New Glutamine-Containing Azaphilone Alkaloids from Deep-Sea-Derived Fungus Chaetomium globosum HDN151398

 ,
,  and
and
Abstract
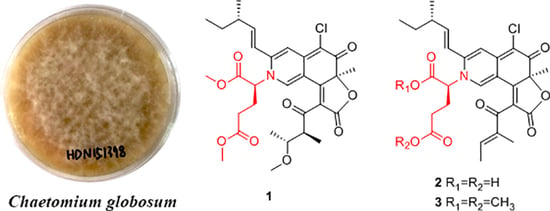
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Fermentation
3.4. Isolation
3.5. Absolute Configuration of Amino Acids
3.6. Degradation of 1–3 by Potassium Hydroxide
3.7. Cytotoxicity Assay
3.8. Amino Acid Incubation Experiment
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wang, Y.T.; Xue, Y.R.; Liu, C.H. A Brief Review of Bioactive Metabolites Derived from Deep-Sea Fungi. Mar. Drugs 2015, 13, 4594–4616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tortorella, E.; Tedesco, P.; Palma Esposito, F.; January, G.G.; Fani, R.; Jaspars, M.; de Pascale, D. Antibiotics from Deep-Sea Microorganisms: Current Discoveries and Perspectives. Mar. Drugs 2018, 16, 355. [Google Scholar] [CrossRef]
- Luo, X.; Lin, X.; Tao, H.; Wang, J.; Li, J.; Yang, B.; Zhou, X.; Liu, Y. Isochromophilones A-F, Cytotoxic Chloroazaphilones from the Marine Mangrove Endophytic Fungus Diaporthe sp. SCSIO 41011. J. Nat. Prod. 2018, 81, 934–941. [Google Scholar] [CrossRef]
- Gao, J.M.; Yang, S.X.; Qin, J.C. Azaphilones: chemistry and biology. Chem. Rev. 2013, 113, 4755–4811. [Google Scholar] [CrossRef]
- Chen, M.; Shen, N.X.; Chen, Z.Q.; Zhang, F.M.; Chen, Y. Penicilones A-D, Anti-MRSA Azaphilones from the Marine-Derived Fungus Penicillium janthinellum HK1-6. J. Nat. Prod. 2017, 80, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Tian, Y.; Yang, S.X.; Zhang, Y.M.; Qin, J.C. Cytotoxic azaphilone alkaloids from Chaetomium globosum TY1. Bioorg. Med. Chem. Lett. 2013, 23, 2945–2947. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.H.; Lin, T.F. Monascus Rice Products. Adv. Food Nutr. Res. 2007, 53, 123–159. [Google Scholar] [PubMed]
- Nam, J.Y.; Kim, H.K.; Kwon, J.Y.; Han, M.Y.; Son, K.H.; Lee, U.C.; Choi, J.D.; Kwon, B.M. 8-O-Methylsclerotiorinamine, antagonist of the Grb2-SH2 domain, isolated from Penicillium multicolor. J. Nat. Prod. 2000, 63, 1303–1305. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.C.; Lee, S.Y.; Searcey, M.; Boger, D.L. The isolation, total synthesis and structure elucidation of chlorofusin, a natural product inhibitor of the p53–MDM2 protein–protein interaction. Nat. Prod. Rep. 2009, 26, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Musso, L.; Dallavalle, S.; Merlini, L.; Bava, A.; Nasini, G.; Penco, S.; Giannini, G.; Giommarelli, C.; De Cesare, A.; Zuco, V.; et al. Natural and semisynthetic azaphilones as a new scaffold for Hsp90 inhibitors. Bioorg. Med. Chem. 2010, 18, 6031–6043. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Lu, Y.; Xu, Y.M.; Zhang, W.; Chen, M.; Lin, M.; Gunatilaka, A.A.; Xu, Y.; Molnar, I. Diversity-Oriented Combinatorial Biosynthesis of Hybrid Polyketide Scaffolds from Azaphilone and Benzenediol Lactone Biosynthons. Org. Lett. 2016, 18, 1262–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makrerougras, M.; Coffinier, R.; Oger, S.; Chevalier, A.; Sabot, C.; Franck, X. Total Synthesis and Structural Revision of Chaetoviridins, A. Org. Lett. 2017, 19, 4146–4149. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Koyama, K.; Natori, S. Four new azaphilones from Chaetomium globosum var. flavo-viridae. Chem. Pharm. Bull. 1990, 38, 625–628. [Google Scholar] [CrossRef]
- Wang, W.; Liao, Y.; Chen, R.; Hou, Y.; Ke, W.; Zhang, B.; Gao, M.; Shao, Z.; Chen, J.; Li, F. Chlorinated Azaphilone Pigments with Antimicrobial and Cytotoxic Activities Isolated from the Deep Sea Derived Fungus Chaetomium sp. NA-S01-R1. Mar. Drugs 2018, 16, 61. [Google Scholar] [CrossRef] [PubMed]
- Steyn, P.S.; Vleggaar, R. The Structure of Dihydrodeoxy-8-epi-austdiol and the Absolute Configuration of the Azaphilones. J. Chem. Soc. Perkin Transactions 1976, 7, 204–206. [Google Scholar] [CrossRef]
- Yamada, T.; Muroga, Y.; Tanaka, R. New azaphilones, seco-chaetomugilins A and D, produced by a marine-fish-derived Chaetomium globosum. Mar. Drugs 2009, 7, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Fujii, K.; Ikai, Y.; Mayumi, T.; Oka, H.; Suzuki, M.; Harada, K.I. A Nonempirical Method Using LC/MS for Determination of the Absolute Configuration of Constituent Amino Acids in a Peptide: Elucidation of Limitations of Marfey’s Method and of Its Separation Mechanism. Anal. Chem. 1997, 69, 549–557. [Google Scholar] [CrossRef]
- Yamada, T.; Doi, M.; Shigeta, H.; Muroga, Y.; Hosoe, S.; Numata, A.; Tanaka, R. Absolute stereostructures of cytotoxic metabolites, chaetomugilins A–C, produced by a Chaetomium species separated from a marine fish. Tetrahedron Lett. 2008, 49, 4192–4195. [Google Scholar] [CrossRef]
- Peng, J.; Gao, H.; Zhang, X.; Wang, S.; Wu, C.; Gu, Q.; Guo, P.; Zhu, T.; Li, D. Psychrophilins E–H and Versicotide C, Cyclic Peptides from the Marine-Derived Fungus Aspergillus versicolor ZLN-60. J. Nat. Prod. 2014, 77, 2218–2223. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; He, X.; Liu, C.; Che, Q.; Zhu, T.; Gu, Q.; Li, D. Clindanones A and B and cladosporols F and G, polyketides from the deep-sea derived fungus Cladosporium cladosporioides HDN14-342. RSC Adv. 2016, 6, 76498–76504. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 a | 2 b | 3 a |
|---|---|---|---|
| 1 | 8.48, s | 7.76, s | 7.81, s |
| 4 | 6.84, s | 6.90, s | 6.72 |
| 9 | 6.25, d (15.4) | 6.46, d (14.2) | 6.17, d (15.4) |
| 10 | 6.40, dd (7.5, 15.4) | 6.41, m | 6.32, dd (7.6, 15.4) |
| 11 | 2.33, m | 2.35, m | 2.28, m |
| 12 | 1.49, m | 1.51, m | 1.44, m |
| 13 | 0.95, t (7.4) | 0.96, t (7.5) | 0.91, t (7.5) |
| 14 | 1.13, d (6.7) | 1.13, d (6.7) | 1.08, d (6.7) |
| 15 | 1.73, s | 1.69, s | 1.69, s |
| 2′ | 5.13, t (7.9) | 5.26, m | 5.05, t (7.3) |
| 3′ | 2.37, m | 2.29, m | 2.47, m |
| 2.68, m | 2.57, m | 2.30, m | |
| 4′ | 2.47, m | 2.44, t (6.6) | 2.42, m |
| 2.57, m | 2.49, m | 2.56, m | |
| 6′ | 3.72, s | 3.67, s | |
| 7′ | 3.83, s | 3.77, s | |
| 4″ | 3.71, m | ||
| 5″ | 3.56, m | 6.64, q (6.9) | 6.55, q (6.3) |
| 6″ | 1.04, d (6.3) | 1.89, d (7.0) | 1.87, d (6.9) |
| 7″ | 1.07, d (6.8) | 1.87, s | 1.83, s |
| 8″ | 2.93, s |
| No. | 1 a | 2 b | 3 a |
|---|---|---|---|
| 1 | 136.9 | 136.3 | 135.4 |
| 2 | |||
| 3 | 148.4 | 150.4 | 148.2 |
| 4 | 111.1 | 110.6 | 110.7 |
| 4a | 144.5 | 145.9 | 143.8 |
| 5 | 101.2 | 99.4 | 101.6 |
| 6 | 182.1 | 181.8 | 182.1 |
| 7 | 88.5 | 88.3 | 88.5 |
| 8 | 165.4 | 161.6 | 162.9 |
| 8a | 112.4 | 112.4 | 112.3 |
| 9 | 119.0 | 119.7 | 119.2 |
| 10 | 150.3 | 150.1 | 149.9 |
| 11 | 39.0 | 38.9 | 38.9 |
| 12 | 29.0 | 28.6 | 29.0 |
| 13 | 11.7 | 10.6 | 11.6 |
| 14 | 16.1 | 17.9 | 19.0 |
| 15 | 26.6 | 25.0 | 26.2 |
| 1′ | 167.9 | 169.4 | 168.3 |
| 2′ | 61.9 | 62.7 | 61.7 |
| 3′ | 27.0 | 26.8 | 27.2 |
| 4′ | 29.2 | 29.2 | 29.3 |
| 5′ | 172.2 | 173.8 | 172.1 |
| 6′ | 52.1 | 52.1 | |
| 7′ | 53.6 | 53.4 | |
| 1″ | 168.4 | 168.3 | 168.1 |
| 2″ | 125.0 | 124.4 | 125.1 |
| 3″ | 199.3 | 190.6 | 190.8 |
| 4″ | 49.3 | 138.0 | 137.6 |
| 5″ | 77.7 | 146.7 | 146.5 |
| 6″ | 16.1 | 14.0 | 15.4 |
| 7″ | 8.4 | 9.2 | 10.7 |
| 8″ | 56.2 |
| No. | 6 b,d | 7 a,e | 8 a,e | 9 a,e | 10 a,e |
|---|---|---|---|---|---|
| 1 | 9.08, s | 8.61, s | 8.53, s | 8.53, s | 8.99, s |
| 4 | 6.87, s | 6.42, s | 6.49, s | 6.78, s | 6.58, s |
| 9 | 6.26, d (15.0) | 6.14, d (12.8) | 6.28, d (12.8) | 6.40, d (15.6) | 6.34, d (15.7) |
| 10 | 6.43, m | 6.01, dd (5.9, 12.8) | 6.14, m | 6.45, dd (7.1, 15.6) | 6.25, m |
| 11 | 2.35, m | 2.06, m | 2.24, m | 2.26, m | 2.26, m |
| 12 | 1.59, m | 1.26, m | 1.39, m | 1.39, m | 1.40, m |
| 13 | 0.96, t (6.6) | 0.76, t (6.2) | 0.86, t (6.2) | 0.83, t (7.4) | 0.85, t (7.4) |
| 14 | 1.14, ov. c | 0.98, d (5.6) | 0.89, d (5.0) | 1.02, d (6.7) | 1.03, d (6.7) |
| 15 | 1.93, s | 1.43, s | 1.54, s | 1.56, s | 1.60, s |
| 2′ | 5.08, m | 5.53, dd (4.1, 8.5) | 5.49, dd (4.3, 8.9) | 4.97, d (18.1) | 5.48, t (7.7) |
| 5.10, d (18.1) | |||||
| 3′ | 1.50, t (6.3) | 3.48, m | 3.17, t (9.4) | 3.49, m | |
| 3.68, dd (4.1, 12.8) | 3.47, dd (4.1,12.3) | ||||
| 4′ | |||||
| 5′ | 6.96, d (7.1) | 7.34, s | |||
| 6′ | 7.49, d (6.6) | 6.58, d (7.1) | |||
| 7′ | 6.91, t (6.2) | 7.41, s | |||
| 8′ | 7.03, t (6.1) | 6.58, d (7.1) | |||
| 9′ | 7.29, d (6.8) | 6.96, d (7.1) | |||
| 11′ | 10.94, s | ||||
| 12′ | 7.14, d (1.9) | ||||
| 4″ | 3.56, m | 3.56, m | 3.56, m | 3.49, m | |
| 5″ | 3.81, m | 3.62, m | 3.61, m | 3.62, m | 6.65, d (6.8) |
| 6″ | 1.07, m | 0.89, d (5.2) | 0.98, d (5.4) | 0.92, d (6.2) | 1.82, d (6.8) |
| 7″ | 1.14, ov. c | 0.92, d (5.6) | 1.01, d (5.6) | 0.96, d (6.7) | 1.77, s |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, C.; Ge, X.; Mudassir, S.; Zhou, L.; Yu, G.; Che, Q.; Zhang, G.; Peng, J.; Gu, Q.; Zhu, T.; et al. New Glutamine-Containing Azaphilone Alkaloids from Deep-Sea-Derived Fungus Chaetomium globosum HDN151398. Mar. Drugs 2019, 17, 253. https://doi.org/10.3390/md17050253
Sun C, Ge X, Mudassir S, Zhou L, Yu G, Che Q, Zhang G, Peng J, Gu Q, Zhu T, et al. New Glutamine-Containing Azaphilone Alkaloids from Deep-Sea-Derived Fungus Chaetomium globosum HDN151398. Marine Drugs. 2019; 17(5):253. https://doi.org/10.3390/md17050253
Chicago/Turabian StyleSun, Chunxiao, Xueping Ge, Shah Mudassir, Luning Zhou, Guihong Yu, Qian Che, Guojian Zhang, Jixing Peng, Qianqun Gu, Tianjiao Zhu, and et al. 2019. "New Glutamine-Containing Azaphilone Alkaloids from Deep-Sea-Derived Fungus Chaetomium globosum HDN151398" Marine Drugs 17, no. 5: 253. https://doi.org/10.3390/md17050253
APA StyleSun, C., Ge, X., Mudassir, S., Zhou, L., Yu, G., Che, Q., Zhang, G., Peng, J., Gu, Q., Zhu, T., & Li, D. (2019). New Glutamine-Containing Azaphilone Alkaloids from Deep-Sea-Derived Fungus Chaetomium globosum HDN151398. Marine Drugs, 17(5), 253. https://doi.org/10.3390/md17050253