Ribbon α-Conotoxin KTM Exhibits Potent Inhibition of Nicotinic Acetylcholine Receptors

 , ,
, , 

Abstract
:
1. Introduction
2. Results
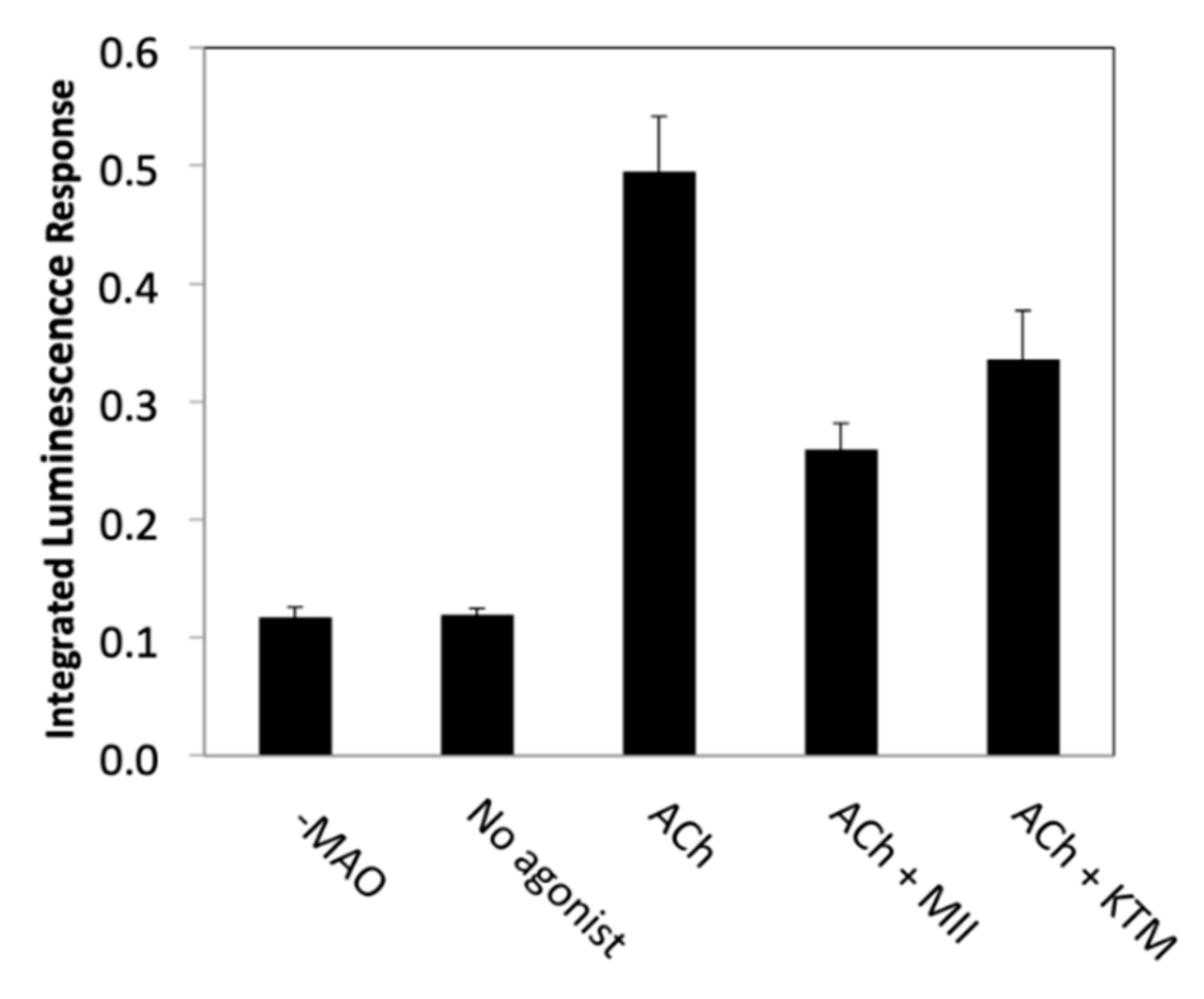
2.1. Bioactivity
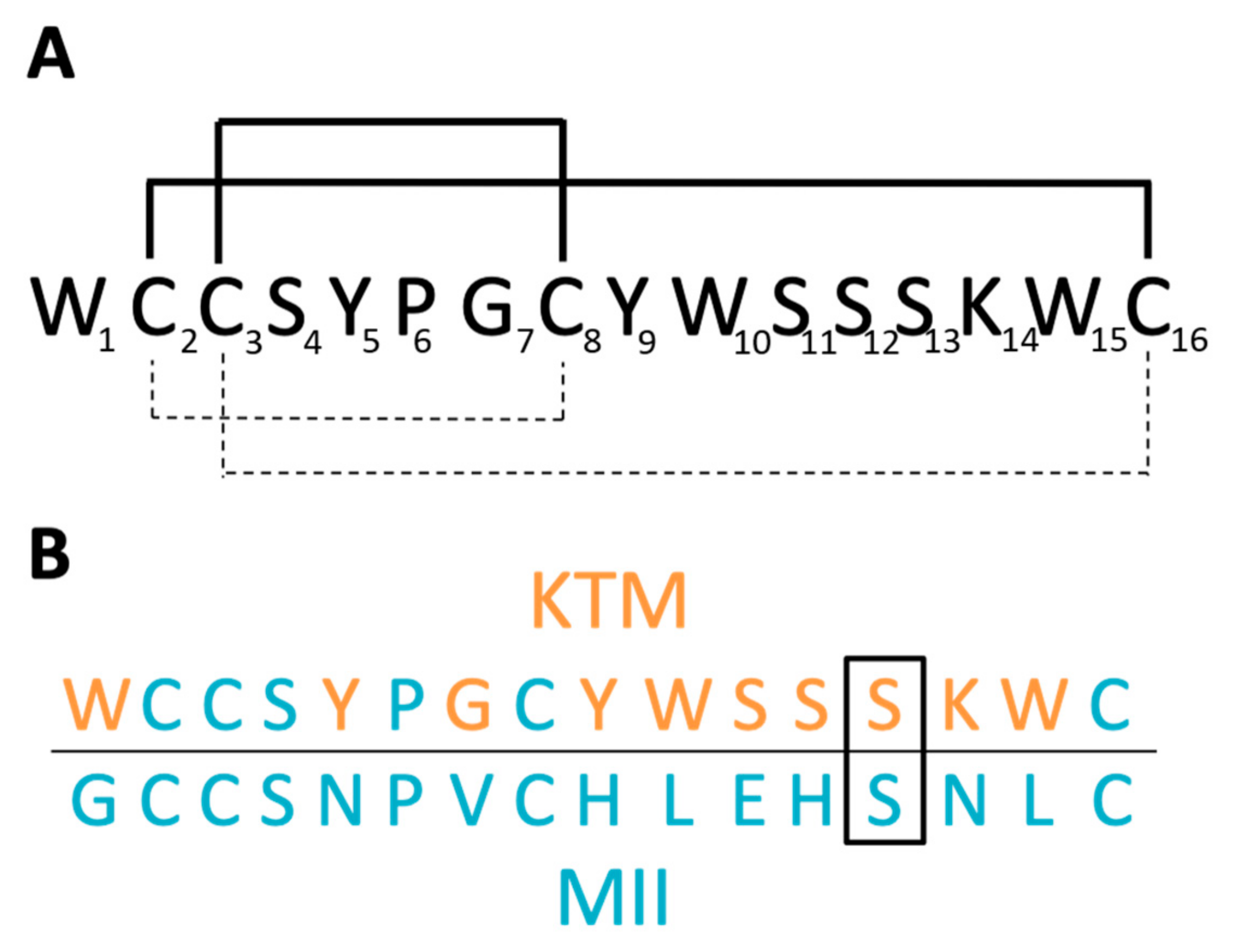
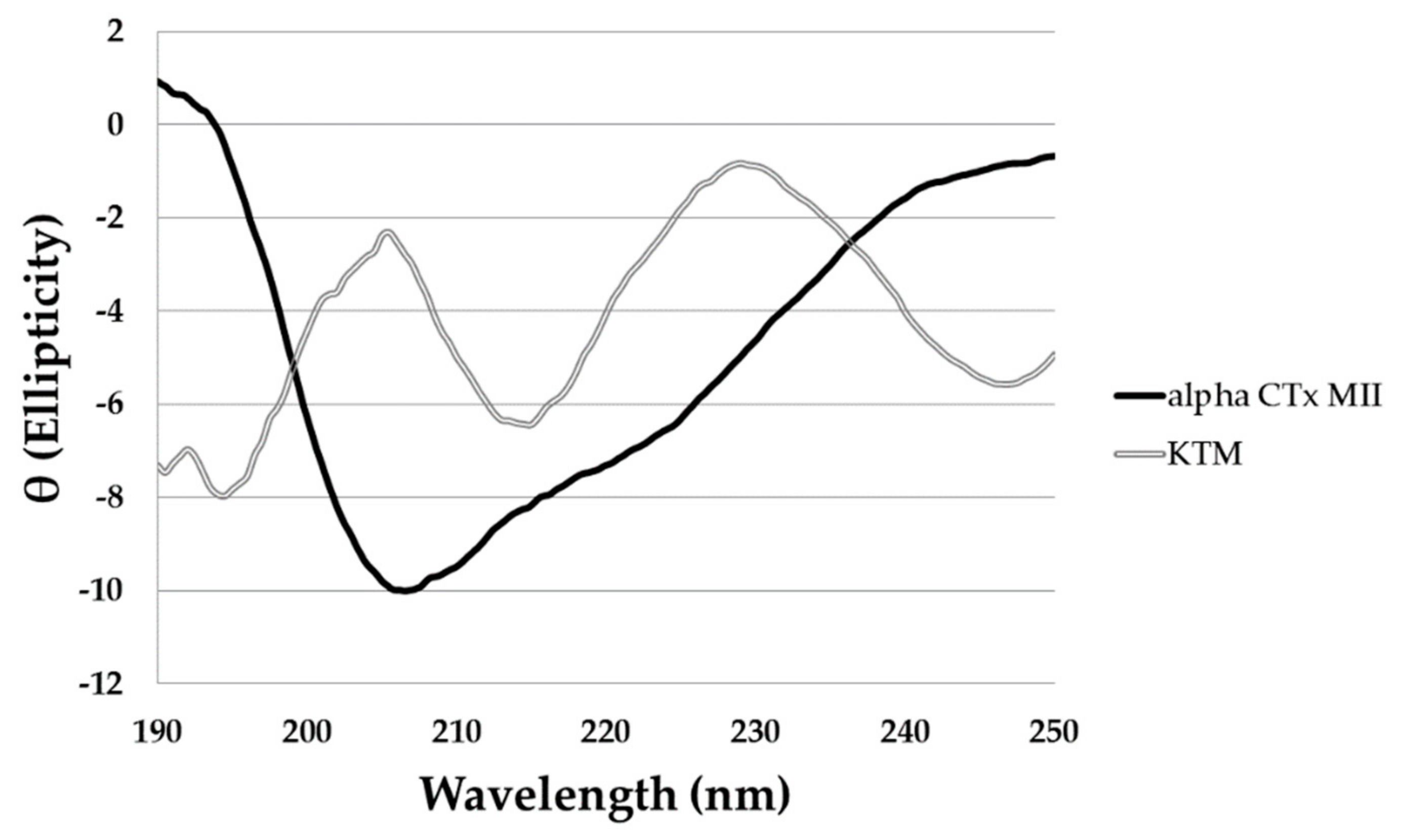
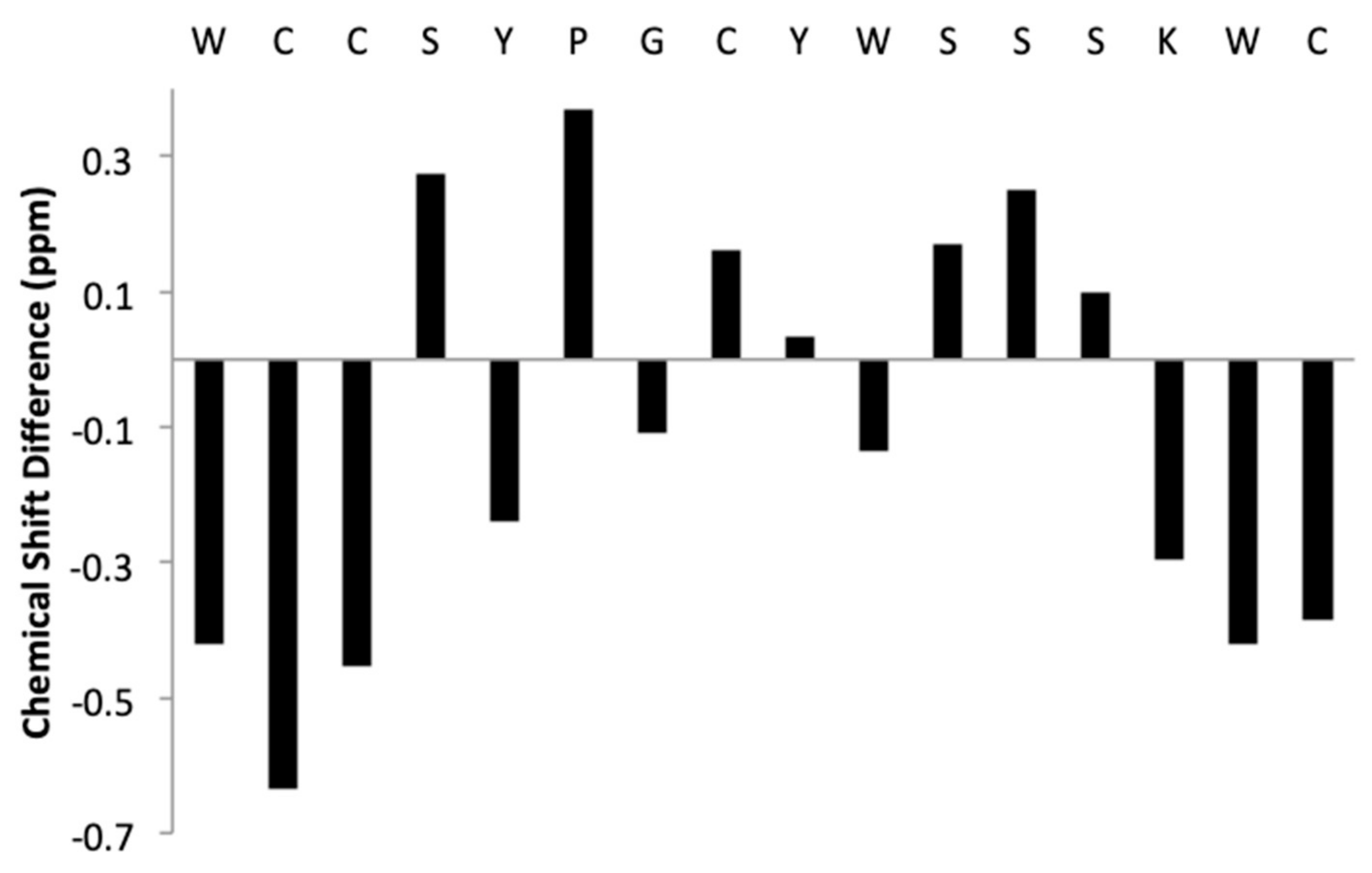
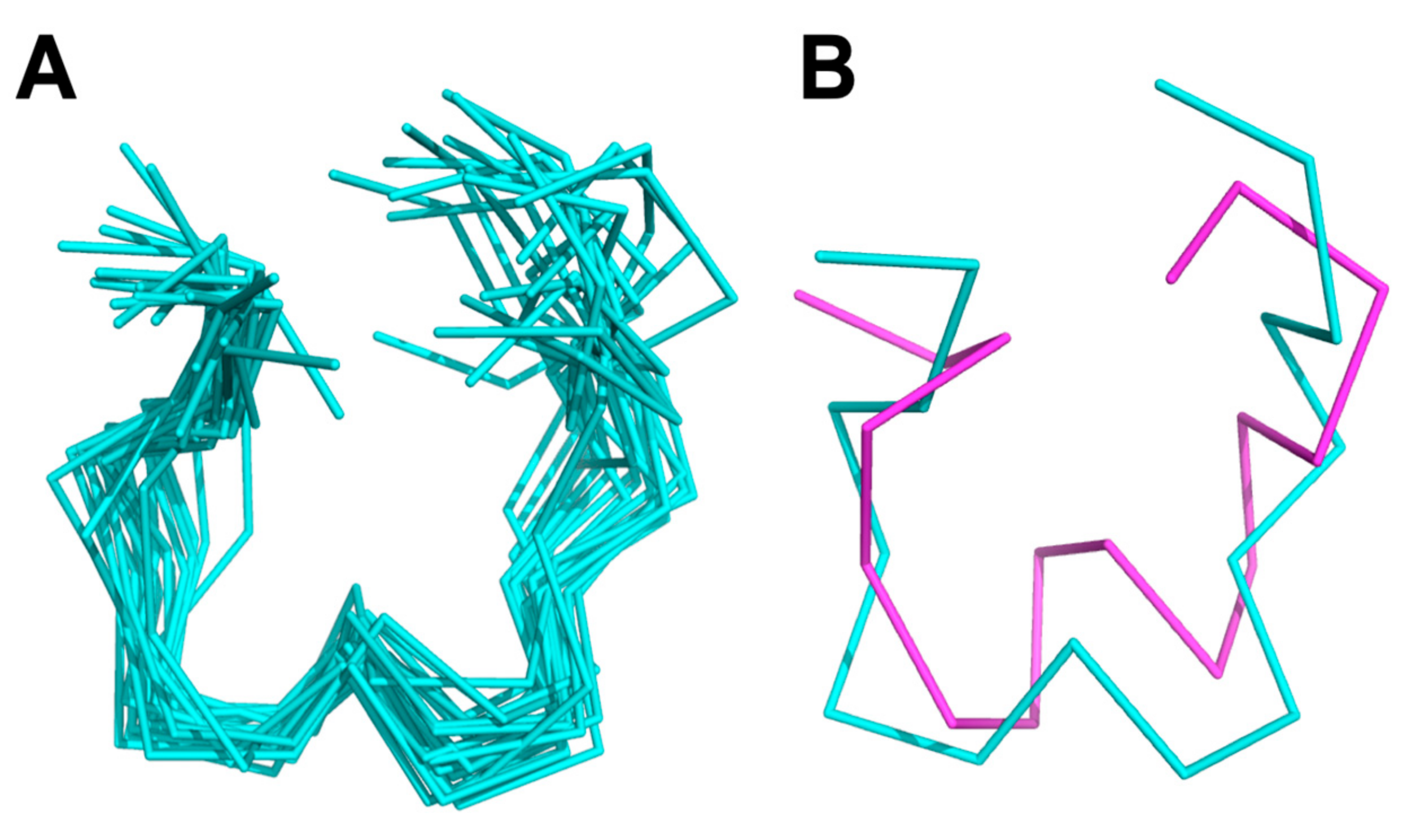
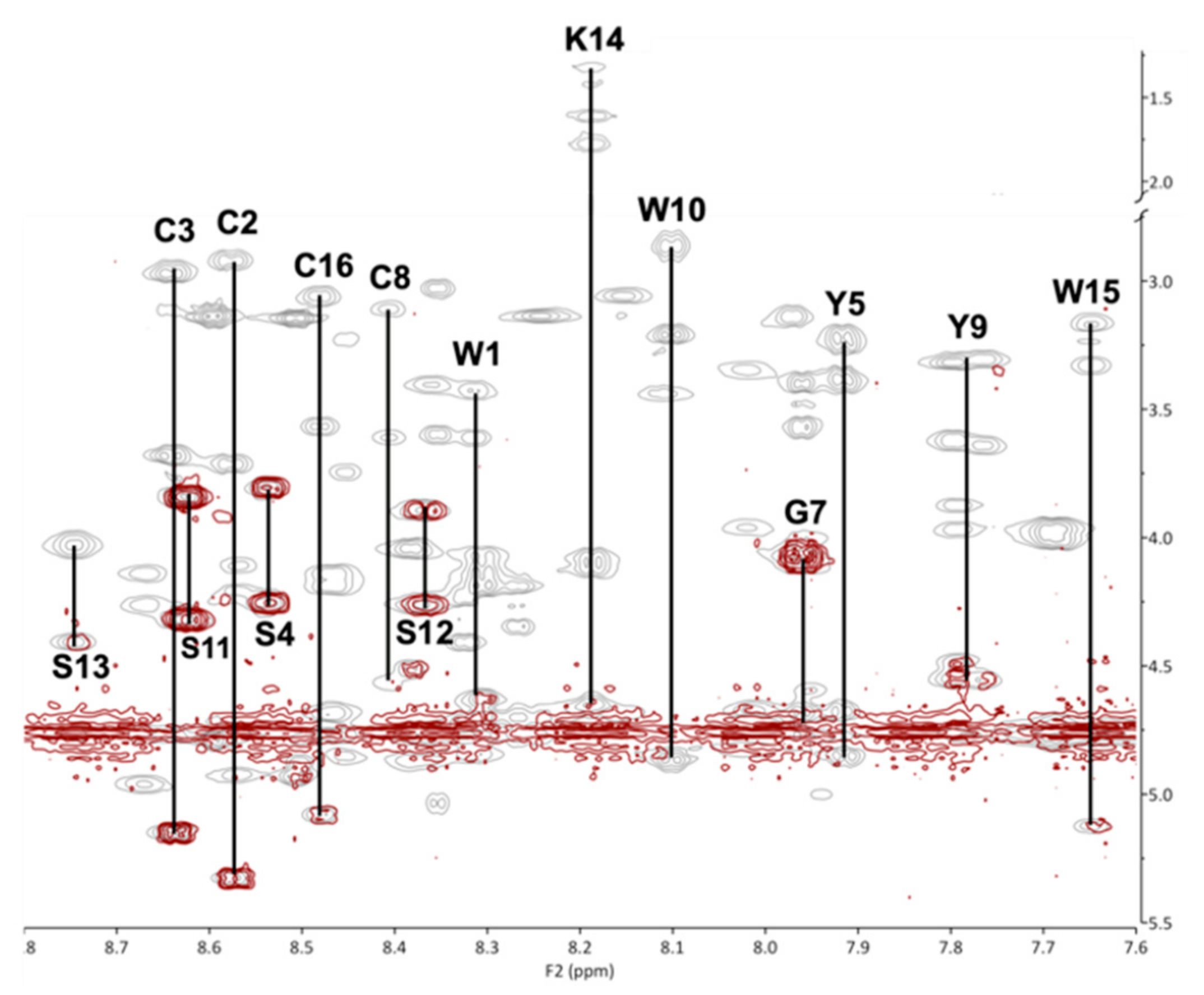
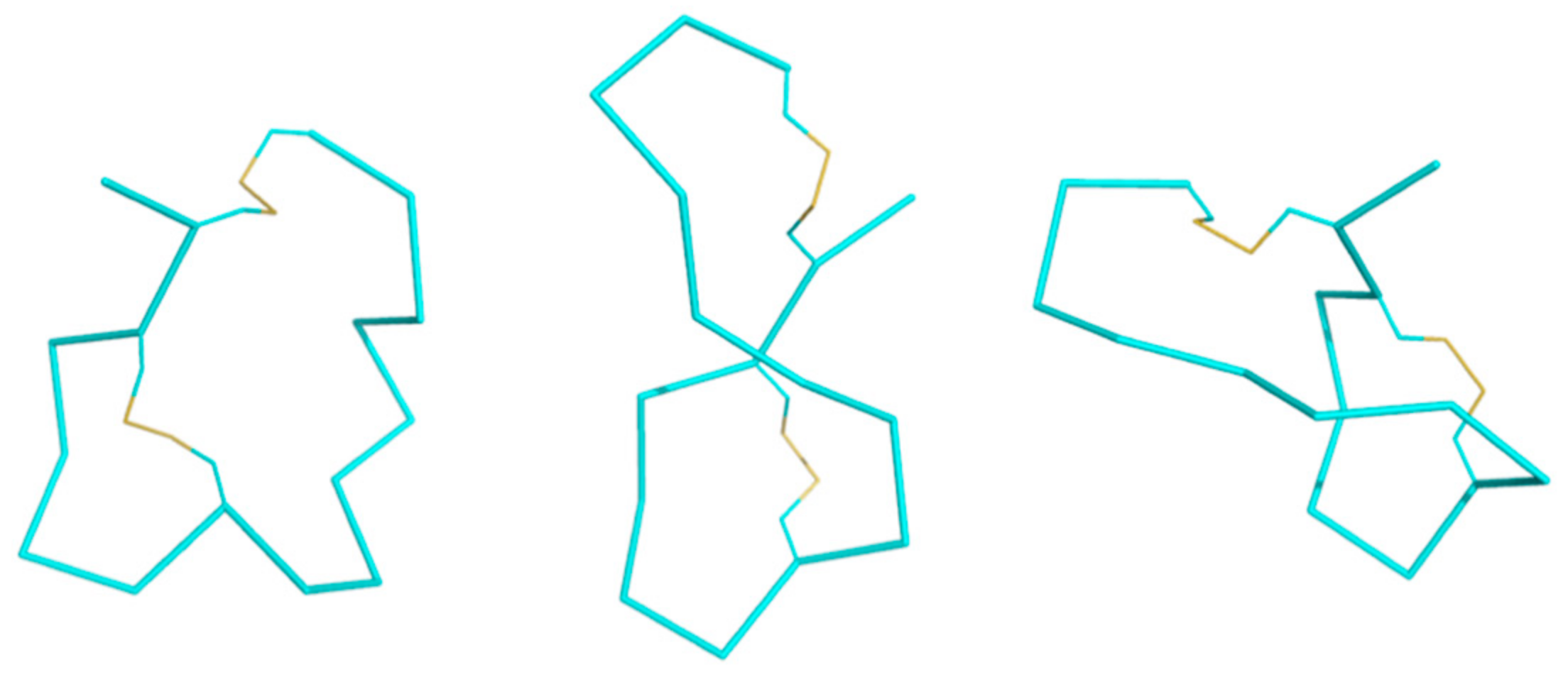
2.2. Structure Determination
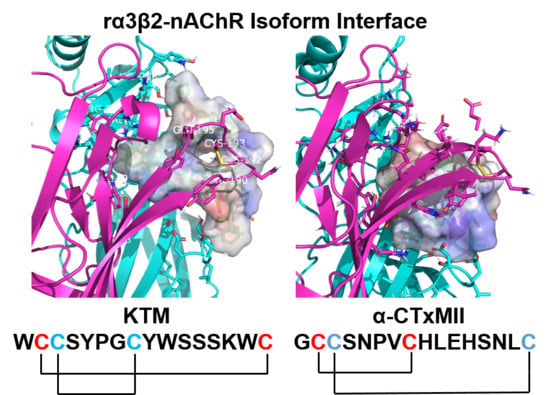
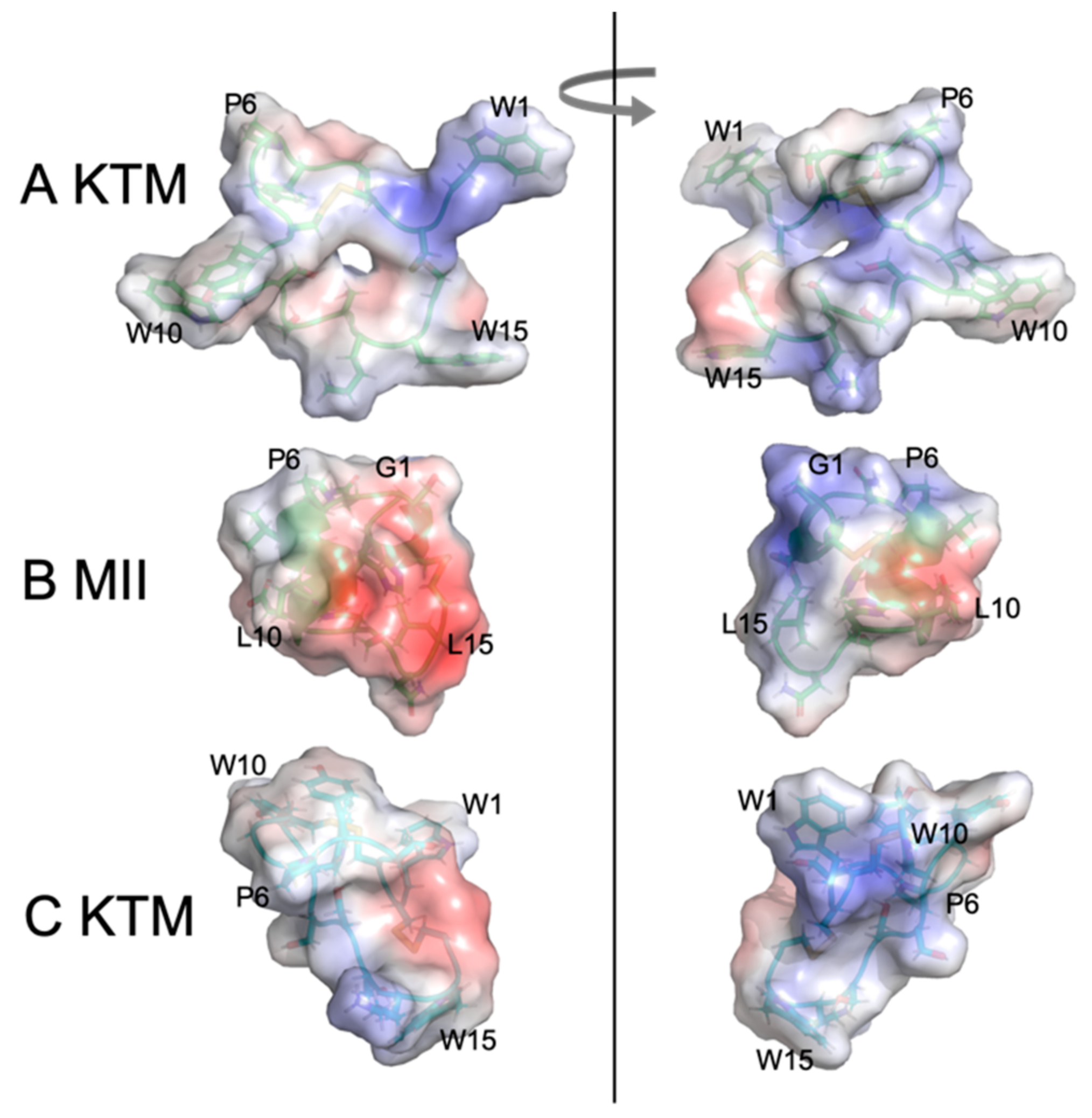
2.3. Molecular Dynamics Simulations
3. Discussion
4. Materials and Methods
4.1. Synthesis
4.2. Disulfide Bond Analysis
4.3. Circular Dichroism Spectropolarimetry
4.4. Nuclear Magnetic Resonance Spectroscopy
4.4.1. Restraint Set Generation
4.4.2. Structure Calculation
4.5. PC12 Assay
4.5.1. Cell Culture
4.5.2. Assay
4.6. Electrophysiology
4.6.1. rα3β2-nAChR expression in Xenopus laevis oocytes
4.6.2. Two-Electrode Voltage Clamp
4.7. Molecular Dynamics Simulations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kaas, Q.; Yu, R.L.; Jin, A.H.; Dutertre, S.; Craik, D.J. ConoServer: Updated content, knowledge, and discovery tools in the conopeptide database. Nucleic Acids Res. 2012, 40, D325–D330. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, E.X.; Pereira, E.F.R.; Alkondon, M.; Rogers, S.W. Mammalian Nicotinic Acetylcholine Receptors: From Structure to Function. Physiol. Rev. 2009, 89, 73–120. [Google Scholar] [CrossRef] [PubMed]
- Quik, M.; Wonnacott, S. alpha 6 beta 2 and alpha 4 beta 2 Nicotinic Acetylcholine Receptors as Drug Targets for Parkinson’s Disease. Pharmacol. Rev. 2011, 63, 938–966. [Google Scholar] [CrossRef] [PubMed]
- Quik, M.; Bordia, T.; O’Leary, K. Nicotinic receptors as CNS targets for Parkinson’s disease. Biochem. Pharmacol. 2007, 74, 1224–1234. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Vijayaraghavan, S. Nicotinic receptors: Role in addiction and other disorders of the brain. Subst. Abus. Res. Treat. 2008, 81–95. [Google Scholar] [CrossRef]
- Young, J.W.; Geyer, M.A. Evaluating the role of the alpha-7 nicotinic acetylcholine receptor in the pathophysiology and treatment of schizophrenia. Biochem. Pharmacol. 2013, 86, 1122–1132. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, S.; Maskos, U. Role of the nicotinic acetylcholine receptor in Alzheimer’s disease pathology and treatment. Neuropharmacology 2015, 96, 255–262. [Google Scholar] [CrossRef]
- Turner, M.W.; Marquart, L.A.; Phillips, P.D.; McDougal, O.M. Mutagenesis of alpha-Conotoxins for Enhancing Activity and Selectivity for Nicotinic Acetylcholine Receptors. Toxins 2019, 11, 29. [Google Scholar] [CrossRef]
- Yu, R.L.; Kompella, S.N.; Adams, D.J.; Craik, D.J.; Kaas, Q. Determination of the alpha-Conotoxin Vc1.1 Binding Site on the alpha 9 alpha 10 Nicotinic Acetylcholine Receptor. J. Med. Chem. 2013, 56, 3557–3567. [Google Scholar] [CrossRef]
- Bordia, T.; Grady, S.R.; McIntosh, J.M.; Quik, M. Nigrostriatal damage preferentially decreases a subpopulation of alpha 6 beta 2 nAChRs in mouse, monkey, and Parkinson’s disease striatum. Mol. Pharmacol. 2007, 72, 52–61. [Google Scholar] [CrossRef]
- Marquart, L.A.; Turner, M.W.; McDougal, O.M. Qualitative assay to detect dopamine release by ligand action on nicotinic acetylcholine receptors. Toxins 2019, 11, 682. [Google Scholar] [CrossRef]
- Kuryatov, A.; Lindstrom, J. Expression of Functional Human alpha 6 beta 2 beta 3 Acetylcholine Receptors in Xenopus laevis Oocytes Achieved through Subunit Chimeras and Concatamers. Mol. Pharmacol. 2011, 79, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Alkondon, M.; Albuquerque, E.X. Nicotinic receptor subtypes in rat hippocampal slices are differentially sensitive to desensitization and early in vivo functional up-regulation by nicotine and to block by bupropion. J. Pharmacol. Exp. Ther. 2005, 313, 740–750. [Google Scholar] [CrossRef] [PubMed]
- Arvin, M.C.; Wokosin, D.L.; Banala, S.; Lavis, L.D.; Drenan, R.M. Probing Nicotinic Acetylcholine Receptor Function in Mouse Brain Slices via Laser Flash Photolysis of Photoactivatable Nicotine. Jove-J. Vis. Exp. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Engle, S.E.; Broderick, H.J.; Drenan, R.M. Local Application of Drugs to Study Nicotinic Acetylcholine Receptor Function in Mouse Brain Slices. Jove J. Vis. Exp. 2012, 8. [Google Scholar] [CrossRef]
- King, M.D.; Long, T.; Pfalmer, D.L.; Andersen, T.L.; McDougal, O.M. SPIDR: Small-molecule peptide-influenced drug repurposing. Bmc Bioinform. 2018, 19, 11. [Google Scholar] [CrossRef]
- King, M.D.; Long, T.; Andersen, T.; McDougal, O.M. Genetic Algorithm Managed Peptide Mutant Screening: Optimizing Peptide Ligands for Targeted Receptor Binding. J. Chem. Inf. Modeling 2016, 56, 2378–2387. [Google Scholar] [CrossRef]
- Long, T.; McDougal, O.M.; Andersen, T. GAMPMS: Genetic algorithm managed peptide mutant screening. J. Comput. Chem. 2015, 36, 1304–1310. [Google Scholar] [CrossRef]
- Bullock, C.; Cornia, N.; Jacob, R.; Remm, A.; Peavey, T.; Weekes, K.; Mallory, C.; Oxford, J.T.; McDougal, O.M.; Andersen, T.L. DockoMatic 2.0: High Throughput Inverse Virtual Screening and Homology Modeling. J. Chem. Inf. Modeling 2013, 53, 2161–2170. [Google Scholar] [CrossRef]
- Jacob, R.B.; Andersen, T.; McDougal, O.M. Accessible High-Throughput Virtual Screening Molecular Docking Software for Students and Educators. PLoS Comput. Biol. 2012, 8, 5. [Google Scholar] [CrossRef]
- Jacob, R.B.; Bullock, C.W.; Andersen, T.; McDougal, O.M. DockoMatic: Automated Peptide Analog Creation for High Throughput Virtual Screening. J. Comput. Chem. 2011, 32, 2936–2941. [Google Scholar] [CrossRef] [PubMed]
- Leffler, A.E.; Kuryatov, A.; Zebroski, H.A.; Powell, S.R.; Filipenko, P.; Hussein, A.K.; Gorson, J.; Heizmann, A.; Lyskov, S.; Tsien, R.W.; et al. Discovery of peptide ligands through docking and virtual screening at nicotinic acetylcholine receptor homology models. Proc. Natl. Acad. Sci. USA 2017, 114, E8100–E8109. [Google Scholar] [CrossRef] [PubMed]
- Kasheverov, I.E.; Chugunov, A.O.; Kudryavtsev, D.S.; Ivanov, I.A.; Zhmak, M.N.; Shelukhina, I.V.; Spirova, E.N.; Tabakmakher, V.M.; Zelepuga, E.A.; Efremov, R.G.; et al. High-Affinity alpha-Conotoxin PnIA Analogs Designed on the Basis of the Protein Surface Topography Method. Sci. Rep. 2016, 6, 11. [Google Scholar] [CrossRef]
- Luo, S.L.; Zhangsun, D.T.; Schroeder, C.I.; Zhu, X.P.; Hu, Y.Y.; Wu, Y.; Weltzin, M.M.; Eberhard, S.; Kaas, Q.; Craik, D.J.; et al. A novel alpha 4/7-conotoxin LvIA from Conus lividus that selectively blocks alpha 3 beta 2 vs. alpha 6/alpha 3 beta 2 beta 3 nicotinic acetylcholine receptors. Faseb J. 2014, 28, 1842–1853. [Google Scholar] [CrossRef] [PubMed]
- Sali, A.; Blundell, T.L. Comparative Protein Modeling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Koromyslova, A.D.; Chugunov, A.O.; Efremov, R.G. Deciphering Fine Molecular Details of Proteins’ Structure and Function with a Protein Surface Topography (PST) Method. J. Chem. Inf. Modeling 2014, 54, 1189–1199. [Google Scholar] [CrossRef]
- Xu, J.; Wang, X.B.; Ensign, B.; Li, M.; Wu, L.; Guia, A.; Xu, J.Q. Ion-channel assay technologies: Quo vadis? Drug Discov. Today 2001, 6, 1278–1287. [Google Scholar] [CrossRef]
- Accardi, M.V.; Pugsley, M.K.; Forster, R.; Troncy, E.; Huang, H.; Authier, S. The emerging role of in vitro electrophysiological methods in CNS safety pharmacology. J. Pharmacol. Toxicol. Methods 2016, 81, 47–59. [Google Scholar] [CrossRef]
- Armstrong, L.C.; Kirsch, G.E.; Fedorov, N.B.; Wu, C.Y.; Kuryshev, Y.A.; Sewell, A.L.; Liu, Z.Q.; Motter, A.L.; Leggett, C.S.; Orr, M.S. High-Throughput Patch Clamp Screening in Human alpha 6-Containing Nicotinic Acetylcholine Receptors. Slas Discov. 2017, 22, 686–695. [Google Scholar] [CrossRef]
- Dunlop, J.; Bowlby, M.; Peri, R.; Vasilyev, D.; Arias, R. High-throughput electrophysiology: An emerging paradigm for ion-channel screening and physiology. Nat. Rev. Drug Discov. 2008, 7, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Jacob, R.B.; Bullock, C. Automatic DockOmatic: Ligand and receptor screening made easy. Abstr. Pap. Am. Chem. Soc. 2010, 240, 1. [Google Scholar]
- King, M.D.; Phillips, P.; Turner, M.W.; Katz, M.; Lew, S.; Bradburn, S.; Andersen, T.; McDougal, O.M. Computational Exploration of a Protein Receptor Binding Space with Student Proposed Peptide Ligands. Biochem. Mol. Biol. Educ. 2016, 44, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Bullock, C.W.; Jacob, R.B.; McDougal, O.M.; Hampikian, G.; Andersen, T. DockoMatic—Automated ligand creation and docking. BMC Res. Notes. 2010, 3, 289–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akondi, K.B.; Muttenthaler, M.; Dutertre, S.; Kaas, Q.; Craik, D.J.; Lewis, R.J.; Alewood, P.F. Discovery, Synthesis, and Structure Activity Relationships of Conotoxins. Chem. Rev. 2014, 114, 5815–5847. [Google Scholar] [CrossRef] [PubMed]
- Grishin, A.A.; Wang, C.I.A.; Muttenthaler, M.; Alewood, P.F.; Lewis, R.J.; Adams, D.J. alpha-Conotoxin AuIB Isomers Exhibit Distinct Inhibitory Mechanisms and Differential Sensitivity to Stoichiometry of alpha 3 beta 4 Nicotinic Acetylcholine Receptors. J. Biol. Chem. 2010, 285, 22254–22263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutton, J.L.; Bansal, P.S.; Hogg, R.C.; Adams, D.J.; Alewood, P.F.; Craik, D.J. A new level of conotoxin diversity, a non-native disulfide bond connectivity in alpha-conotoxin AuIB reduces structural definition but increases biological activity. J. Biol. Chem. 2002, 277, 48849–48857. [Google Scholar] [CrossRef] [Green Version]
- Shinohara, H.; Wang, F.F.; Hossain, S.M.Z. A convenient, high-throughput method for enzyme-luminescence detection of dopamine released from PC12 cells. Nat. Protoc. 2008, 3, 1639–1644. [Google Scholar] [CrossRef]
- Mir, T.A.; Shinohara, H.; Shimizu, Y. Enzyme-luminescence method: Tool for evaluation of neuronal differentiation based on real-time monitoring of dopamine release response from PC12 cells. Anal. Methods 2011, 3, 837–841. [Google Scholar] [CrossRef]
- Wüthrich, K. NMR of Proteins and Nucleic Acids; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Güntert, P. Automated NMR Structure Calculation with CYANA. In Protein NMR Techniques; Downing, A.K., Ed.; Humana Press: New York, NY, USA, 2004; Volume 278, pp. 353–378. [Google Scholar]
- Wishart, D.S.; Sykes, B.D.; Richards, F.M. The chemical-shift index—A fast and simple method for the assignment of protein secondary structure through nmr-spectroscopy. Biochemistry 1992, 31, 1647–1651. [Google Scholar] [CrossRef]
- Robinson, S.D.; Norton, R.S. Conotoxin Gene Superfamilies. Mar. Drugs 2014, 12, 6058–6101. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System; 2.1.1; Schrödinger, LLC: New York, NY, USA, 2019.
- Gray, W.R. Disulfide Structures of Highly Bridged Peptides—A New Strategy for Analysis. Protein Sci. 1993, 2, 1732–1748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.H.; Yang, J.T. New Approach to Calculation of Secondary Structures of Globular Proteins by Optical Rotatory Dispersion and Circular Dichroism. Biochem. Biophys. Res. Commun. 1971, 44, 1285–1291. [Google Scholar] [CrossRef]
- Everhart, D.; Cartier, G.E.; Malhotra, A.; Gomes, A.V.; McIntosh, J.M.; Luetje, C.W. Determinants of potency on alpha-conotoxin MII, a peptide antagonist of neuronal nicotinic receptors. Biochemistry 2004, 43, 2732–2737. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.W.; Cort, J.R.; McDougal, O.M. alpha-Conotoxin Decontamination Protocol Evaluation: What Works and What Doesn’t. Toxins 2017, 9, 10. [Google Scholar] [CrossRef] [Green Version]
- Wishart, D.S.; Bigam, C.G.; Holm, A.; Hodges, R.S.; Sykes, B.D. H-1, C-13 and N-15 Random Coil NMR Chemical-Shifts of the Common Amino-Acids 1. Investigations of Nearest-Neighbor Effects. J. Biomol. Nmr 1995, 5, 332. [Google Scholar] [CrossRef]
- Wishart, D.S.; Sykes, B.D.; Richards, F.M. Simple techniques for the quantification of protein secondary structure by H-1-NMR spectroscopy. Febs. Lett. 1991, 293, 72–80. [Google Scholar] [CrossRef] [Green Version]
- Bax, A. Two-dimensional NMR and protein structure. Annu. Rev. Biochem. 1989, 58, 223–256. [Google Scholar] [CrossRef]
- Jeener, J.; Meier, B.H.; Bachmann, P.; Ernst, R.R. Investigation of exchange processes by 2-dimensional NMR-spectroscopy. J. Chem. Phys. 1979, 71, 4546–4553. [Google Scholar] [CrossRef]
- Braunschweiler, L.; Ernst, R.R. Coherence transfer by isotropic mixing—Application to proton correlation spectroscopy. J. Magn. Reson. 1983, 53, 521–528. [Google Scholar] [CrossRef]
- McDougal, O.M.; Turner, M.W.; Ormond, A.J.; Poulter, C.D. Three-dimensional structure of conotoxin tx3a: An m-1 branch peptide of the M-superfamily. Biochemistry 2008, 47, 2826–2832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vranken, W.F.; Boucher, W.; Stevens, T.J.; Fogh, R.H.; Pajon, A.; Llinas, P.; Ulrich, E.L.; Markley, J.L.; Ionides, J.; Laue, E.D. The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins Struct. Funct. Bioinform. 2005, 59, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Vriend, G. WHAT IF—A molecular modeling and drug design program. J. Mol. Graph. 1990, 8, 52–56. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Rullmann, J.A.C.; MacArthur, M.W.; Kaptein, R.; Thornton, J.M. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. Nmr 1996, 8, 477–486. [Google Scholar] [CrossRef]
- Eisenberg, D.; Luthy, R.; Bowie, J.U. VERIFY3D: Assessment of protein models with three-dimensional profiles. Macromol. Crystallogr. Pt B 1997, 277, 396–404. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D-Struct. Biol. 2010, 66, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Sambasivarao, S.V.; Roberts, J.; Bharadwaj, V.S.; Slingsby, J.G.; Rohleder, C.; Mallory, C.; Groome, J.R.; McDougal, O.M.; Maupin, C.M. Acetylcholine Promotes Binding of alpha-Conotoxin MII at alpha(3)beta(2) Nicotinic Acetylcholine Receptors. Chembiochem 2014, 15, 413–424. [Google Scholar] [CrossRef] [Green Version]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.M.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef]
- Van der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]











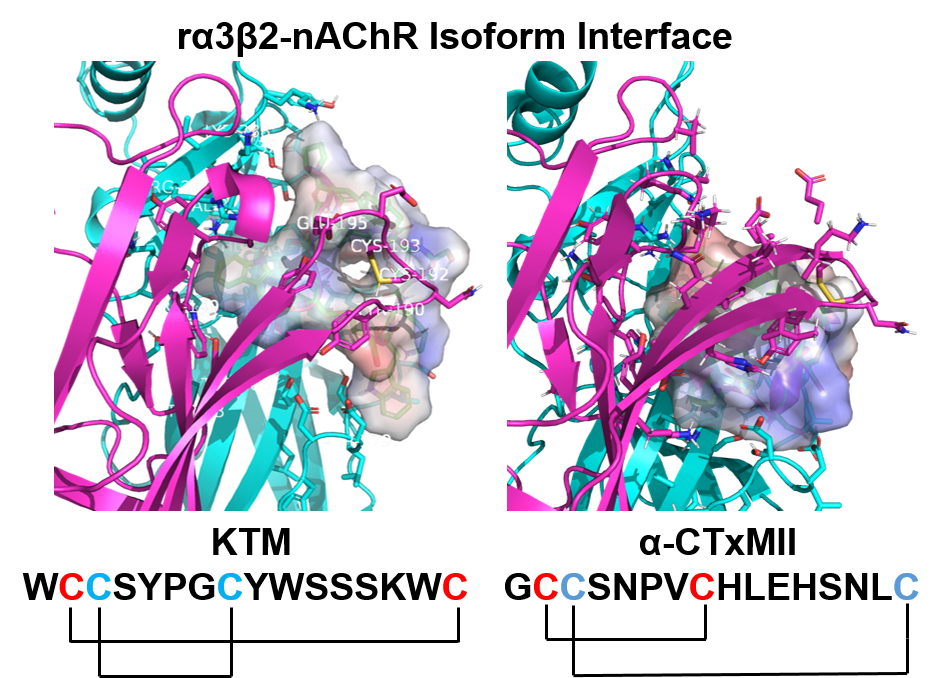{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
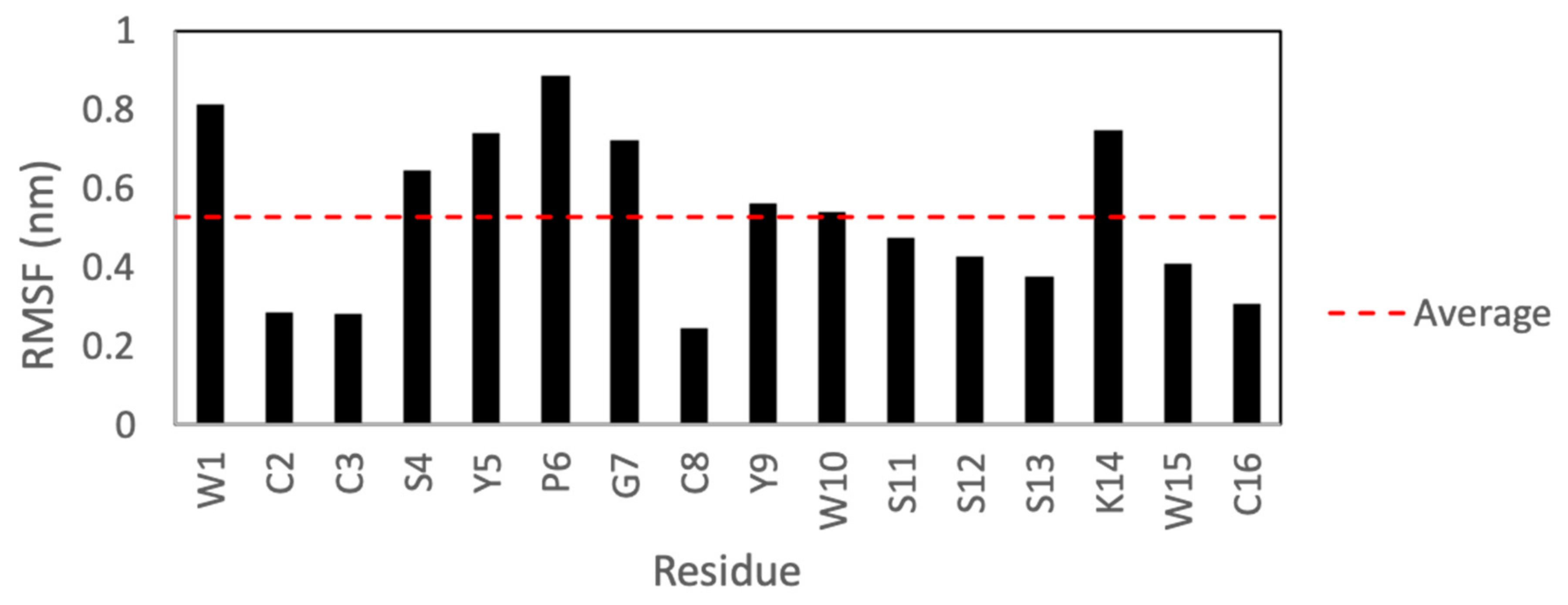{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Residue | NH | αH | βH | Other |
|---|---|---|---|---|
| W1 | 8.31 | 4.62 | 3.61,3.44 | 2H 7.37, N1H 10.23 |
| C2 | 8.57 | 5.33 | 3.71,2.92 | |
| C3 | 8.63 | 5.14 | 2.96,3.67 | |
| S4 | 8.53 | 4.23 | 3.97 | |
| Y5 | 7.91 | 4.84 | 3.39,3.23 | |
| P6 | 4.07 | 1.99,2.20 | δH 3.70,3.58 | |
| G7 | 7.96 | 4.08 | ||
| C8 | 8.40 | 4.53 | 3.62,3.11 | |
| Y9 | 7.78 | 4.57 | 3.32,3.60 | |
| W10 | 8.09 | 4.84 | 3.21 | 2H 7.74, N1H 10.52, 7H 7.88 |
| S11 | 8.62 | 4.33 | 3.83 | |
| S12 | 8.38 | 4.25 | 3.88 | |
| S13 | 8.74 | 4.40 | 4.01 | |
| K14 | 8.19 | 4.66 | 1.72,1.56 | δH 1.39, γH 1.29, εH 2.91, N2H 8.00 |
| W15 | 7.64 | 5.12 | 3.13,3.31 | 2H 7.48, N1H 10.33 |
| C16 | 8.47 | 5.08 | 3.57,3.04 |
| Experimental Data | |
|---|---|
| Distance Restraints | |
| Total NOE | 32 |
| Intra-residue | 12 |
| Inter-residue | 20 |
| Sequential | 18 |
| Short range | 30 |
| Medium range | 2 |
| Long range | 0 |
| ϕ Dihedral angle restraints | 4 |
| Disulfide restraints | 2 |
| Total NOE violations exceeding 0.3 Å | 0 |
| Total NOE violations exceeding 0.3 Å | 0 |
| Structure Statistics | |
| Average pairwise RMSD (Å) | |
| Backbone atoms (residues 1–16) | 1.7 ± 0.5 |
| Heavy atoms (residues 1–16) | 3.0 ± 0.7 |
| Ramachandran statistics | |
| %Favored and allowed regions | 100 |
| %Disallowed regions | 0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marquart, L.A.; Turner, M.W.; Warner, L.R.; King, M.D.; Groome, J.R.; McDougal, O.M. Ribbon α-Conotoxin KTM Exhibits Potent Inhibition of Nicotinic Acetylcholine Receptors. Mar. Drugs 2019, 17, 669. https://doi.org/10.3390/md17120669
Marquart LA, Turner MW, Warner LR, King MD, Groome JR, McDougal OM. Ribbon α-Conotoxin KTM Exhibits Potent Inhibition of Nicotinic Acetylcholine Receptors. Marine Drugs. 2019; 17(12):669. https://doi.org/10.3390/md17120669
Chicago/Turabian StyleMarquart, Leanna A., Matthew W. Turner, Lisa R. Warner, Matthew D. King, James R. Groome, and Owen M. McDougal. 2019. "Ribbon α-Conotoxin KTM Exhibits Potent Inhibition of Nicotinic Acetylcholine Receptors" Marine Drugs 17, no. 12: 669. https://doi.org/10.3390/md17120669
APA StyleMarquart, L. A., Turner, M. W., Warner, L. R., King, M. D., Groome, J. R., & McDougal, O. M. (2019). Ribbon α-Conotoxin KTM Exhibits Potent Inhibition of Nicotinic Acetylcholine Receptors. Marine Drugs, 17(12), 669. https://doi.org/10.3390/md17120669