Isolation and Characterization of Antimicrobial Peptides with Unusual Disulfide Connectivity from the Colonial Ascidian Synoicum turgens

 , , , , , , , and
, , , , , , , and
Abstract
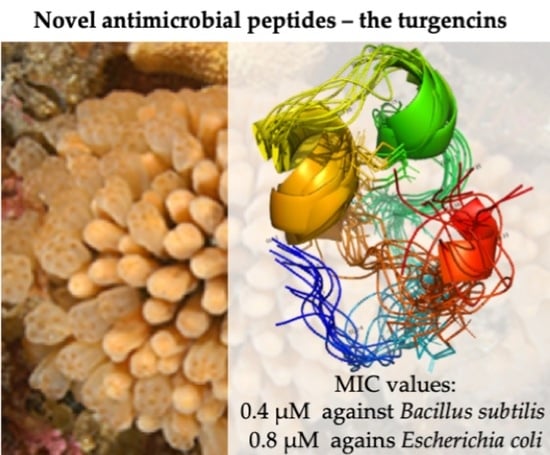
:
1. Introduction
2. Results and Discussion
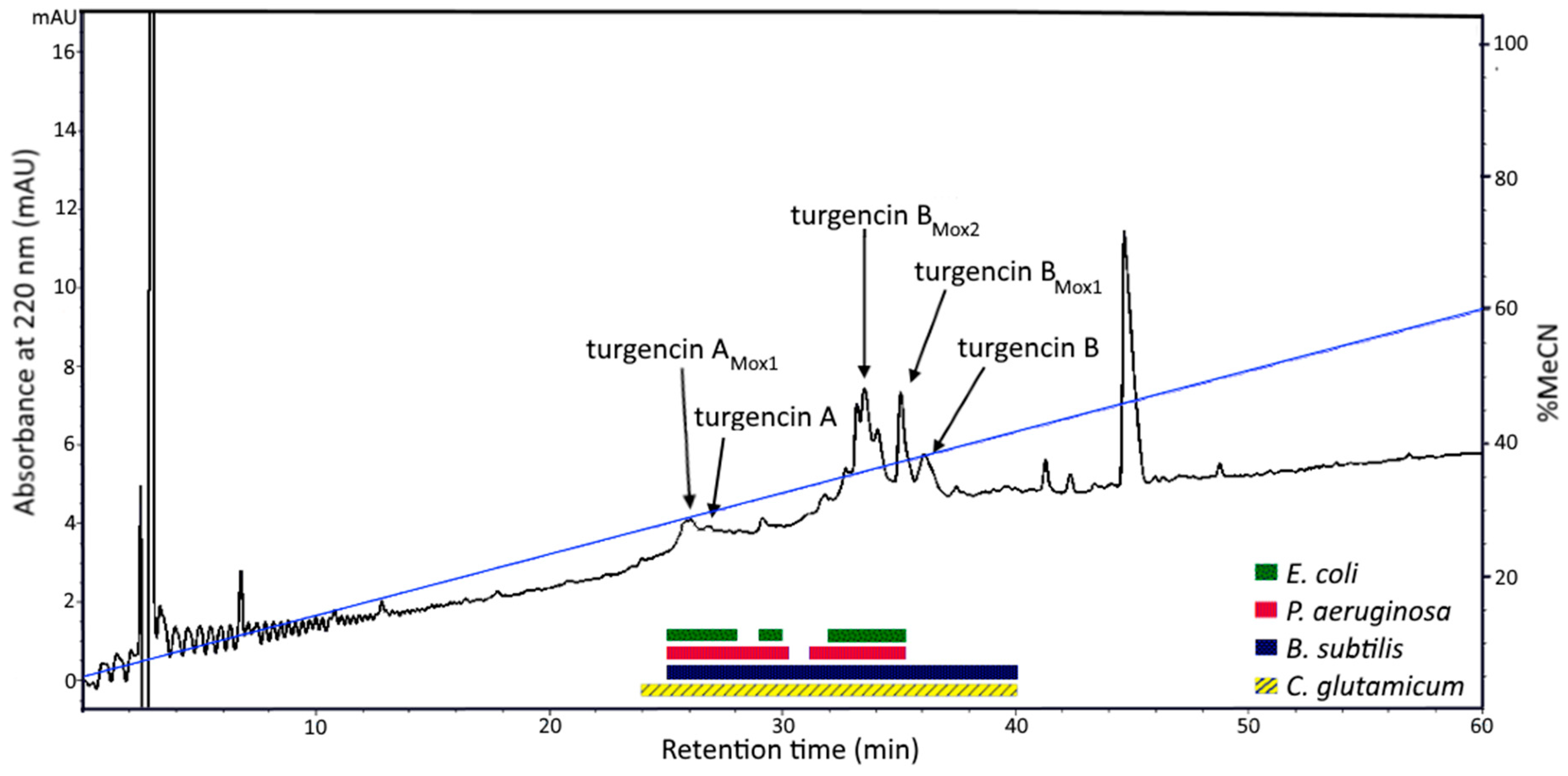
2.1. Peptide Purification and Mass Spectrometry Analysis
2.2. Sequence Analysis
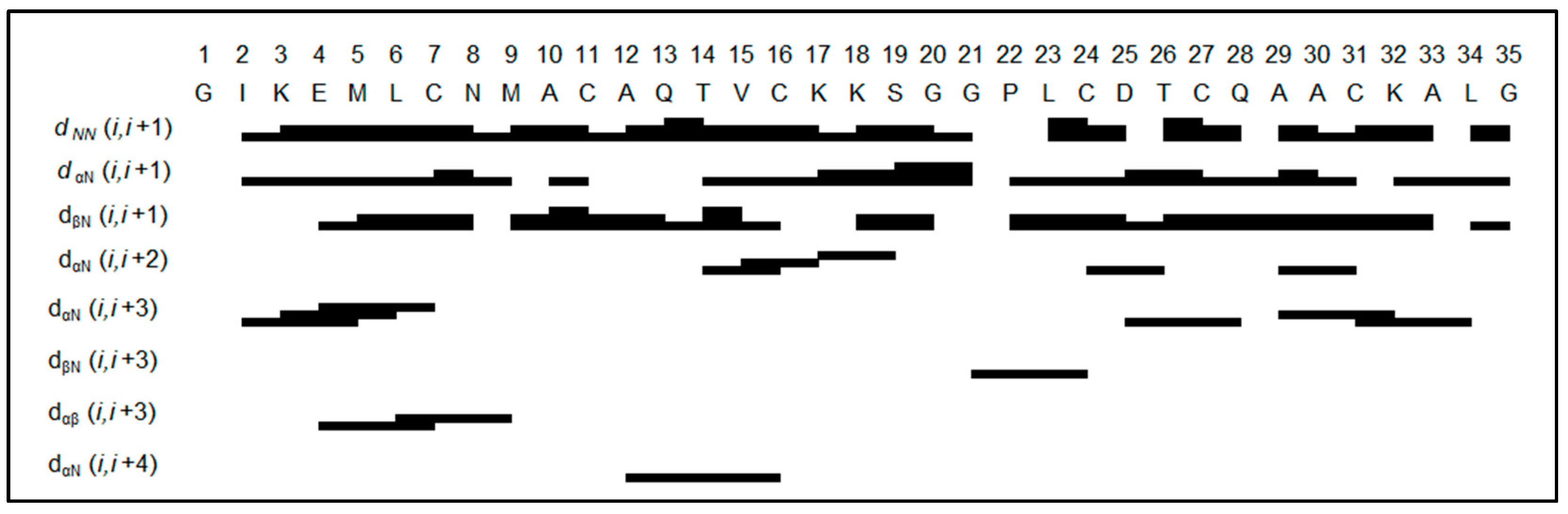
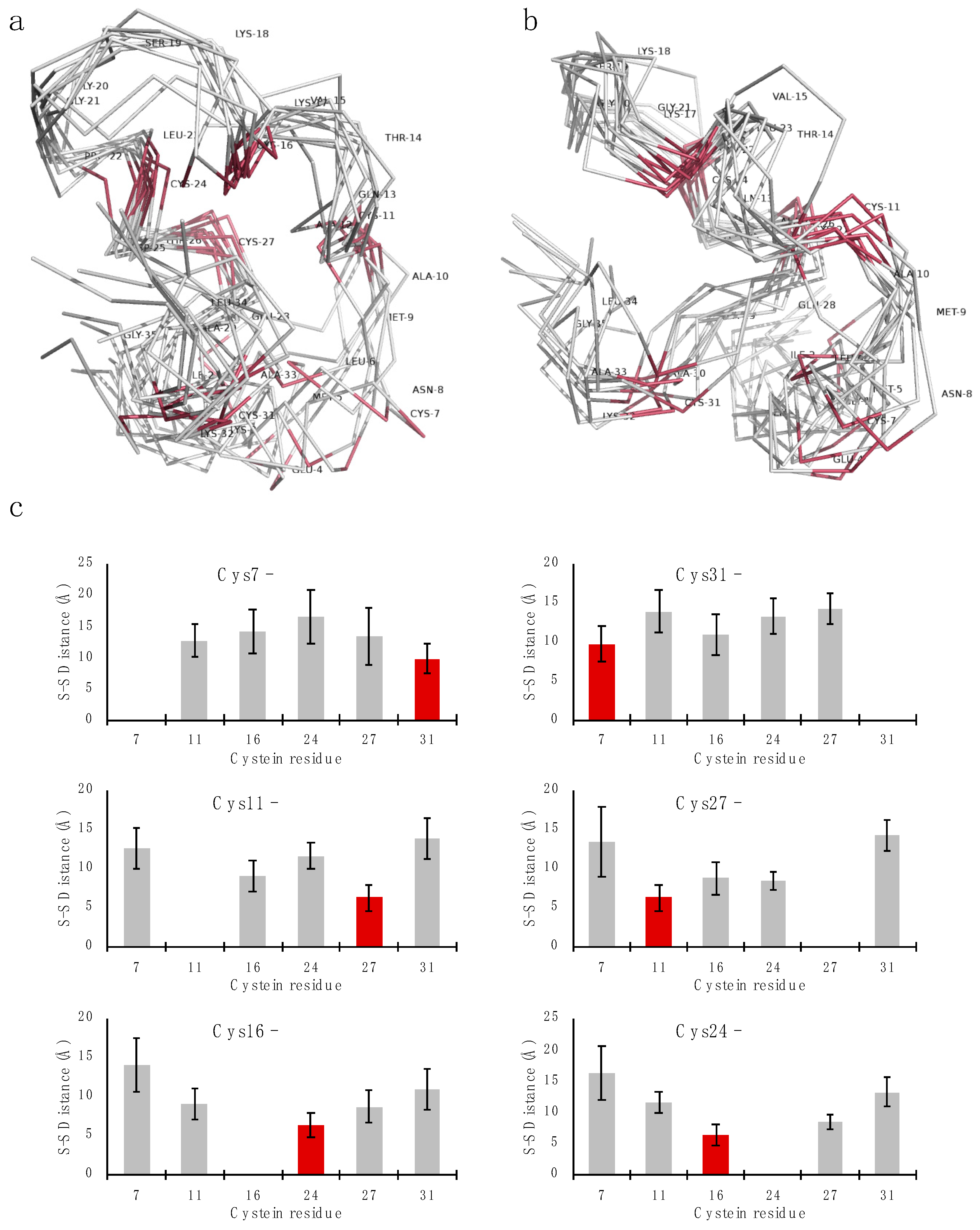
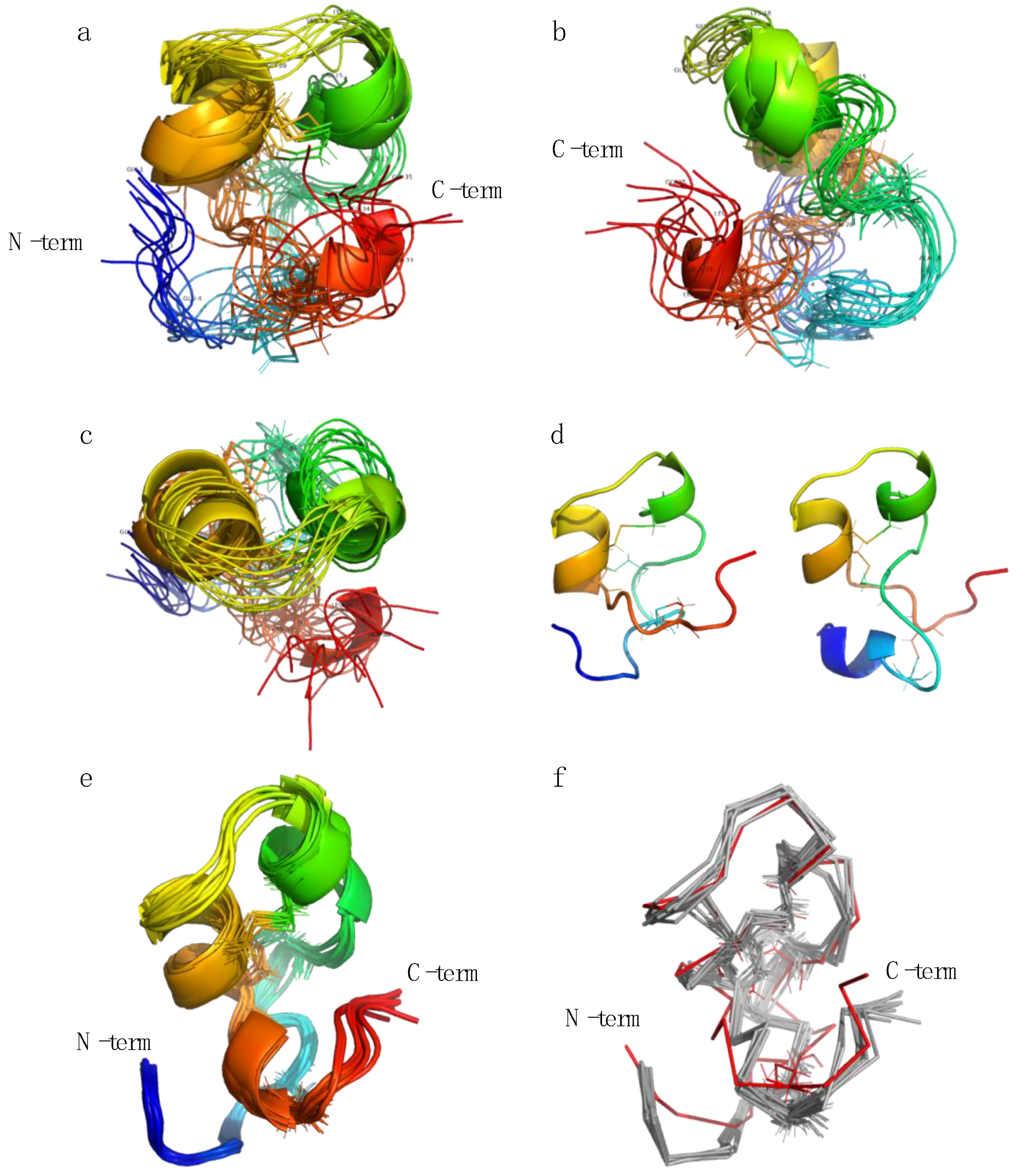
2.3. Structure Determination
2.4. Biological Activity
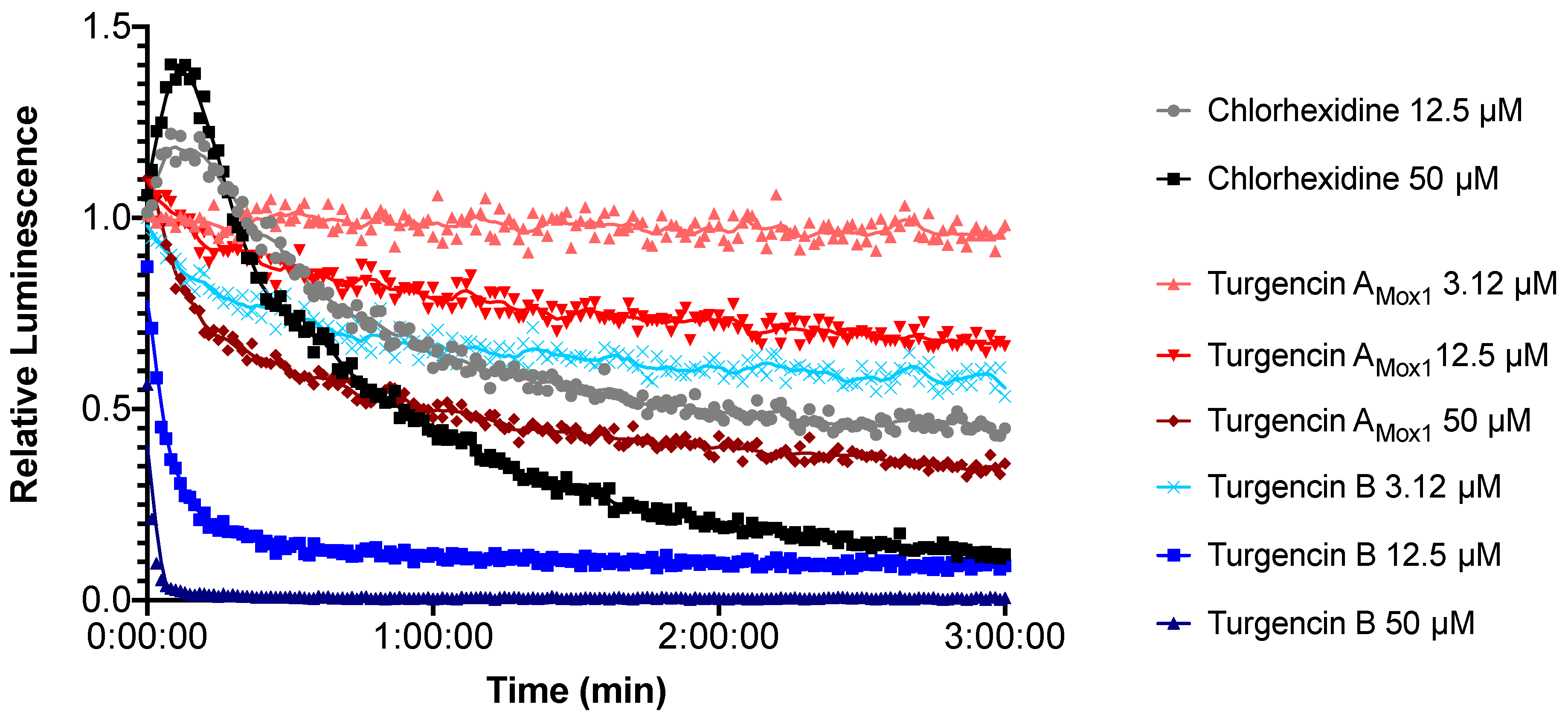
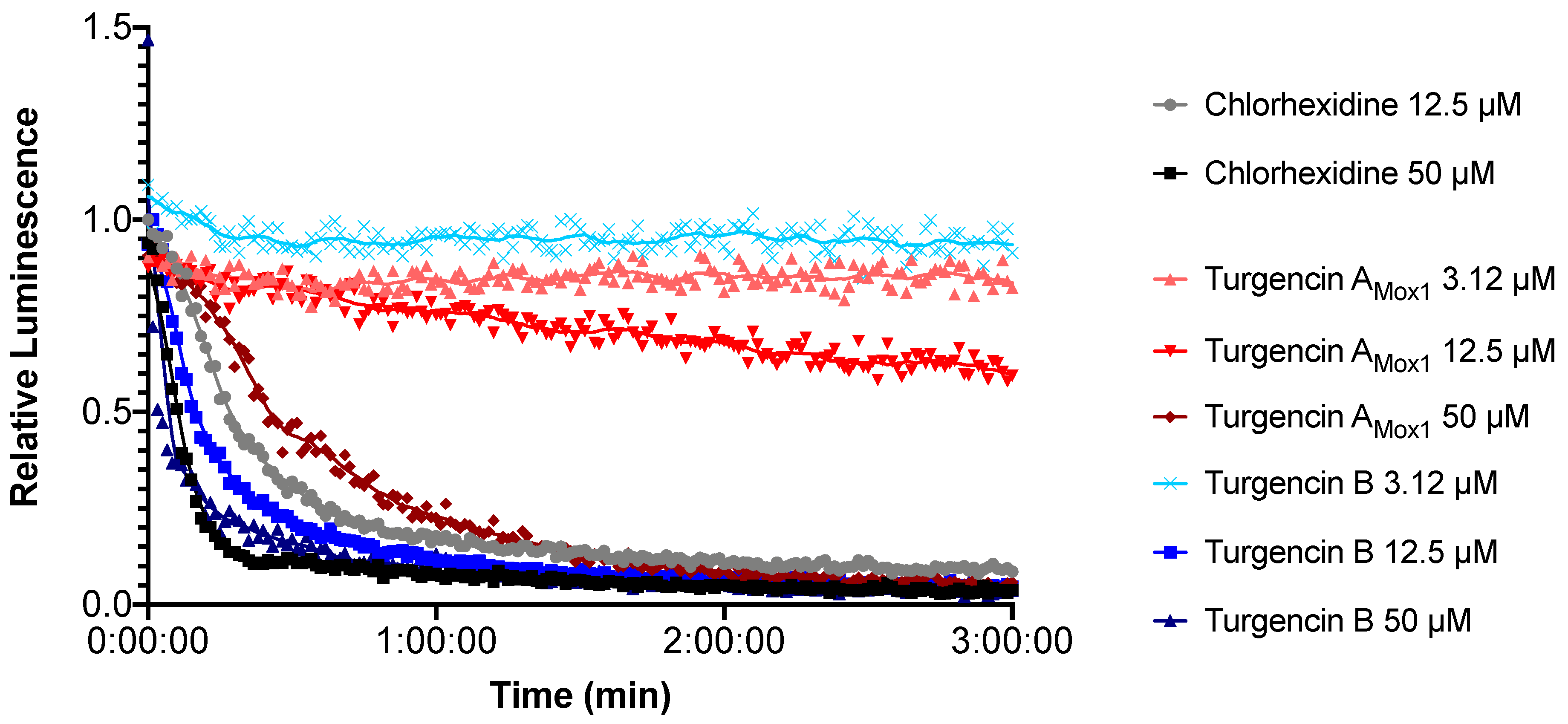
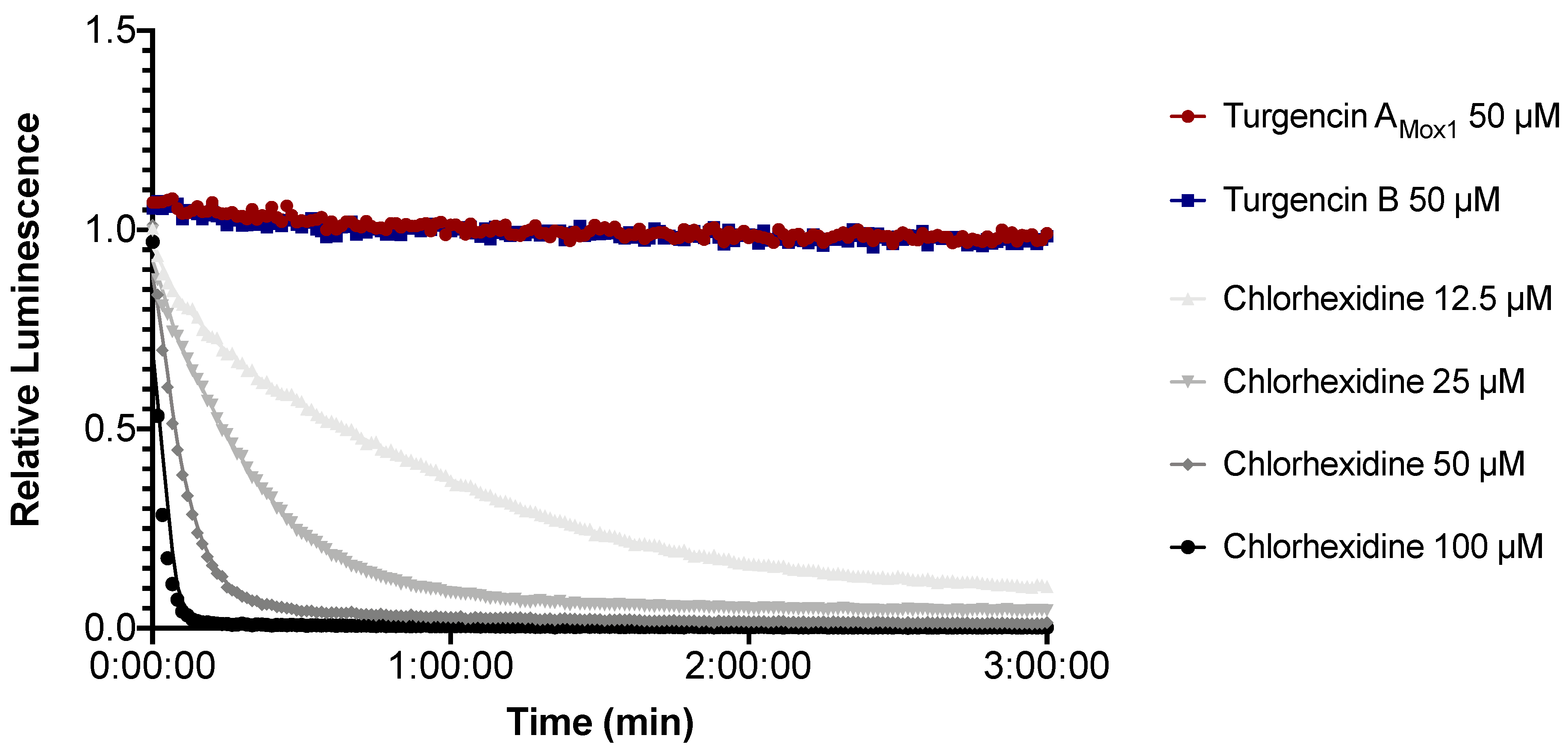
2.5. Real-time Measurement of Immediate Effect on Membrane Integrity and Viability
3. Materials and Methods
3.1. Materials
3.2. Extraction of Antimicrobial Peptides
3.3. Peptide Sequencing
3.4. Partial Reduction and Alkylation
3.5. NMR Spectroscopy and Calculations
3.6. Microbial Strains and Antibacterial Activity Assays
3.7. Real-time Assay Measuring Immediate Membrane Disruption
3.8. Real-time Assay Measuring Bacterial Cell Viability
3.9. Human Cell Viability Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global Action Plan on Antimicrobial Resistance. Available online: http://www.who.int/antimicrobial-resistance/publications/global-action-plan/en/ (accessed on 14 September 2019).
- Wright, G.D. The antibiotic resistome: The nexus of chemical and genetic diversity. Nat. Rev. Microbiol. 2007, 5, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Magiorakos, A.P.; Srinivasan, A.; Carey, R.B.; Carmeli, Y.; Falagas, M.E.; Giske, C.G.; Harbarth, S.; Hindler, J.F.; Kahlmeter, G.; Olsson-Liljequist, B.; et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: An international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infec. 2012, 18, 268–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Projan, S.J. Why is big Pharma getting out of antibacterial drug discovery? Curr. Opin. Microbiol. 2003, 6, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Brogan, D.M.; Mossialos, E. Systems, not pills: The options market for antibiotics seeks to rejuvenate the antibiotic pipeline. Soc. Sci. Med. 2016, 151, 167–172. [Google Scholar] [CrossRef]
- Hancock, R.; Patrzykat, A. Clinical development of cationic antimicrobial peptides: From natural to novel antibiotics. Curr. Drug Targets Infect. Disord. 2002, 2, 79–83. [Google Scholar] [CrossRef]
- Hancock, R.E.W.; Sahl, H.-G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef]
- Ganz, T. The role of antimicrobial peptides in innate immunity. Integr. Comp. Biol. 2003, 43, 300–304. [Google Scholar] [CrossRef] [Green Version]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Haney, E.F.; Vogel, H.J. The expanding scope of antimicrobial peptide structures and their modes of action. Trends Biotechnol. 2011, 29, 464–472. [Google Scholar] [CrossRef]
- Peach, K.C.; Bray, W.M.; Winslow, D.; Linington, P.F.; Linington, R.G. Mechanism of action-based classification of antibiotics using high-content bacterial image analysis. Mol. Biosyst. 2013, 9, 1837–1848. [Google Scholar] [CrossRef]
- Pasupuleti, M.; Schmidtchen, A.; Malmsten, M. Antimicrobial peptides: Key components of the innate immune system. Crit. Rev. Biotechnol. 2012, 32, 143–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuzaki, K. Why and how are peptide–lipid interactions utilized for self-defense? Magainins and tachyplesins as archetypes. BBA Biomembr. 1999, 1462, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Pushpanathan, M.; Gunasekaran, P.; Rajendhran, J. Antimicrobial peptides: Versatile biological properties. Int. J. Pept. 2013, 2013, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef] [Green Version]
- Deslouches, B.; Di, Y.P. Antimicrobial peptides with selective antitumor mechanisms: Prospect for anticancer applications. Oncotarget 2017, 8, 46635–46651. [Google Scholar] [CrossRef] [Green Version]
- Millero, F.J. Descriptive oceanography. In Chemical Oceanography, 4th ed.; CRC Press Taylor & Francis Group: Boca Raton, FL, USA, 2013. [Google Scholar]
- Buchmann, K. Evolution of innate immunity: Clues from invertebrates via fish to mammals. Front. Immunol. 2014, 5, 459. [Google Scholar] [CrossRef] [Green Version]
- Falanga, A.; Lombardi, L.; Franci, G.; Vitiello, M.; Iovene, M.R.; Morelli, G.; Galdiero, M.; Galdiero, S. Marine antimicrobial peptides: Nature provides templates for the design of novel compounds against pathogenic bacteria. Int. J. Mol. Sci. 2016, 17, 785. [Google Scholar] [CrossRef] [Green Version]
- Fedders, H.; Michalek, M.; Grötzinger, J.; Leippe, M. An exceptional salt-tolerant antimicrobial peptide derived from a novel gene family of haemocytes of the marine invertebrate Ciona intestinalis. Biochem. J. 2008, 416, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Venier, P.; Varotto, L.; Rosani, U.; Millino, C.; Celegato, B.; Bernante, F.; Lanfranchi, G.; Novoa, B.; Roch, P.; Figueras, A.; et al. Insights into the innate immunity of the Mediterranean mussel Mytilus Galloprovincialis. BMC Genom. 2011, 12, 69. [Google Scholar] [CrossRef] [Green Version]
- Stensvåg, K.; Haug, T.; Sperstad, S.V.; Rekdal, Ø.; Indrevoll, B.; Styrvold, O.B. Arasin 1, a proline–arginine-rich antimicrobial peptide isolated from the spider crab, Hyas araneus. Dev. Comp. Immunol. 2008, 32, 275–285. [Google Scholar] [CrossRef]
- Wiens, M.; Schröder, H.C.; Korzhev, M.; Wang, X.-H.; Batel, R.; Müller, W.E.G. Inducible ASABF-type antimicrobial peptide from the sponge Suberites domuncula: Microbicidal and hemolytic activity in vitro and toxic effect on molluscs in vivo. Mar. Drugs 2011, 9, 1969–1994. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Dupiol, J.; Ladrière, O.; Destoumieux-Garzón, D.; Sautière, P.-E.; Meistertzheim, A.-L.; Tambutté, E.; Tambutté, S.; Duval, D.; Fouré, L.; Adjeroud, M.; et al. Innate immune responses of a scleractinian coral to vibriosis. J. Biol. Chem. 2011, 286, 22688–22698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, I.H.; Cho, Y.; Lehrer, R.I. Styelins, broad-spectrum antimicrobial peptides from the solitary tunicate, Styela Clava. Comp. Biochem. Phys. B 1997, 118, 515–521. [Google Scholar] [CrossRef]
- Lee, I.H.; Zhao, C.; Cho, Y.; Harwig, S.S.L.; Cooper, E.L.; Lehrer, R.I. Clavanins, α-helical antimicrobial peptides from tunicate hemocytes. FEBS Lett. 1997, 400, 158–162. [Google Scholar] [CrossRef] [Green Version]
- Galinier, R.; Roger, E.; Sautiere, P.-E.; Aumelas, A.; Banaigs, B.; Mitta, G. Halocyntin and papillosin, two new antimicrobial peptides isolated from hemocytes of the solitary tunicate, Halocynthia Papillosa. J. Pept. Sci. 2009, 15, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.H.; Lee, Y.S.; Kim, C.H.; Kim, C.R.; Hong, T.; Menzel, L.; Boo, L.M.; Pohl, J.; Sherman, M.A.; Waring, A.; et al. Dicynthaurin: An antimicrobial peptide from hemocytes of the solitary tunicate. Halocynthia aurantium. BBA Gen. Subj. 2001, 1527, 141–148. [Google Scholar] [CrossRef]
- Jang, W.S.; Kim, K.N.; Lee, Y.S.; Nam, M.H.; Lee, I.H. Halocidin: A new antimicrobial peptide from hemocytes of the solitary tunicate, Halocynthia aurantium. FEBS Lett. 2002, 521, 81–86. [Google Scholar] [CrossRef] [Green Version]
- Diyabalanage, T.; Amsler, C.D.; McClintock, J.B.; Baker, B.J. Palmerolide A, a cytotoxic macrolide from the Antarctic tunicate Synoicum adareanum. J. Am. Chem. Soc. 2006, 128, 5630–5631. [Google Scholar] [CrossRef]
- Noguez, J.H.; Diyabalanage, T.K.K.; Miyata, Y.; Xie, X.-S.; Valeriote, F.A.; Amsler, C.D.; McClintock, J.B.; Baker, B.J. Palmerolide macrolides from the Antarctic tunicate Synoicum adareanum. Bioorgan. Med. Chem. 2011, 19, 6608–6614. [Google Scholar] [CrossRef]
- Buck, M.L.; Evans, D.M.; Evans, V.A.; Herkt, D.; Sharp, H.; Hollinshead, J.; Mitschang, F.; Murphy, P.J.; Nash, R.J.; Wenbourne, J. A synthesis of tiruchanduramine and a reinvestigation of its glycosidase inhibitory activity. Tetrahedron 2017, 73, 4545–4548. [Google Scholar] [CrossRef]
- Ravinder, K.; Vijender Reddy, A.; Krishnaiah, P.; Ramesh, P.; Ramakrishna, S.; Laatsch, H.; Venkateswarlu, Y. Isolation and synthesis of a novel β-carboline guanidine derivative tiruchanduramine from the Indian ascidian Synoicum macroglossum. Tetrahedron Lett. 2005, 46, 5475–5478. [Google Scholar] [CrossRef]
- Tadesse, M.; Strøm, M.B.; Svenson, J.; Jaspars, M.; Milne, B.F.; Tørfoss, V.; Andersen, J.H.; Hansen, E.; Stensvåg, K.; Haug, T. Synoxazolidinones A and B: Novel bioactive alkaloids from the ascidian Synoicum pulmonaria. Org. Lett. 2010, 12, 4752–4755. [Google Scholar] [CrossRef] [PubMed]
- Tadesse, M.; Svenson, J.; Jaspars, M.; Strøm, M.B.; Abdelrahman, M.H.; Andersen, J.H.; Hansen, E.; Kristiansen, P.E.; Stensvåg, K.; Haug, T. Synoxazolidinone C; a bicyclic member of the synoxazolidinone family with antibacterial and anticancer activities. Tetrahedron Lett. 2011, 52, 1804–1806. [Google Scholar] [CrossRef]
- Tadesse, M.; Svenson, J.; Sepčić, K.; Trembleau, L.; Engqvist, M.; Andersen, J.H.; Jaspars, M.; Stensvåg, K.; Haug, T. Isolation and synthesis of pulmonarins A and B, acetylcholinesterase inhibitors from the colonial ascidian Synoicum pulmonaria. J. Nat. Prod. 2014, 77, 364–369. [Google Scholar] [CrossRef] [Green Version]
- Trepos, R.; Cervin, G.; Hellio, C.; Pavia, H.; Stensen, W.; Stensvåg, K.; Svendsen, J.-S.; Haug, T.; Svenson, J. Antifouling compounds from the sub-arctic ascidian Synoicum pulmonaria: Synoxazolidinones A and C, pulmonarins A and B, and synthetic analogues. J. Nat. Prod. 2014, 77, 2105–2113. [Google Scholar] [CrossRef]
- Gu, Y.; Shih, P.-H. Salt-induced phase separation can effectively remove the acetonitrile from the protein sample after the preparative RP-HPLC. Enzym. Microb. Technol. 2004, 35, 592–597. [Google Scholar] [CrossRef]
- Lao, Y.W.; Gungormusler-Yilmaz, M.; Shuvo, S.; Verbeke, T.; Spicer, V.; Krokhin, O.V. Chromatographic behavior of peptides containing oxidized methionine residues in proteomic LC–MS experiments: Complex tale of a simple modification. J. Proteom. 2015, 125, 131–139. [Google Scholar] [CrossRef]
- Waghu, F.H.; Barai, R.S.; Idicula-Thomas, S. Leveraging family-specific signatures for AMP discovery and high-throughput annotation. Sci. Rep. 2016, 6, 24684. [Google Scholar] [CrossRef] [Green Version]
- Squier, T.C.; Bigelow, D.J. Protein oxidation and age-dependent alterations in calcium homeostasis. Front. Biosci. 2000, 5, d504–d526. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.; Weiss, S.J.; Levine, R.L. Methionine oxidation and reduction in proteins. BBA Gen. Subj. 2014, 1840, 901–905. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, G.K.T.; Lim, W.H.; Nguyen, P.Q.T.; Tam, J.P. Novel cyclotides and uncyclotides with highly shortened precursors from Chassalia chartacea and effects of methionine oxidation on bioactivities. J. Biol. Chem. 2012, 287, 17598–17607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, S.; Kodera, Y.; Saito, T.; Fujimoto, K.; Momozono, A.; Hayashi, A.; Kamata, Y.; Shichiri, M. Methionine sulfoxides in serum proteins as potential clinical biomarkers of oxidative stress. Sci. Rep. 2016, 6, 38299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Nip, A.M.; Wishart, D.S. A simple method to quantitatively measure polypeptide JHNHα coupling constants from TOCSY or NOESY spectra. J. Biomol. NMR 1997, 10, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Ludvigsen, S.; Andersen, K.V.; Poulsen, F.M. Accurate measurements of coupling constants from two-dimensional nuclear magnetic resonance spectra of proteins and determination of φ-angles. J. Mol. Biol. 1991, 217, 731–736. [Google Scholar] [CrossRef]
- Schneider, J.J.; Unholzer, A.; Schaller, M.; Schäfer-Korting, M.; Korting, H.C. Human defensins. J. Mol. Med. 2005, 83, 587–595. [Google Scholar] [CrossRef]
- Romagnoli, S.; Ugolini, R.; Fogolari, F.; Schaller, G.; Urech, K.; Giannattasio, M.; Ragona, L.; Molinari, H. NMR structural determination of viscotoxin A3 from Viscum album L. Biochem. J. 2000, 350, 569–577. [Google Scholar] [CrossRef]
- Pal, A.; Debreczeni, J.E.; Sevvana, M.; Gruene, T.; Kahle, B.; Zeeck, A.; Sheldrick, G.M. Structures of viscotoxins A1 and B2 from European mistletoe solved using native data alone. Acta Crystallogr. D 2008, 64, 985–992. [Google Scholar] [CrossRef]
- Ahn, H.-C.; Juranić, N.; Macura, S.; Markley, J.L. Three-dimensional structure of the water-insoluble protein crambin in dodecylphosphocholine micelles and its minimal solvent-exposed surface. J. Am. Chem. Soc. 2006, 128, 4398–4404. [Google Scholar] [CrossRef] [Green Version]
- Teeter, M.M. Water structure of a hydrophobic protein at atomic resolution: Pentagon rings of water molecules in crystals of crambin. Proc. Natl. Acad. Sci. USA 1984, 81, 6014–6018. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.C.H.; Hanson, B.L.; Fisher, S.Z.; Langan, P.; Kovalevsky, A.Y. Direct observation of hydrogen atom dynamics and interactions by ultrahigh resolution neutron protein crystallography. Proc. Natl. Acad. Sci. USA 2012, 109, 15301–15306. [Google Scholar] [CrossRef] [Green Version]
- Chattopadhyay, S.; Sinha, N.K.; Banerjee, S.; Roy, D.; Chattopadhyay, D.; Roy, S. Small cationic protein from a marine turtle has β-defensin-like fold and antibacterial and antiviral activity. Proteins 2006, 64, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Lakshminarayanan, R.; Vivekanandan, S.; Samy, R.P.; Banerjee, Y.; Chi-Jin, E.O.; Teo, K.W.; Jois, S.D.S.; Kini, R.M.; Valiyaveettil, S. Structure, self-assembly, and dual role of a β-defensin-like peptide from the Chinese soft-shelled turtle eggshell matrix. J. Am. Chem. Soc. 2008, 130, 4660–4668. [Google Scholar] [CrossRef] [PubMed]
- Niederhafner, P.; Bednárová, L.; Buděšínský, M.; Šafařík, M.; Ehala, S.; Ježek, J.; Borovičková, L.; Fučík, V.; Čeřovský, V.; Slaninová, J. Melectin MAPs: The influence of dendrimerization on antimicrobial and hemolytic activity. Amino Acids 2010, 39, 1553–1561. [Google Scholar] [CrossRef] [PubMed]
- Virta, M.; Åkerman, K.E.O.; Saviranta, P.; Oker-Blom, C.; Karp, M.T. Real-time measurement of cell permeabilization with low-molecular-weight membranolytic agents. J. Antimicrob. Chemoth. 1995, 36, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Kuyyakanond, T.; Quesnel, L.B. The mechanism of action of chlorhexidine. FEMS Microbiol. Lett. 1992, 100, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Karp, M. Whole cell strategies based on lux genes for high throughput applications toward new antimicrobials. Comb. Chem. High Throughput Scr. 2006, 9, 501–514. [Google Scholar]
- Galluzzi, L.; Karp, M. Intracellular redox equilibrium and growth phase affect the performance of luciferase-based biosensors. J. Biotechnol. 2007, 127, 188–198. [Google Scholar] [CrossRef]
- Haug, T.; Kjuul, A.K.; Stensvåg, K.; Sandsdalen, E.; Styrvold, O.B. Antibacterial activity in four marine crustacean decapods. Fish Shellfish Immunol. 2002, 12, 371–385. [Google Scholar] [CrossRef]
- Solstad, R.G.; Li, C.; Isaksson, J.; Johansen, J.; Svenson, J.; Stensvåg, K.; Haug, T. Novel antimicrobial peptides EeCentrocins 1, 2 and EeStrongylocin 2 from the edible sea urchin Echinus esculentus have 6-br-trp post-translational modifications. PLoS ONE 2016, 11, e0151820. [Google Scholar] [CrossRef] [Green Version]
- Vesterlund, S.; Paltta, J.; Lauková, A.; Karp, M.; Ouwehand, A.C. Rapid screening method for the detection of antimicrobial substances. J. Microbiol. Meth. 2004, 57, 23–31. [Google Scholar] [CrossRef]
- Radeck, J.; Kraft, K.; Bartels, J.; Cikovic, T.; Dürr, F.; Emenegger, J.; Kelterborn, S.; Sauer, C.; Fritz, G.; Gebhard, S.; et al. The Bacillus BioBrick Box: Generation and evaluation of essential genetic building blocks for standardized work with Bacillus subtilis. J. Biol. Eng. 2013, 7, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frackman, S.; Anhalt, M.; Nealson, K.H. Cloning, organization, and expression of the bioluminescence genes of Xenorhabdus luminescens. J Bacteriol 1990, 172, 5767–5773. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Antimicrobial Activity (MIC; µM) | Cytotoxic Activity (IC50; μM) | |||||
|---|---|---|---|---|---|---|---|
| C. g. | B. s. | S. a. | E. c. | P. a. | A2058 | MRC-5 | |
| Turgencin AMox1 (72% Mox1, 28% Mox0) | 0.4 | 0.4 | 6.3 | 0.8 | 1.6 | 1.4 | 4.8 |
| Turgencin B (67% Mox0, 29% Mox1, 4% Mox2) | 1.6 | 1.6 | >100.0 | 12.5 | 25.0 | 4.1 | 7.5 |
| Turgencin BMox1 (88% Mox1, 10% Mox2, 2% Mox0) | 1.6 | 3.1 | >100.0 | 25.0 | >100.0 | 27.4 | >50.0 |
| Turgencin BMox2 (97% Mox2, 3% Mox1) | 25.0 | 25.0 | >100.0 | >100.0 | >100.0 | >50.0 | >50.0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hansen, I.K.Ø.; Isaksson, J.; Poth, A.G.; Hansen, K.Ø.; Andersen, A.J.C.; Richard, C.S.M.; Blencke, H.-M.; Stensvåg, K.; Craik, D.J.; Haug, T. Isolation and Characterization of Antimicrobial Peptides with Unusual Disulfide Connectivity from the Colonial Ascidian Synoicum turgens. Mar. Drugs 2020, 18, 51. https://doi.org/10.3390/md18010051
Hansen IKØ, Isaksson J, Poth AG, Hansen KØ, Andersen AJC, Richard CSM, Blencke H-M, Stensvåg K, Craik DJ, Haug T. Isolation and Characterization of Antimicrobial Peptides with Unusual Disulfide Connectivity from the Colonial Ascidian Synoicum turgens. Marine Drugs. 2020; 18(1):51. https://doi.org/10.3390/md18010051
Chicago/Turabian StyleHansen, Ida K. Ø., Johan Isaksson, Aaron G. Poth, Kine Ø. Hansen, Aaron J. C. Andersen, Céline S. M. Richard, Hans-Matti Blencke, Klara Stensvåg, David J. Craik, and Tor Haug. 2020. "Isolation and Characterization of Antimicrobial Peptides with Unusual Disulfide Connectivity from the Colonial Ascidian Synoicum turgens" Marine Drugs 18, no. 1: 51. https://doi.org/10.3390/md18010051
APA StyleHansen, I. K. Ø., Isaksson, J., Poth, A. G., Hansen, K. Ø., Andersen, A. J. C., Richard, C. S. M., Blencke, H. -M., Stensvåg, K., Craik, D. J., & Haug, T. (2020). Isolation and Characterization of Antimicrobial Peptides with Unusual Disulfide Connectivity from the Colonial Ascidian Synoicum turgens. Marine Drugs, 18(1), 51. https://doi.org/10.3390/md18010051