation period. Nature 2004, 432, 1032–1036. [Google Scholar] [CrossRef] [PubMed]
- Mathiassen, S.G.; De Zio, D.; Cecconi, F. Autophagy and the Cell Cycle: A Complex Landscape. Front. Oncol. 2017, 7, 51. [Google Scholar] [CrossRef] [PubMed]
- Maskey, D.; Yousefi, S.; Schmid, I.; Zlobec, I.; Perren, A.; Friis, R.; Simon, H.U. ATG5 is induced by DNA-damaging agents and promotes mitotic catastrophe independent of autophagy. Nat. Commun. 2013, 4, 2130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.H.; Lin, S.M.; Chung, Y.; Vonderfecht, S.; Camden, J.M.; Flodby, P.; Borok, Z.; Limesand, K.H.; Mizushima, N.; Ann, D.K. Dynamic involvement of ATG5 in cellular stress responses. Cell Death Dis. 2014, 5, e1478. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, E.; Maiuri, M.C.; Tajeddine, N.; Vitale, I.; Criollo, A.; Vicencio, J.M.; Hickman, J.A.; Geneste, O.; Kroemer, G. Cell cycle-dependent induction of autophagy, mitophagy and reticulophagy. Cell Cycle 2007, 6, 2263–2267. [Google Scholar] [CrossRef] [PubMed]
- Kaminskyy, V.; Abdi, A.; Zhivotovsky, B. A quantitative assay for the monitoring of autophagosome accumulation in different phases of the cell cycle. Autophagy 2011, 7, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Young, A.R.; Narita, M.; Ferreira, M.; Kirschner, K.; Sadaie, M.; Darot, J.F.; Tavare, S.; Arakawa, S.; Shimizu, S.; Watt, F.M.; et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009, 23, 798–803. [Google Scholar] [CrossRef] [PubMed]
- Shoji-Kawata, S.; Sumpter, R.; Leveno, M.; Campbell, G.R.; Zou, Z.; Kinch, L.; Wilkins, A.D.; Sun, Q.; Pallauf, K.; MacDuff, D.; et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature 2013, 494, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Shoji-Kawata, S.; Sumpter, R.M., Jr.; Wei, Y.; Ginet, V.; Zhang, L.; Posner, B.; Tran, K.A.; Green, D.R.; Xavier, R.J.; et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, 20364–20371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Levine, B. Autosis and autophagic cell death: The dark side of autophagy. Cell Death Differ. 2015, 22, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Hommel, U.; Weber, H.P.; Oberer, L.; Naegeli, H.U.; Oberhauser, B.; Foster, C.A. The 3D-structure of a natural inhibitor of cell adhesion molecule expression. FEBS Lett. 1996, 379, 69–73. [Google Scholar] [CrossRef]
- Besemer, J.; Harant, H.; Wang, S.; Oberhauser, B.; Marquardt, K.; Foster, C.A.; Schreiner, E.P.; de Vries, J.E.; Dascher-Nadel, C.; Lindley, I.J. Selective inhibition of cotranslational translocation of vascular cell adhesion molecule 1. Nature 2005, 436, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Garrison, J.L.; Kunkel, E.J.; Hegde, R.S.; Taunton, J. A substrate-specific inhibitor of protein translocation into the endoplasmic reticulum. Nature 2005, 436, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Sarfo, F.S.; Phillips, R.; Wansbrough-Jones, M.; Simmonds, R.E. Recent advances: Role of mycolactone in the pathogenesis and monitoring of Mycobacterium ulcerans infection/Buruli ulcer disease. Cell. Microbiol. 2016, 18, 17–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thornburg, C.C.; Cowley, E.S.; Sikorska, J.; Shaala, L.A.; Ishmael, J.E.; Youssef, D.T.; McPhail, K.L. Apratoxin H and apratoxin A sulfoxide from the Red Sea cyanobacterium Moorea producens. J. Nat. Prod. 2013, 76, 1781–1788. [Google Scholar] [CrossRef] [PubMed]












© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wan, X.; Serrill, J.D.; Humphreys, I.R.; Tan, M.; McPhail, K.L.; Ganley, I.G.; Ishmael, J.E. ATG5 Promotes Death Signaling in Response to the Cyclic Depsipeptides Coibamide A and Apratoxin A. Mar. Drugs 2018, 16, 77. https://doi.org/10.3390/md16030077
Wan X, Serrill JD, Humphreys IR, Tan M, McPhail KL, Ganley IG, Ishmael JE. ATG5 Promotes Death Signaling in Response to the Cyclic Depsipeptides Coibamide A and Apratoxin A. Marine Drugs. 2018; 16(3):77. https://doi.org/10.3390/md16030077
Chicago/Turabian StyleWan, Xuemei, Jeffrey D. Serrill, Ian R. Humphreys, Michelle Tan, Kerry L. McPhail, Ian G. Ganley, and Jane E. Ishmael. 2018. "ATG5 Promotes Death Signaling in Response to the Cyclic Depsipeptides Coibamide A and Apratoxin A" Marine Drugs 16, no. 3: 77. https://doi.org/10.3390/md16030077
APA StyleWan, X., Serrill, J. D., Humphreys, I. R., Tan, M., McPhail, K. L., Ganley, I. G., & Ishmael, J. E. (2018). ATG5 Promotes Death Signaling in Response to the Cyclic Depsipeptides Coibamide A and Apratoxin A. Marine Drugs, 16(3), 77. https://doi.org/10.3390/md16030077