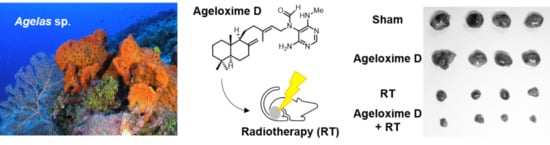
Therapeutic Potential of (−)-Agelamide D, a Diterpene Alkaloid from the Marine Sponge Agelas sp., as a Natural Radiosensitizer in Hepatocellular Carcinoma Models

 , and
, and
Abstract
:
1. Introduction
2. Results
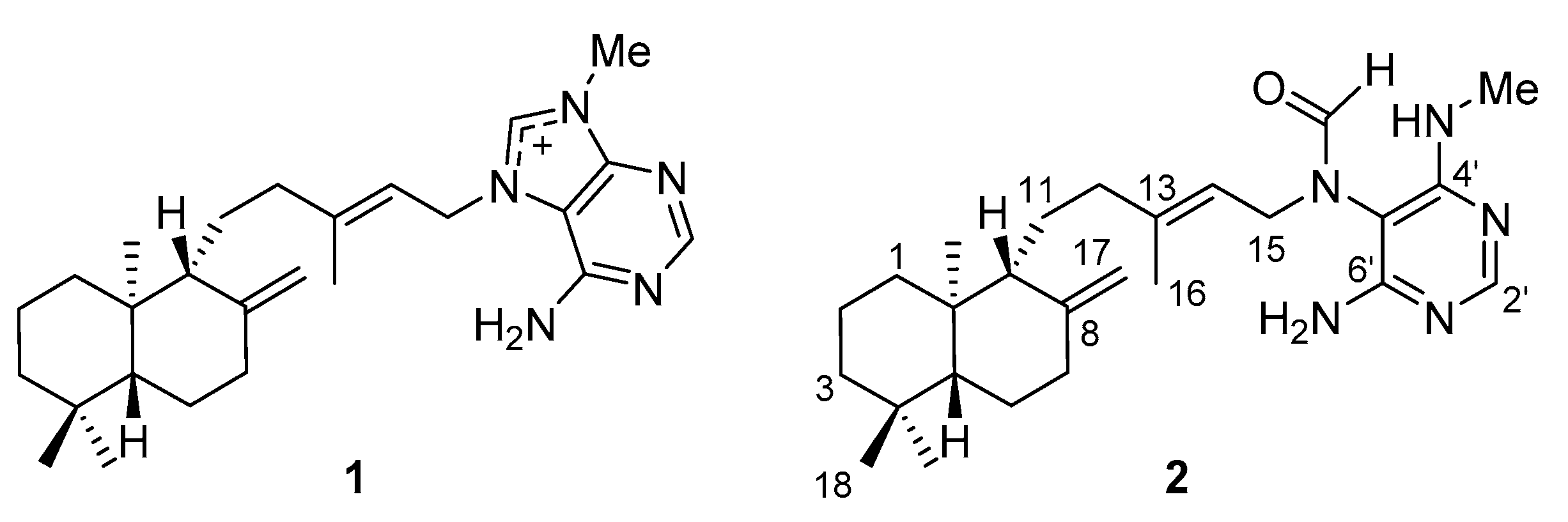
2.1. Separation and Identification of Active Ingredients
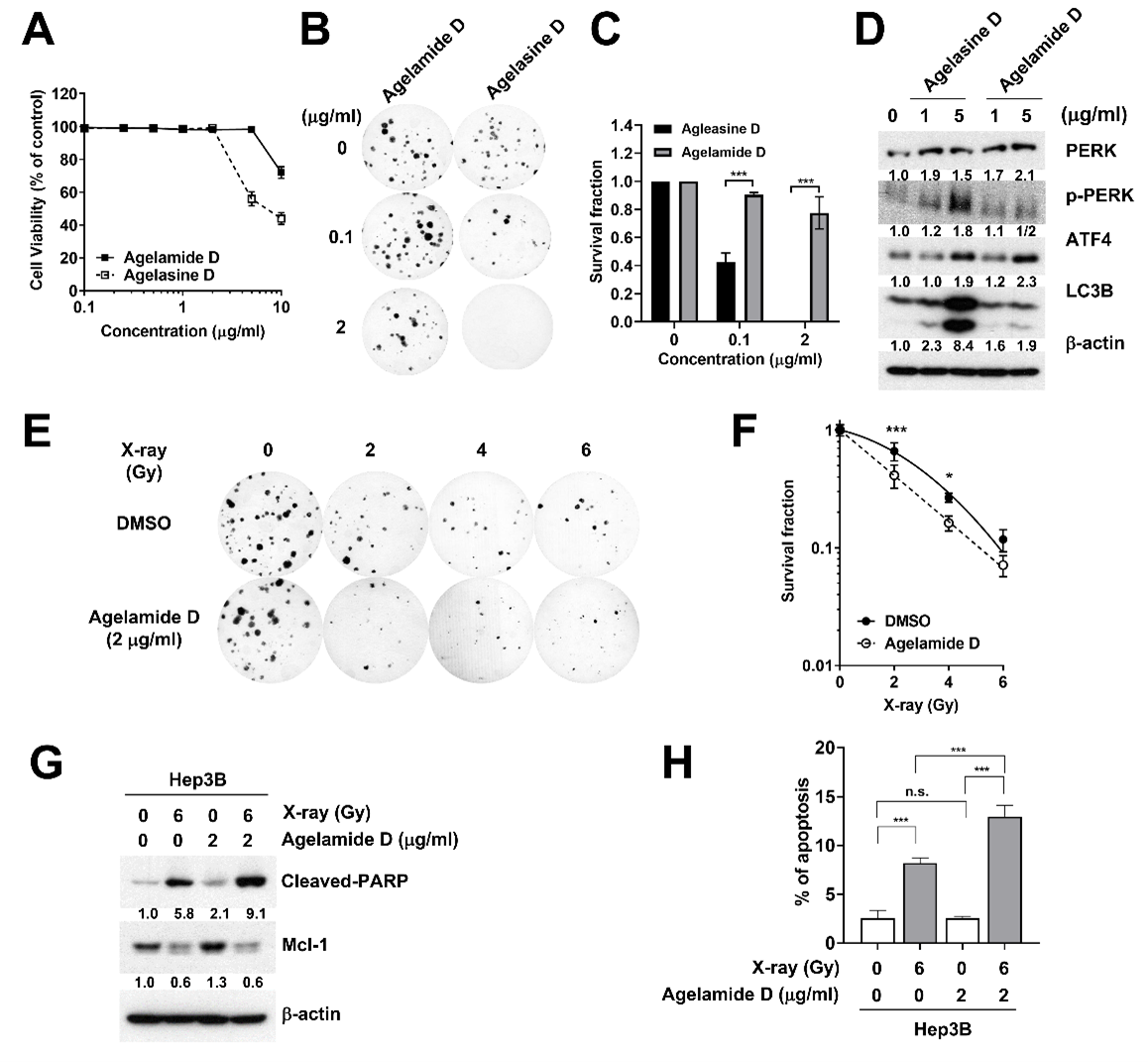
2.2. (−)-Agelamide D Exerts a Radiosensitizing Effect on Hep3B Cells
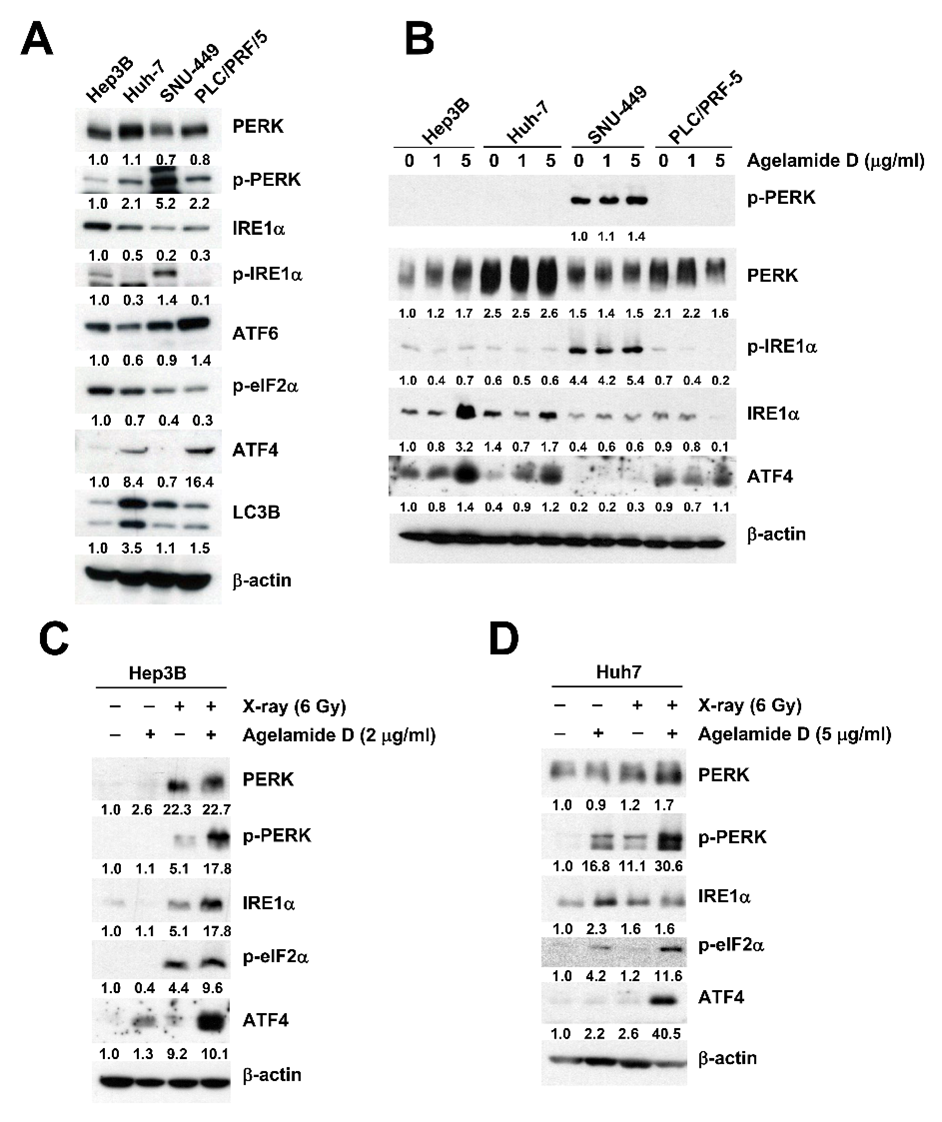
2.3. (−)-Agelamide D Augments Radiation-Induced ER Stress
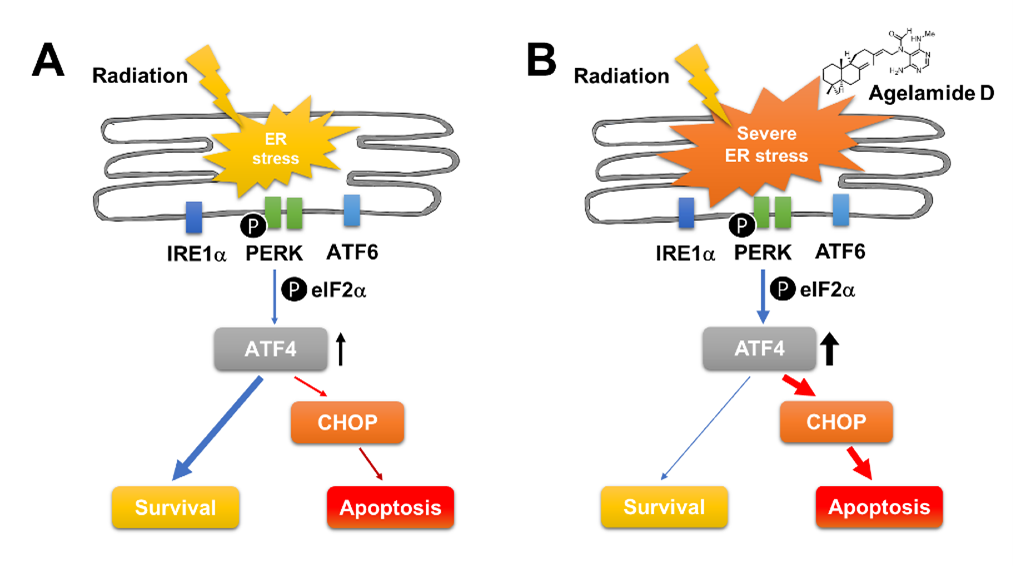
2.4. (−)-Agelamide-D-Induced Radiosensitization Mediated by the PERK/ATF4 Axis
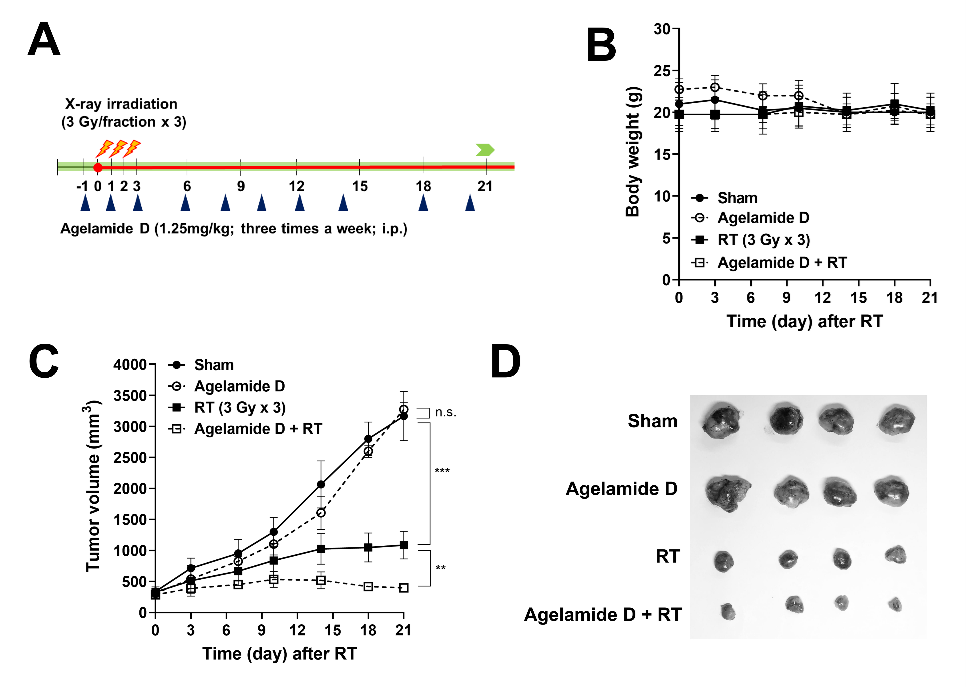
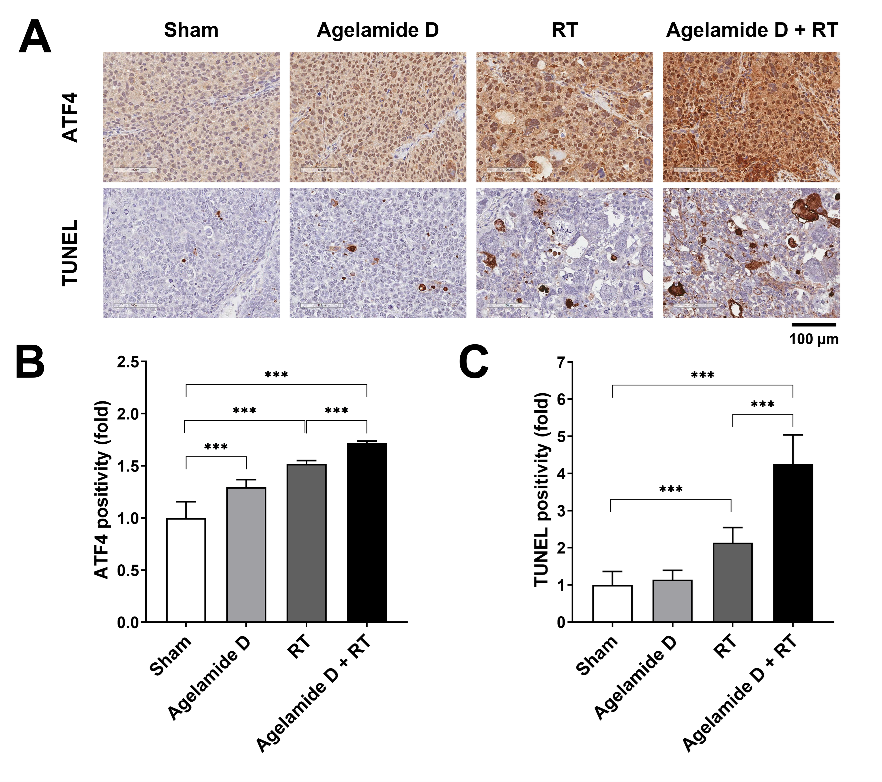
2.5. (−)-Agelamide D Enhances the Efficacy of Radiotherapy in an HCC Xenograft Mouse Model
3. Discussion
4. Materials and Methods
4.1. Isolation and Structure Elucidation of Active Compounds from Agelas sp.
4.2. Cell Proliferation Assay.
4.3. Irradiation Experiments
4.4. Clonogenic Assay
4.5. Apoptosis Assay
4.6. Western Blot Analysis
4.7. Animal Experiments
4.8. TUNEL Assay and Immunohistochemistry
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Higgins, G.S.; O’Cathail, S.M.; Muschel, R.J.; McKenna, W.G. Drug radiotherapy combinations: Review of previous failures and reasons for future optimism. Cancer Treat. Rev. 2015, 41, 105–113. [Google Scholar] [CrossRef]
- Wang, H.; Mu, X.; He, H.; Zhang, X.-D. Cancer radiosensitizers. Trends Pharmacol. Sci. 2018, 39, 24–48. [Google Scholar] [CrossRef]
- Mistry, I.N.; Thomas, M.; Calder, E.D.D.; Conway, S.J.; Hammond, E.M. Clinical advances of hypoxia-activated prodrugs in combination with radiation therapy. Int. J. Radiat. Oncol. Biol. Phys. 2017, 98, 1183–1196. [Google Scholar] [CrossRef] [Green Version]
- Abdjul, D.B.; Yamazaki, H.; Kanno, S.-I.; Takahashi, O.; Kirikoshi, R.; Ukai, K.; Namikoshi, M. Structures and biological evaluations of agelasines isolated from the Okinawan marine sponge Agelas nakamurai. J. Nat. Prod. 2015, 78, 1428–1433. [Google Scholar] [CrossRef]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Azarnia, N.; Shin, D.M.; Cohen, R.B.; Jones, C.U.; Sur, R.; Raben, D.; Jassem, J.; et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2006, 354, 567–578. [Google Scholar] [CrossRef] [Green Version]
- Choi, C.; Yoo, G.S.; Cho, W.K.; Park, H.C. Optimizing radiotherapy with immune checkpoint blockade in hepatocellular carcinoma. World J. Gastroenterol. 2019, 25, 2416–2429. [Google Scholar] [CrossRef]
- Surh, Y.-J. Cancer chemoprevention with dietary phytochemicals. Nat. Rev. Cancer 2003, 3, 768–780. [Google Scholar] [CrossRef]
- Zoberi, I.; Bradbury, C.M.; Curry, H.A.; Bisht, K.S.; Goswami, P.C.; Roti Roti, J.L.; Gius, D. Radiosensitizing and anti-proliferative effects of resveratrol in two human cervical tumor cell lines. Cancer Lett. 2002, 175, 165–173. [Google Scholar] [CrossRef]
- Papazisis, K.T.; Zambouli, D.; Kimoundri, O.T.; Papadakis, E.S.; Vala, V.; Geromichalos, G.D.; Voyatzi, S.; Markala, D.; Destouni, E.; Boutis, L.; et al. Protein tyrosine kinase inhibitor, genistein, enhances apoptosis and cell cycle arrest in K562 cells treated with γ-irradiation. Cancer Lett. 2000, 160, 107–113. [Google Scholar] [CrossRef]
- Chendil, D.; Ranga, R.S.; Meigooni, D.; Sathishkumar, S.; Ahmed, M.M. Curcumin confers radiosensitizing effect in prostate cancer cell line PC-3. Oncogene 2004, 23, 1599–1607. [Google Scholar] [CrossRef] [Green Version]
- Liang, P.-S.; Haff, R.P.; Ovchinnikova, I.; Light, D.M.; Mahoney, N.E.; Kim, J.H. Curcumin and quercetin as potential radioprotectors and/or radiosensitizers for X-ray-based sterilization of male navel orangeworm larvae. Sci. Rep. 2019, 9, 2016. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, J.; Chie, E.; DaYoung, P.; Kim, I.; Kim, I. DNMT (DNA methyltransferase) inhibitors radiosensitize human cancer cells by suppressing DNA repair activity. Radia. Oncol. 2012, 7, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labay, E.; Mauceri, H.J.; Efimova, E.V.; Flor, A.C.; Sutton, H.G.; Kron, S.J.; Weichselbaum, R.R. Repurposing cephalosporin antibiotics as pro-senescent radiosensitizers. Oncotarget 2016, 7, 33919–33933. [Google Scholar] [CrossRef] [Green Version]
- Usoltseva, R.V.; Shevchenko, N.M.; Malyarenko, O.S.; Anastyuk, S.D.; Kasprik, A.E.; Zvyagintsev, N.V.; Ermakova, S.P. Fucoidans from brown algae Laminaria longipes and Saccharina cichorioides: Structural characteristics, anticancer and radiosensitizing activity in vitro. Carbohydr. Polym. 2019, 221, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.; Son, A.; Lee, H.-S.; Lee, Y.-J.; Park, H.C. Radiosensitization by marine sponge Agelas sp. extracts in hepatocellular carcinoma cells with autophagy induction. Sci. Rep. 2018, 8, 6317. [Google Scholar] [CrossRef]
- Nakamura, H.; Wu, H.; Ohizumi, Y.; Hirata, Y. Agelasine-A, -B, -C and -D, novel bicyclic diterpenoids with a 9-methyladeninium unit possessing inhibitory effects on Na, K-atpase from the okinawa sea sponge Agelas sp. Tetrahedron Lett. 1984, 25, 2989–2992. [Google Scholar] [CrossRef]
- Hertiani, T.; Edrada-Ebel, R.; Ortlepp, S.; van Soest, R.W.M.; de Voogd, N.J.; Wray, V.; Hentschel, U.; Kozytska, S.; Mueller, W.E.G.; Proksch, P. From anti-fouling to biofilm inhibition: New cytotoxic secondary metabolites from two Indonesian Agelas sponges. Bioorg. Med. Chem. 2010, 18, 1297–1311. [Google Scholar] [CrossRef]
- Paulsen, B.; Fredriksen, K.A.; Petersen, D.; Maes, L.; Matheeussen, A.; Naemi, A.-O.; Scheie, A.A.; Simm, R.; Ma, R.; Wan, B.; et al. Synthesis and antimicrobial activities of N6-hydroxyagelasine analogs and revision of the structure of ageloximes. Bioorg. Med. Chem. 2019, 27, 620–629. [Google Scholar] [CrossRef]
- Laursen, J.S.; Engel-Andreasen, J.; Fristrup, P.; Harris, P.; Olsen, C.A. Cis—Trans amide bond rotamers in β-peptoids and peptoids: Evaluation of stereoelectronic effects in backbone and side chains. J. Am. Chem. Soc. 2013, 135, 2835–2844. [Google Scholar] [CrossRef] [Green Version]
- de Koning, C.B.; van Otterlo, W.A.L.; Michael, J.P. Amide rotamers of N-acetyl-1,3-dimethyltetrahydroisoquinolines: Synthesis, variable temperature NMR spectroscopy and molecular modelling. Tetrahedron 2003, 59, 8337–8345. [Google Scholar] [CrossRef]
- Ohtani, I.; Kusumi, T.; Kakisawa, H.; Kashman, Y.; Hirsh, S. Structure and chemical properties of ptilomycalin A. J. Am. Chem. Soc. 1992, 114, 8472–8479. [Google Scholar] [CrossRef]
- Campos, P.-E.; Wolfender, J.-L.; Queiroz, E.F.; Marcourt, L.; Al-Mourabit, A.; Frederich, M.; Bordignon, A.; De Voogd, N.; Illien, B.; Gauvin-Bialecki, A. Unguiculin A and ptilomycalins E–H, antimalarial guanidine alkaloids from the marine sponge Monanchora unguiculata. J. Nat. Prod. 2017, 80, 1404–1410. [Google Scholar] [CrossRef] [PubMed]
- García, P.A.; Valles, E.; Díez, D.; Castro, M.-Á. Marine alkylpurines: A promising group of bioactive marine natural products. Mar. Drugs 2018, 16, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vik, A.; Proszenyák, Á.; Vermeersch, M.; Cos, P.; Maes, L.; Gundersen, L.-L. Screening of agelasine D and analogs for inhibitory activity against pathogenic protozoa; Identification of hits for visceral leishmaniasis and Chagas disease. Molecules 2009, 14, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell. Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Siwecka, N.; Rozpedek, W.; Pytel, D.; Wawrzynkiewicz, A.; Dziki, A.; Dziki, L.; Diehl, J.A.; Majsterek, I. Dual role of endoplasmic reticulum stress-mediated unfolded protein response signaling pathway in carcinogenesis. Int. J. Mol. Sci. 2019, 20, 4354. [Google Scholar] [CrossRef] [Green Version]
- Riha, R.; Gupta-Saraf, P.; Bhanja, P.; Badkul, S.; Saha, S. Stressed out—Therapeutic implications of ER stress related cancer research. Oncomedicine 2017, 2, 156–167. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Han, C.; Jiang, Y.; Han, N.; Zhang, M.; Li, G.; Qiao, Q. Inhibition of GRP78 abrogates radioresistance in oropharyngeal carcinoma cells after EGFR inhibition by cetuximab. PLoS ONE 2017, 12, e0188932. [Google Scholar] [CrossRef]
- Gopal, U.; Mowery, Y.; Young, K.; Pizzo, S.V. Targeting cell surface GRP78 enhances pancreatic cancer radiosensitivity through YAP/TAZ protein signaling. J. Biol. Chem. 2019, 294, 13939–13952. [Google Scholar] [CrossRef]
- Dadey, D.Y.A.; Kapoor, V.; Hoye, K.; Khudanyan, A.; Collins, A.; Thotala, D.; Hallahan, D.E. Antibody targeting GRP78 enhances the efficacy of radiation therapy in human glioblastoma and non-small cell lung cancer cell lines and tumor models. Clin. Cancer Res. 2017, 23, 2556–2564. [Google Scholar] [CrossRef] [Green Version]
- Dadey, D.Y.; Kapoor, V.; Khudanyan, A.; Urano, F.; Kim, A.H.; Thotala, D.; Hallahan, D.E. The ATF6 pathway of the ER stress response contributes to enhanced viability in glioblastoma. Oncotarget 2016, 7, 2080–2092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.S.; Rodriguez, G.A.; Musick, A.; Walters, W.M.; de Cordoba, N.; Barbarite, E.; Marlow, M.M.; Marples, B.; Prince, J.S.; Komotar, R.J.; et al. Targeting glioblastoma stem cells with 2-deoxy-D-glucose (2-DG) potentiates radiation-induced unfolded protein response (UPR). Cancers 2019, 11, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Ji, W.; Shergalis, A.; Xu, J.; Delaney, A.M.; Calcaterra, A.; Pal, A.; Ljungman, M.; Neamati, N.; Rehemtulla, A. Activation of the unfolded protein response via inhibition of protein disulfide isomerase decreases the capacity for DNA repair to sensitize glioblastoma to radiotherapy. Cancer Res. 2019, 79, 2923–2932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oommen, D.; Prise, K.M. Down-regulation of PERK enhances resistance to ionizing radiation. Biochem. Biophys. Res. Commun. 2013, 441, 31–35. [Google Scholar] [CrossRef]
- Nagelkerke, A.; Bussink, J.; van der Kogel, A.J.; Sweep, F.C.; Span, P.N. The PERK/ATF4/LAMP3-arm of the unfolded protein response affects radioresistance by interfering with the DNA damage response. Radiother. Oncol. 2013, 108, 415–421. [Google Scholar] [CrossRef]
- Wortel, I.M.N.; van der Meer, L.T.; Kilberg, M.S.; van Leeuwen, F.N. Surviving stress: Modulation of ATF4-mediated stress responses in normal and malignant cells. Trends Endocrinol. Metab. 2017, 28, 794–806. [Google Scholar] [CrossRef]
- Lebeaupin, C.; Vallee, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic reticulum stress signaling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 927–947. [Google Scholar] [CrossRef]
- Shuda, M.; Kondoh, N.; Imazeki, N.; Tanaka, K.; Okada, T.; Mori, K.; Hada, A.; Arai, M.; Wakatsuki, T.; Matsubara, O.; et al. Activation of the ATF6, XBP1 and grp78 genes in human hepatocellular carcinoma: A possible involvement of the ER stress pathway in hepatocarcinogenesis. J. Hepatol. 2003, 38, 605–614. [Google Scholar] [CrossRef]
- Feng, Y.H.; Tung, C.L.; Su, Y.C.; Tsao, C.J.; Wu, T.F. Proteomic profile of sorafenib resistance in hepatocellular carcinoma; GRP78 expression is associated with inferior response to sorafenib. Cancer Genom. Proteom. 2019, 16, 569–576. [Google Scholar] [CrossRef] [Green Version]
- Lei, Y.; Wang, S.; Ren, B.; Wang, J.; Chen, J.; Lu, J.; Zhan, S.; Fu, Y.; Huang, L.; Tan, J. CHOP favors endoplasmic reticulum stress-induced apoptosis in hepatocellular carcinoma cells via inhibition of autophagy. PLoS ONE 2017, 12, e0183680. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Yin, J.; Zhang, C.; Liang, N.; Bai, N.; Chang, A.; Liu, Y.; Li, Z.; Tan, X.; Li, N.; et al. Activating transcription factor 4 increases chemotherapeutics resistance of human hepatocellular carcinoma. Cancer Biol. Ther. 2012, 13, 435–442. [Google Scholar] [CrossRef] [Green Version]
- Wahl, D.R.; Stenmark, M.H.; Tao, Y.; Pollom, E.L.; Caoili, E.M.; Lawrence, T.S.; Schipper, M.J.; Feng, M. Outcomes after stereotactic body radiotherapy or radiofrequency ablation for hepatocellular carcinoma. J. Clin. Oncol. 2016, 34, 452–459. [Google Scholar] [CrossRef] [Green Version]
- Choi, C.; Son, A.; Lee, G.H.; Shin, S.W.; Park, S.; Ahn, S.H.; Chung, Y.; Yu, J.I.; Park, H.C. Targeting DNA-dependent protein kinase sensitizes hepatocellular carcinoma cells to proton beam irradiation through apoptosis induction. PLoS ONE 2019, 14, e0218049. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.W.; Jung, W.; Choi, C.; Kim, S.Y.; Son, A.; Kim, H.; Lee, N.; Park, H.C. Fucoidan-manganese dioxide nanoparticles potentiate radiation therapy by co-targeting tumor hypoxia and angiogenesis. Mar. Drugs 2018, 16, 510. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δC a | δH (J in Hz) | COSY | HMBC |
|---|---|---|---|---|
| 1a | 40.3 | 0.98, m | 1.61 | - |
| 1b | 1.51, m | - | 15.1 | |
| 2a | 20.4 | 1.49, m | - | - |
| 2b | 1.61, ddd (13.3, 3.2, 3.2) | 0.98 | - | |
| 3a | 43.3 | 1.22, m | 1.40 | - |
| 3b | 1.40, brd (13.3) | 1.22 | 56.4, 40.7 | |
| 4 | 40.7 | - | - | - |
| 5 | 56.4 | 1.11, dd (12.6, 3.0) | 1.31 | 40.7, 57.7, 34.5, 15.1 |
| 6a | 25.6 | 1.31, m | 1.74, 2.37, 1.11, 1.96 | - |
| 6b | 1.74, m | - | 56.4, 149.8 | |
| 7a | 39.4 | 1.96, m | 2.37, 1.31 | 149.8, 106.9, 25.6 |
| 7b | 2.37, m | 1.96, 1.31 | 149.8, 106.9, 56.4, 25.6 | |
| 8 | 149.8 | - | - | - |
| 9 | 57.8/57.7 | 1.52, m/1.52, m | 4.81, 4.46, 4.44 | 23.0, 106.9, 149.8 |
| 10 | 34.5 | - | - | - |
| 11a | 23.0/23.1 | 1.52, m | 2.05 | - |
| 11b | 1.29, m | - | ||
| 12a | 39.6/39.7 | 2.05, m | 1.52, 4.14 | 16.0, 23.0, 118.0, 144.3 |
| 12b | 1.74, m | - | 144.4, 118.0 | |
| 13 | 144.4/144.3 | - | - | - |
| 14 | 118.4/118.0 | 5.29, t (7.8)/5.21, t (7.8) | 4.14, 2.05/4.23 | 16.0, 39.6, 46.0/16.2, 39.7 |
| 15 | 46.0/41.6 | 4.14, m/4.23, m | 5.29 5.21 | 97.3, 118.4,144.4, 165.9/99.2, 118.0,144.3, 166.5 |
| 16 | 16.0/16.2 | 1.49, s/1.56, s | - | 39.6, 118.4, 144.4/39.7, 118.0, 144.3 |
| 17a | 106.9/106.9 | 4.81, | 1.52 | 57.8, 39.4 |
| 17b | 4.46, brs/4.44, brs | 1.52/1.52 | 39.4, 57.8, 149.8 | |
| 18 | 34.1 | 0.89, s | - | 56.4, 43.3, 40.7, 22.2 |
| 19 | 22.2 | 0.82, s | - | 56.4, 43.3, 34.1 |
| 20 | 15.1 | 0.69 | - | 57.7, 40.3, |
| 2′ | 157.9/157.5 | 7.92, s/7.92, s | - | 161.6 |
| 4′ | 162.0/161.6 | - | - | - |
| 5′ | 97.3/99.2 | - | - | - |
| 6′ | 160.6/160.0 | - | - | - |
| NCHO | 165.9/166.5 | 8.20, s/7.91, s | - | 97.3, 46.0 /99.2, 41.6 |
| NMe | 28.2 | 2.88, s | - | 161.6 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, C.; Cho, Y.; Son, A.; Shin, S.-W.; Lee, Y.-J.; Park, H.C. Therapeutic Potential of (−)-Agelamide D, a Diterpene Alkaloid from the Marine Sponge Agelas sp., as a Natural Radiosensitizer in Hepatocellular Carcinoma Models. Mar. Drugs 2020, 18, 500. https://doi.org/10.3390/md18100500
Choi C, Cho Y, Son A, Shin S-W, Lee Y-J, Park HC. Therapeutic Potential of (−)-Agelamide D, a Diterpene Alkaloid from the Marine Sponge Agelas sp., as a Natural Radiosensitizer in Hepatocellular Carcinoma Models. Marine Drugs. 2020; 18(10):500. https://doi.org/10.3390/md18100500
Chicago/Turabian StyleChoi, Changhoon, Yeonwoo Cho, Arang Son, Sung-Won Shin, Yeon-Ju Lee, and Hee Chul Park. 2020. "Therapeutic Potential of (−)-Agelamide D, a Diterpene Alkaloid from the Marine Sponge Agelas sp., as a Natural Radiosensitizer in Hepatocellular Carcinoma Models" Marine Drugs 18, no. 10: 500. https://doi.org/10.3390/md18100500
APA StyleChoi, C., Cho, Y., Son, A., Shin, S.-W., Lee, Y.-J., & Park, H. C. (2020). Therapeutic Potential of (−)-Agelamide D, a Diterpene Alkaloid from the Marine Sponge Agelas sp., as a Natural Radiosensitizer in Hepatocellular Carcinoma Models. Marine Drugs, 18(10), 500. https://doi.org/10.3390/md18100500