Makaluvamine G from the Marine Sponge Zyzzia fuliginosa Inhibits Muscle nAChR by Binding at the Orthosteric and Allosteric Sites

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Makaluvamine G Shows Properties of an Un-Competitive Blocker at theMuscle nAChR
2.2. Makaluvamine G Does Not Have Channel Blocker Activity
2.3. Makaluvamine G Binds at the Orthosteric Sites of the Muscle-Type nAChR
2.4. Docking of Acetylcholine and Makaluvamine G witha Model of the Α+Δ− Subunit Interface of Torpedo californica Muscle-Type nAChR
2.5. Makaluvamine G Is More Effective against the Gain-Of-Function Muscle nAChRMutant Α1(G153S) Than against the Wild-Type Receptor
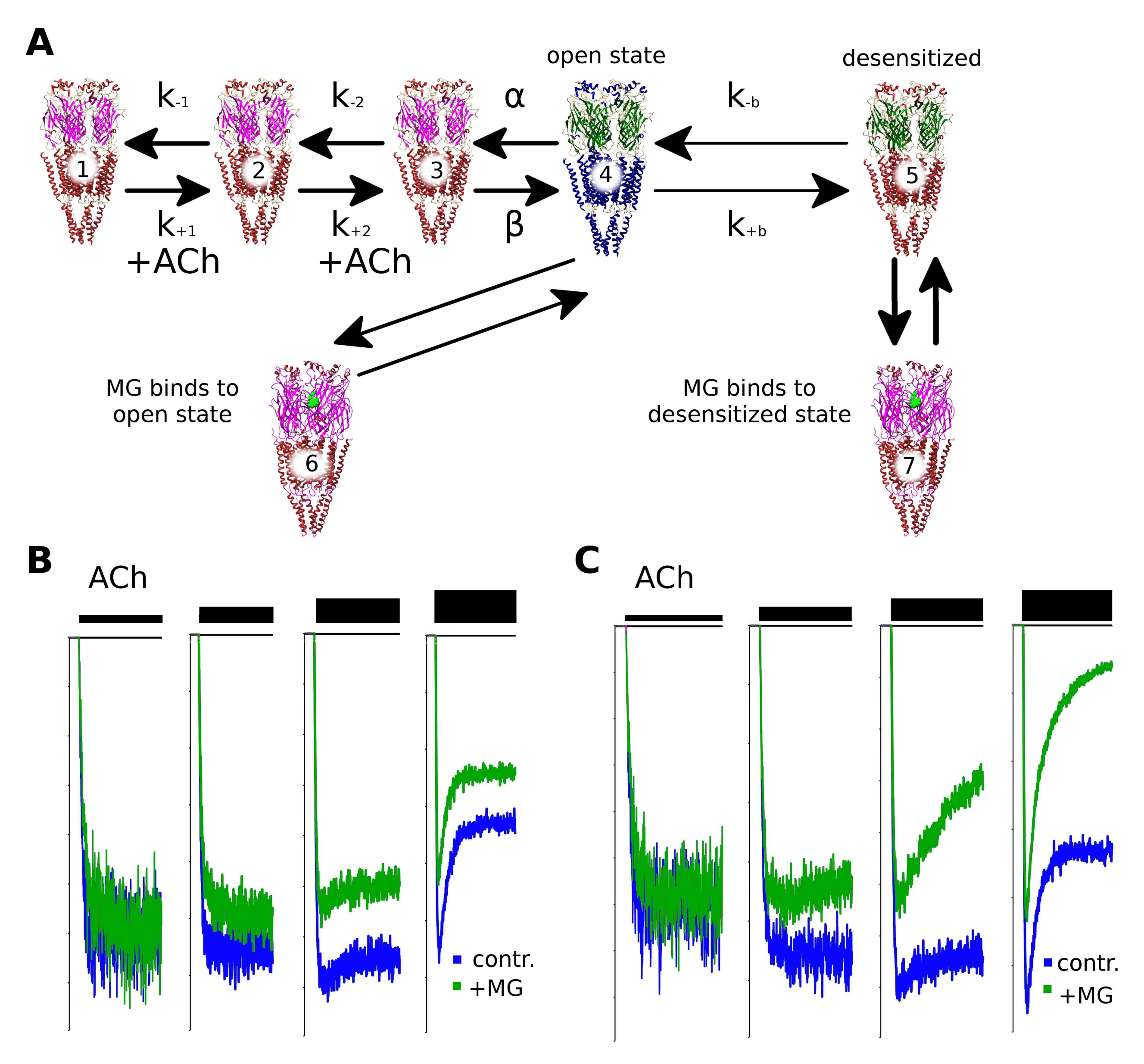
2.6. Makaluvamine G Mode of Action Is Due to the Distinct Channel States
2.7. Makaluvamine G at Higher Micromolar Concentration Inhibits the Α4β2 nAChR
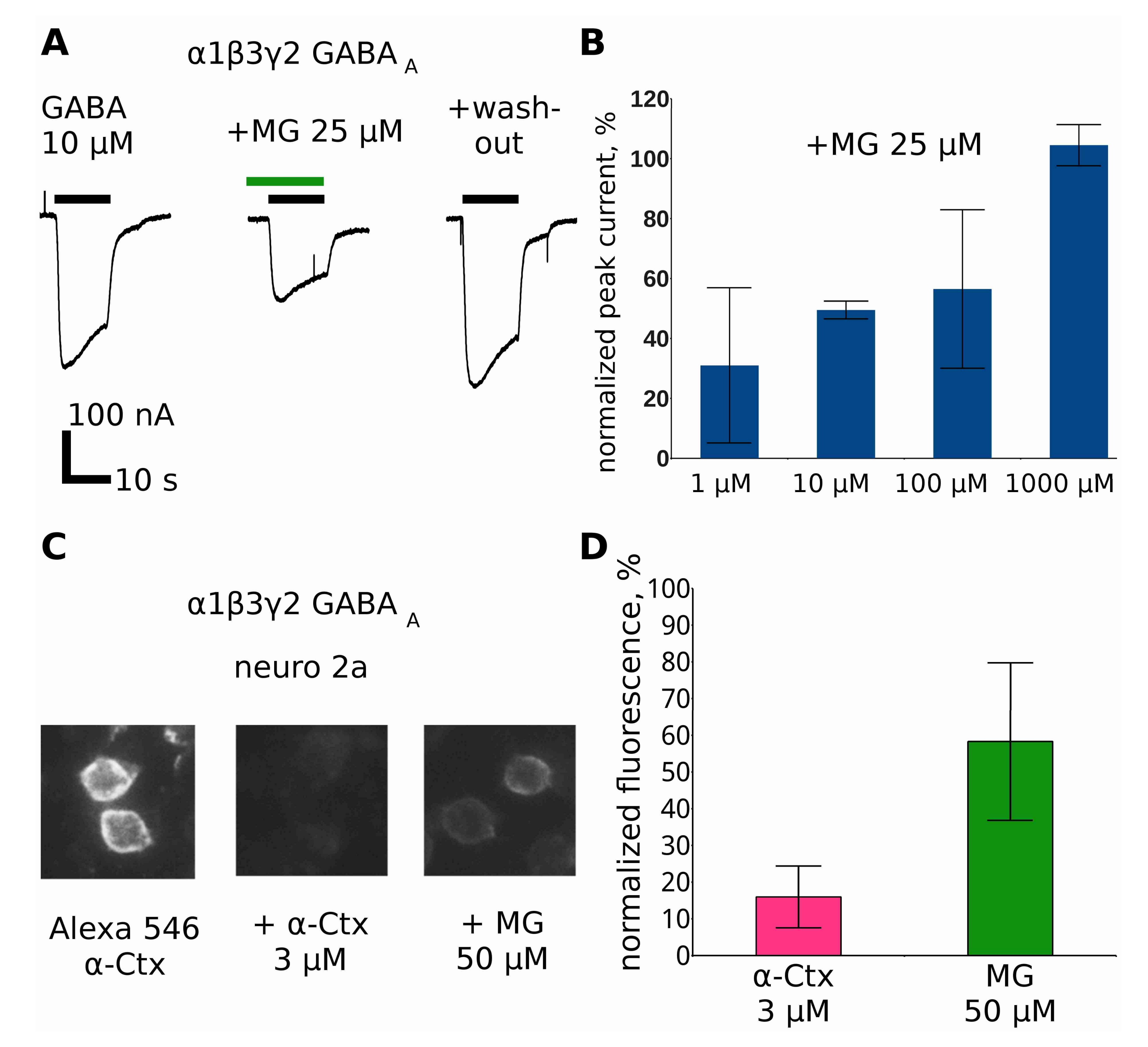
2.8. Makaluvamine G at Higher Concentrations Inhibits GABAAR
2.9. Discussion
3. Experimental Section
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Changeux, J.P. The nicotinic acetylcholine receptor: The founding father of the pentameric ligand-gated ion channel superfamily. J. Biol. Chem. 2012, 287, 40207–40215. [Google Scholar] [CrossRef] [PubMed]
- Flood, P. The importance of myorelaxants in anesthesia. Curr. Opin. Pharmacol. 2005, 5, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, S.; Nicke, A.; Tsetlin, V.I. Nicotinic acetylcholine receptor inhibitors derived from snake and snail venoms. Neuropharmacology 2017. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.T.T.; Newton, E.K.; Mount, V.A.H.; Lee, J.S.; Mansour, C.; Wells, G.A.; Perry, J.J. Rocuronium vs. succinylcholine for rapid sequence intubation: A Cochrane systematic review. Anaesthesia 2017, 72, 765–777. [Google Scholar] [CrossRef] [PubMed]
- Magorian, T.; Flannery, K.B.; Miller, R.D. Comparison of rocuronium, succinylcholine, and vecuronium for rapid-sequence induction of anesthesia in adult patients. Anesthesiology 1993, 79, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Auerbach, A. Thinking in cycles: MWC is a good model for acetylcholine receptor-channels. J. Physiol. 2012, 590, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Harper, C.M. Congenital myasthenic syndromes. Semin. Neurol. 2004, 24, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Fukudome, T.; Ohno, K.; Brengman, J.M.; Engel, A.G. Quinidine normalizes the open duration of slow-channel mutants of the acetylcholine receptor. Neuroreport 1998, 9, 1907–1911. [Google Scholar] [CrossRef] [PubMed]
- Harper, C.M.; Engel, A.G. Quinidine sulfate therapy for the slow-channel congenital myasthenic syndrome. Ann. Neurol. 1998, 43, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Colomer, J.; Müller, J.S.; Vernet, A.; Nascimento, A.; Pons, M.; Gonzalez, V.; Abicht, A.; Lochmüller, H. Long-term improvement of slow-channel congenital myasthenic syndrome with fluoxetine. Neuromuscul. Disord. 2006, 16, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Sieb, J.P.; Milone, M.; Engel, A.G. Effects of the quinoline derivatives quinine, quinidine, and chloroquine on neuromuscular transmission. Brain Res. 1996, 712, 179–189. [Google Scholar] [CrossRef]
- Garcia-Colunga, J.; Awad, J.N.; Miledi, R. Blockage of muscle and neuronal nicotinic acetylcholine receptors by fluoxetine (Prozac). Proc. Natl. Acad. Sci. USA 1997, 94, 2041–2044. [Google Scholar] [CrossRef] [PubMed]
- Carney, J.R.; Scheuer, P.J.; Kelly-Borges, M. Makaluvamine G, a cytotoxic pigment from an an Indonesian Sponge Histodermella sp. Tetrahedron 1993, 49, 8483–8486. [Google Scholar] [CrossRef]
- Kudryavtsev, D.; Makarieva, T.; Utkina, N.; Santalova, E.; Kryukova, E.; Methfessel, C.; Tsetlin, V.; Stonik, V.; Kasheverov, I. Marine natural products acting on the acetylcholine-binding protein and nicotinic receptors: From computer modeling to binding studies and electrophysiology. Mar. Drugs 2014, 12, 1859–1875. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.S.; Lipton, S.A. Mechanism of memantine block of NMDA-activated channels in rat retinal ganglion cells: Uncompetitive antagonism. J. Physiol. 1997, 499 Pt 1, 27–46. [Google Scholar] [CrossRef] [PubMed]
- Alberola-Die, A.; Fernández-Ballester, G.; González-Ros, J.M.; Ivorra, I.; Morales, A. Muscle-Type Nicotinic Receptor Modulation by 2,6-Dimethylaniline, a Molecule Resembling the Hydrophobic Moiety of Lidocaine. Front. Mol. Neurosci. 2016, 9, 127. [Google Scholar] [CrossRef] [PubMed]
- Van Arnam, E.B.; Dougherty, D.A. Functional Probes of Drug–Receptor Interactions Implicated by Structural Studies: Cys-Loop Receptors Provide a Fertile Testing Ground. J. Med. Chem. 2014, 57, 6289–6300. [Google Scholar] [CrossRef] [PubMed]
- Puskar, N.L.; Lester, H.A.; Dougherty, D.A. Probing the effects of residues located outside the agonist binding site on drug-receptor selectivity in the nicotinic receptor. ACS Chem. Biol. 2012, 7, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Otero-Cruz, J.D.; Báez-Pagán, C.A.; Dorna-Pérez, L.; Grajales-Reyes, G.E.; Ramírez-Ordoñez, R.T.; Luciano, C.A.; Gómez, C.M.; Lasalde-Dominicci, J.A. Decoding pathogenesis of slow-channel congenital myasthenic syndromes using recombinant expression and mice models. P. R. Health Sci. J. 2010, 29, 4–17. [Google Scholar] [PubMed]
- Shelukhina, I.; Spirova, E.; Kudryavtsev, D.; Ojomoko, L.; Werner, M.; Methfessel, C.; Hollmann, M.; Tsetlin, V. Calcium imaging with genetically encoded sensor Case12: Facile analysis of α7/α9 nAChR mutants. PLoS ONE 2017, 12, e0181936. [Google Scholar] [CrossRef] [PubMed]
- Sine, S.M.; Claudio, T.; Sigworth, F.J. Activation of Torpedo acetylcholine receptors expressed in mouse fibroblasts. Single channel current kinetics reveal distinct agonist binding affinities. J. Gen. Physiol. 1990, 96, 395–437. [Google Scholar] [CrossRef] [PubMed]
- Nicolai, C.; Sachs, F. Solving Ion Channel Kinetics with the QuB Software. Biophys. Rev. Lett. 2013, 8, 191–211. [Google Scholar] [CrossRef]
- Schwartz, R.D.; Mindlin, M.C. Inhibition of the GABA receptor-gated chloride ion channel in brain by noncompetitive inhibitors of the nicotinic receptor-gated cation channel. J. Pharmacol. Exp. Ther. 1988, 244, 963–970. [Google Scholar] [PubMed]
- Baker, E.R.; Zwart, R.; Sher, E.; Millar, N.S. Pharmacological Properties of α9α10 Nicotinic Acetylcholine Receptors Revealed by Heterologous Expression of Subunit Chimeras. Mol. Pharmacol. 2004, 65, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Dionisio, L.; Bergé, I.; Bravo, M.; del Esandi, M.C.; Bouzat, C. Neurotransmitter GABA Activates Muscle but Not α7 Nicotinic Receptors. Mol. Pharmacol. 2015, 87, 391–400. [Google Scholar] [CrossRef] [PubMed]
- TOXNET. Available online: https://toxnet.nlm.nih.gov/cgi-bin/sis/search2/r?dbs+hsdb:@term+@rn+@rel+57-95-4 (accessed on 27 October 2017).
- Olsen, J.A.; Balle, T.; Gajhede, M.; Ahring, P.K.; Kastrup, J.S. Molecular recognition of the neurotransmitter acetylcholine by an acetylcholine binding protein reveals determinants of binding to nicotinic acetylcholine receptors. PLoS ONE 2014, 9, e91232. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, Y.; Akk, G.; Sine, S.; Auerbach, A. Activation kinetics of recombinant mouse nicotinic acetylcholine receptors: Mutations of alpha-subunit tyrosine 190 affect both binding and gating. Biophys. J. 1995, 69, 849–859. [Google Scholar] [CrossRef]
- Molles, B.E.; Rezai, P.; Kline, E.F.; McArdle, J.J.; Sine, S.M.; Taylor, P. Identification of residues at the alpha and epsilon subunit interfaces mediating species selectivity of Waglerin-1 for nicotinic acetylcholine receptors. J. Biol. Chem. 2002, 277, 5433–5440. [Google Scholar] [CrossRef] [PubMed]
- Tsetlin, V.I. Three-finger snake neurotoxins and Ly6 proteins targeting nicotinic acetylcholine receptors: Pharmacological tools and endogenous modulators. Trends Pharmacol. Sci. 2015, 36, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Lyukmanova, E.N.; Shulepko, M.A.; Kudryavtsev, D.; Bychkov, M.L.; Kulbatskii, D.S.; Kasheverov, I.E.; Astapova, M.V.; Feofanov, A.V.; Thomsen, M.S.; Mikkelsen, J.D.; et al. Human Secreted Ly-6/uPAR Related Protein-1 (SLURP-1) Is a Selective Allosteric Antagonist of α7 Nicotinic Acetylcholine Receptor. PLoS ONE 2016, 11, e0149733. [Google Scholar] [CrossRef] [PubMed]
- Durek, T.; Shelukhina, I.V.; Tae, H.-S.; Thongyoo, P.; Spirova, E.N.; Kudryavtsev, D.S.; Kasheverov, I.E.; Faure, G.; Corringer, P.-J.; Craik, D.J.; et al. Interaction of Synthetic Human SLURP-1 with the Nicotinic Acetylcholine Receptors. Sci. Rep. 2017, 7, 16606. [Google Scholar] [CrossRef] [PubMed]
- Guex, N.; Peitsch, M.C.; Schwede, T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 2009, 30. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A Free Tool to Discover Chemistry for Biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef] [PubMed]
- Mayer, C.; Dimopoulos, S.; Rudolf, F.; Stelling, J.; Mayer, C.; Dimopoulos, S.; Rudolf, F.; Stelling, J. Using CellX to Quantify Intracellular Events. In Current Protocols in Molecular Biology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; ISBN 9780471142720. [Google Scholar]
- Hamilton, N. Quantification and its Applications in Fluorescent Microscopy Imaging. Traffic 2009, 10, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Utkina, N.R.; Makarchenko, A.E.; Denisenko, V.A.; Dmitrenok, P.S. Zyzzyanone A, a novel pyrrolo[3,2-f]indole alkaloid from the Australian marine sponge Zyzzya filiginosa. Tetrahedron Lett. 2004, 45, 7491–7494. [Google Scholar] [CrossRef]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kudryavtsev, D.S.; Spirova, E.N.; Shelukhina, I.V.; Son, L.V.; Makarova, Y.V.; Utkina, N.K.; Kasheverov, I.E.; Tsetlin, V.I. Makaluvamine G from the Marine Sponge Zyzzia fuliginosa Inhibits Muscle nAChR by Binding at the Orthosteric and Allosteric Sites. Mar. Drugs 2018, 16, 109. https://doi.org/10.3390/md16040109
Kudryavtsev DS, Spirova EN, Shelukhina IV, Son LV, Makarova YV, Utkina NK, Kasheverov IE, Tsetlin VI. Makaluvamine G from the Marine Sponge Zyzzia fuliginosa Inhibits Muscle nAChR by Binding at the Orthosteric and Allosteric Sites. Marine Drugs. 2018; 16(4):109. https://doi.org/10.3390/md16040109
Chicago/Turabian StyleKudryavtsev, Denis S., Ekaterina N. Spirova, Irina V. Shelukhina, Lina V. Son, Yana V. Makarova, Natalia K. Utkina, Igor E. Kasheverov, and Victor I. Tsetlin. 2018. "Makaluvamine G from the Marine Sponge Zyzzia fuliginosa Inhibits Muscle nAChR by Binding at the Orthosteric and Allosteric Sites" Marine Drugs 16, no. 4: 109. https://doi.org/10.3390/md16040109
APA StyleKudryavtsev, D. S., Spirova, E. N., Shelukhina, I. V., Son, L. V., Makarova, Y. V., Utkina, N. K., Kasheverov, I. E., & Tsetlin, V. I. (2018). Makaluvamine G from the Marine Sponge Zyzzia fuliginosa Inhibits Muscle nAChR by Binding at the Orthosteric and Allosteric Sites. Marine Drugs, 16(4), 109. https://doi.org/10.3390/md16040109