Pharmacokinetics of Marine-Derived Drugs

 , and
, and
Abstract
:1. Introduction
2. Analytical Methods Used in Pharmacokinetic Studies
3. Pharmacokinetics Studies in Animals
3.1. Absorption
3.1.1. Algal-Derived Compounds
3.1.2. Crustacean-Derived Compounds
3.1.3. Sea Cucumber-Derived Compounds
3.1.4. Sea Fungus-Derived Compounds
3.1.5. Sea Urchin-Derived Compounds
3.1.6. Marine Compounds Derived from Other Species
3.2. Distribution
3.2.1. Algal-Derived Compounds
3.2.2. Crustacean-Derived Compounds
3.2.3. Sea Cucumber-Derived Compounds
3.2.4. Sea Fungus-Derived Compounds
3.2.5. Marine Sponge-Derived Compounds
3.2.6. Sea Urchin-Derived Compounds
3.2.7. Marine Compounds Derived from Other Species
3.3. Metabolism
3.3.1. Algal-Derived Compounds
3.3.2. Crustacean-Derived Compounds
3.3.3. Sea Cucumber-Derived Compounds
3.3.4. Sea Fungus-Derived Compounds
3.3.5. Marine Sponge-Derived Compounds
3.3.6. Sea Urchin-Derived Compounds
3.3.7. Marine Compounds Derived from Other Species
3.4. Elimination
3.4.1. Algal-Derived Compounds
3.4.2. Crustacean-Derived Compounds
3.4.3. Sea Cucumber-Derived Compounds
3.4.4. Sea Fungus-Derived Compounds
3.4.5. Marine Sponge-Derived Compounds
3.4.6. Sea Urchin-Derived Compounds
3.4.7. Marine Compounds Derived from Other Species
4. Pharmacokinetics Studies in Humans
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Da Nóbrega Alves, R.R. Use of marine turtles in zootherapy in Northeast Brazil. Mar. Turt. Newsl. 2006, 112, 16–17. [Google Scholar]
- Gopal, R.; Vijayakumaran, M.; Venkatesan, R.; Kathiroli, S. Marine organisms in Indian medicine and their future prospects. Nat. Prod. Radiance 2008, 7, 139–145. [Google Scholar]
- Chatterji, A.; Kassim, Z.; Hassan, A.; Therwath, A.; Shaharom, F. Marine living resources in the practice of traditional medicine. J. Coast. Environ. 2010, 1, 41–52. [Google Scholar]
- Bordbar, S.; Anwar, F.; Saari, N. High-Value components and bioactives from sea cucumbers for functional foods—A review. Mar. Drugs 2011, 9, 1761–1805. [Google Scholar] [CrossRef] [Green Version]
- Fu, X.-M.; Zhang, M.-Q.; Shao, C.-L.; Li, G.-Q.; Bai, H.; Dai, G.-L.; Chen, Q.-W.; Kong, W.; Fu, X.-J.; Wang, C.-Y. Chinese Marine Materia Medica resources: Status and potential. Mar. Drugs 2016, 14, 46. [Google Scholar] [CrossRef] [Green Version]
- Khotimchenko, Y. Pharmacological potential of sea cucumbers. Int. J. Mol. Sci. 2018, 19, 1342. [Google Scholar]
- The British Pharmacopoeia; General Medical Council: London, UK, 2014.
- European Pharmacopoeia, 10th ed.; Council of Europe: Strasbourg, France, 2020.
- Prokopov, I.A.; Kovaleva, E.L.; Minaeva, E.D.; Pryakhina, E.A.; Savin, E.V.; Gamayunova, A.V.; Pozharitskaya, O.N.; Makarov, V.G.; Shikov, A.N. Animal-derived medicinal products in Russia: Current nomenclature and specific aspects of quality control. J. Ethnopharmacol. 2019, 240, 111933. [Google Scholar] [CrossRef]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2016, 33, 382–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2017, 34, 235–294. [Google Scholar] [CrossRef] [Green Version]
- Blunt, J.W.; Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2018, 35, 8–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2019, 36, 122–173. [Google Scholar] [CrossRef] [Green Version]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine Natural Products. Nat. Prod. Rep. 2020, 37, 175–223. [Google Scholar] [CrossRef] [PubMed]
- Proksch, P.; Putz, A.; Ortlepp, S.; Kjer, J.; Bayer, M. Bioactive natural products from marine sponges and fungal endophytes. Phytochem. Rev. 2010, 9, 475–489. [Google Scholar]
- Fedoreyev, S.A.; Krylova, N.V.; Mishchenko, N.P.; Vasileva, E.A.; Pislyagin, E.A.; Iunikhina, O.V.; Lavrov, V.F.; Svitich, O.A.; Ebralidze, L.K.; Leonova, G.N. Antiviral and antioxidant properties of echinochrome A. Mar. Drugs 2018, 16, 509. [Google Scholar] [CrossRef] [Green Version]
- Shikov, A.N.; Pozharitskaya, O.N.; Krishtopina, A.S.; Makarov, V.G. Naphthoquinone pigments from sea urchins: Chemistry and pharmacology. Phytochem. Rev. 2018, 17, 509–534. [Google Scholar] [CrossRef]
- Katanaev, V.L.; Di Falco, S.; Khotimchenko, Y. The anticancer drug discovery potential of marine invertebrates from Russian Pacific. Mar. Drugs 2019, 17, 474. [Google Scholar] [CrossRef] [Green Version]
- Cao, Q.; Zhao, J.; Xing, M.; Xiao, H.; Zhang, Q.; Liang, H.; Ji, A.; Song, S. Current research landscape of marine-derived anti-atherosclerotic substances. Mar. Drugs 2020, 18, 440. [Google Scholar] [CrossRef]
- Kuznetsova, T.A.; Smolina, T.P.; Makarenkova, I.D.; Ivanushko, L.A.; Persiyanova, E.V.; Ermakova, S.P.; Silchenko, A.S.; Zaporozhets, T.S.; Besednova, N.N.; Fedyanina, L.N.; et al. Immunoadjuvant Activity of Fucoidans from the Brown Alga Fucus evanescens. Mar. Drugs 2020, 18, 155. [Google Scholar] [CrossRef] [Green Version]
- Landon, R.; Gueguen, V.; Petite, H.; Letourneur, D.; Pavon-Djavid, G.; Anagnostou, F. Impact of astaxanthin on diabetes pathogenesis and chronic complications. Mar. Drugs 2020, 18, 357. [Google Scholar] [CrossRef]
- Mayer, A.M.S.; Guerrero, A.J.; Rodríguez, A.D.; Taglialatela-Scafati, O.; Nakamura, F.; Fusetani, N. Marine Pharmacology in 2014–2015: Marine compounds with antibacterial, antidiabetic, antifungal, anti-inflammatory, antiprotozoal, antituberculosis, antiviral, and anthelmintic activities; affecting the immune and nervous systems, and other miscellaneous mechanisms of action. Mar. Drugs 2020, 18, 5. [Google Scholar] [CrossRef] [Green Version]
- Bian, C.; Wang, J.; Zhou, X.; Wu, W.; Guo, R. Recent advances on marine alkaloids from sponges. Chem. Biodivers. 2020, 17, e2000186. [Google Scholar] [CrossRef] [PubMed]
- Hochhaus, G.; Barrett, J.S.; Derendorf, H. Evolution of pharmacokinetics and pharmacokinetic/dynamic correlations during the 20th century. J. Clin. Pharmacol. 2000, 40, 908–917. [Google Scholar] [CrossRef] [PubMed]
- Talevi, A.; Quiroga, P.A. (Eds.) ADME Processes in Pharmaceutical Sciences: Dosage, Design, and Pharmacotherapy Success, 1st ed.; Springer International Publishing: Cham, Switzerland, 2018; 362p. [Google Scholar] [CrossRef]
- Kerns, E.H.; Li, D. Drug-Like Properties: Concept, Structure Design and Methods; Elsevier: San Diego, CA, USA, 2008; 552p. [Google Scholar]
- Pozharitskaya, O.N.; Shikov, A.N.; Faustova, N.M.; Obluchinskaya, E.D.; Kosman, V.M.; Vuorela, H.; Makarov, V.G. Pharmacokinetic and tissue distribution of fucoidan from Fucus vesiculosus after oral administration to rats. Mar. Drugs 2018, 16, 132. [Google Scholar] [CrossRef] [Green Version]
- Pozharitskaya, O.N.; Shikov, A.N.; Obluchinskaya, E.D.; Vuorela, H. The Pharmacokinetics of fucoidan after topical application to rats. Mar. Drugs 2019, 17, 687. [Google Scholar] [CrossRef] [Green Version]
- Kosman, V.M.; Faustova, N.M.; Urakova, I.N.; Karlina, M.N.; Makarov, V.G. Dypeptydylpeptidase IV activity ingibition after oral administration to rabbits of Strongylocentrotus droebahiensis gonads extract as possible biomarker of pharmakokinetics. Drug Dev. Regist. 2020, 9, 158–165. [Google Scholar] [CrossRef]
- Shikov, A.N.; Pozharitskaya, O.N.; Faustova, N.M.; Kosman, V.M.; Makarov, V.G.; Razzazi-Fazeli, E.; Novak, J. Pharmacokinetic study of bioactive glycopeptide from Strongylocentrotus droebachiensis after intranasal administration to rats using biomarker approach. Mar. Drugs 2019, 17, 577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irhimeh, M.R.; Fitton, J.H.; Lowenthal, R.M.; Kongtawelert, P. A quantitative method to detect fucoidan in human plasma using a novel antibody. Methods Find Exp. Clin. Pharmacol. 2005, 27, 705–710. [Google Scholar] [CrossRef]
- Bai, X.; Zhang, E.; Hu, B.; Liang, H.; Song, S.; Ji, A. Study on absorption mechanism and tissue distribution of fucoidan. Molecules 2020, 25, 1087. [Google Scholar] [CrossRef] [Green Version]
- Egorin, M.; Sentz, D.; Rosen, D.; Ballesteros, M.F.; Kearns, C.M.; Callery, P.S.; Eiseman, J.L. Plasma pharmacokinetics, bioavailability, and tissue distribution in CD2F1 mice of halomon, an antitumor halogenated monoterpene isolated from the red algae Portieria hornemannii. Cancer Chemother. Pharmacol. 1996, 39, 51–60. [Google Scholar] [CrossRef]
- Zhao, X.; Guo, F.; Hu, J.; Zhang, L.; Xue, C.; Zhang, Z.; Li, B. Antithrombotic activity of oral administered low molecular weight fucoidan from Laminaria japonica. Thromb. Res. 2016, 144, 46–52. [Google Scholar] [CrossRef]
- Zhang, E.; Chu, F.; Zhao, T.; Chai, Y.; Liang, H.; Song, S.; Ji, A. Determination of fucoidan in rat plasma by HPLC and its application in pharmacokinetics. Pak. J. Pharm. Sci. 2020, 33, 1–9. [Google Scholar]
- Zhang, W.; Sun, D.; Zhao, X.; Jin, W.; Wang, J.; Zhang, Q. Microanalysis and preliminary pharmacokinetic studies of a sulfated polysaccharide from Laminaria japonica. Chin. J. Oceanolog. Limnolog. 2016, 34, 177–185. [Google Scholar] [CrossRef]
- Su, T.W.; Wu, W.H.; Yan, T.; Zhang, C.Y.; Zhu, Q.G.; Bao, B. Pharmacokinetics and tissue distribution of a novel marine fibrinolytic compound in Wistar rat following intravenous administrations. J. Chromatogr. B 2013, 942–943, 77–82. [Google Scholar] [CrossRef]
- Li, S.; Wang, Y.; Jiang, T.; Wang, H.; Yang, S.; Lv, Z. Absorption and transport of sea cucumber saponins from Apostichopus japonicus. Mar. Drugs 2016, 14, 114. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Cong, P.; Xu, J.; Li, G.; Liu, X.; Li, Z.; Xue, C.; Xue, Y.; Wang, Y. Absorption and pharmacokinetic study of two sulphated triterpenoid saponins in rat after oral and intravenous administration of saponin extracts of Pearsonothuria graeffei by HPLC-MS. J. Funct. Foods 2016, 25, 62–69. [Google Scholar] [CrossRef]
- Ma, Z.; Guo, R.; Elango, J.; Bao, B.; Wu, W. Evaluation of marine diindolinonepyrane in vitro and in vivo: Permeability characterization in Caco-2 cells monolayer and pharmacokinetic properties in beagle dogs. Mar. Drugs 2019, 17, 651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Shemaili, J.; Parekh, K.A.; Newman, R.A.; Hellman, B.; Woodward, C.; Adem, A.; Collin, P.; Adrian, T.E. Pharmacokinetics in mouse and comparative effects of frondosides in pancreatic cancer. Mar. Drugs 2016, 14, 115. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wu, H.; Wen, H.; Fang, H.; Hong, Z.; Yi, R.; Liu, R. Simultaneous determination of fucoxanthin and its deacetylated metabolite fucoxanthinol in rat plasma by liquid chromatography-tandem mass spectrometry. Mar. Drugs 2015, 13, 6521–6536. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Yokose, T.; Yamamoto, Y.; Yamaguchi, K.; Oda, T. Detection and pharmacokinetics of alginate oligosaccharides in mouse plasma and urine after oral administration by a liquid chromatography/tandem mass spectrometry (LC-MS/MS) method. Biosci. Biotechnol. Biochem. 2008, 72, 2184–2190. [Google Scholar] [CrossRef] [Green Version]
- Celli, N.; Gallardo, A.M.; Rossi, C.; Zucchetti, M.; D’Incalci, M.; Rotilio, D. Analysis of aplidine (dehydrodidemnin B), a new marine-derived depsipeptide, in rat biological fluids by liquid chromatography–tandem mass spectrometry. J. Chromatogr. B Biomed. Sci. Appl. 1999, 731, 335–343. [Google Scholar] [CrossRef]
- Tian, W.; Yang, L.; Wu, D.; Deng, Z.; Hong, K. Toxicity, pharmacokinetics, and gut microbiome of oral administration of sesterterpene MHO7 derived from a marine fungus. Mar. Drugs 2019, 17, 667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, H.; Noh, K.; Park, I.; Na, M.; Oh, S.; Shin, B.S.; Kang, W. Stereo-selective pharmacokinetics of ilimaquinone epimers extracted from a marine sponge in rats. Mar. Drugs 2019, 17, 171. [Google Scholar] [CrossRef] [Green Version]
- Pislyagin, E.A.; Dmitrenok, P.S.; Gorpenchenko, T.Y.; Avilov, S.A.; Silchenko, A.S.; Aminin, D.L. Determination of cucumarioside A2-2 in mouse spleen by radiospectroscopy, MALDI-MS and MALDI-IMS. Eur. J. Pharm. Sci. 2013, 49, 461–467. [Google Scholar] [CrossRef]
- Dorr, F.A.; Kuhn, J.G.; Phillips, J.; von Hoff, D.D. Phase I clinical and pharmacokinetic investigation of didemnin B, a cycli depsipeptide. Eur. J. Cancer Clin. Oncol. 1988, 24, 1699–1706. [Google Scholar] [CrossRef]
- Tokita, Y.; Nakajima, K.; Mochida, H.; Iha, M.; Nagamine, T. Development of a fucoidan-specific antibody and measurement of fucoidan in serum and urine by sandwich ELISA. Biosci. Biotechnol. Biochem. 2010, 74, 350–357. [Google Scholar] [CrossRef]
- Cotas, J.; Leandro, A.; Monteiro, P.; Pacheco, D.; Figueirinha, A.; Gonçalves, A.M.; da Silva, G.J.; Pereira, L. Seaweed phenolics: From extraction to applications. Mar. Drugs 2020, 18, 384. [Google Scholar] [CrossRef] [PubMed]
- Barton, C.; Kouokam, J.C.; Hurst, H.; Palmer, K.E. Pharmacokinetics of the antiviral lectin griffithsin administered by different routes indicates multiple potential uses. Viruses 2016, 8, 331. [Google Scholar] [CrossRef]
- Choi, H.D.; Kang, H.E.; Yang, S.H.; Lee, M.G.; Shin, W.G. Pharmacokinetics and first-pass metabolism of astaxanthin in rats. Br. J. Nutr. 2010, 105, 220–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chae, S.Y.; Jang, M.K.; Nah, J.W. Influence of molecular weight on oral absorption of water soluble chitosans. J. Control. Release 2005, 102, 383–394. [Google Scholar] [CrossRef]
- Zeng, L.; Qin, C.; Wang, W.; Chi, W.; Li, W. Absorption and distribution of chitosan in mice after oral administration. Carbohydr. Polym. 2008, 71, 435–440. [Google Scholar] [CrossRef]
- Aminin, D.L. Molecular Mechanisms of the Immunomodulatory Action of Cucumarioside A2-2 and the Drug Coumazid Created on Its Basis; Institute of Chemical Biology and Fundamental Medicine SB RAS: Novosibirsk/Vladivostok, Russia, 2018. [Google Scholar]
- Yousaf, M.; Hammond, N.L.; Peng, J.; Wahyuono, S.; McIntosh, K.A.; Charman, W.N.; Mayer, A.M.S.; Hamann, M.T. New manzamine alkaloids from an Indo-Pacific sponge. Pharmacokinetics, oral availability, and the significant activity of several manzamines against HIV-I, AIDS opportunistic infections, and inflammatory diseases. J. Med. Chem. 2004, 47, 3512–3517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faircloth, G.; del Marchante, M.C.C. Kahalalide F and ES285: Potent anticancer agents from marine molluscs. In Molluscs. Progress in Molecular and Subcellular Biology; Cimino, G., Gavagnin, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; Volume 43, pp. 363–379. [Google Scholar] [CrossRef]
- Newman, R.A.; Fuentes, A.; Covey, J.M.; Benvenuto, J.A. Preclinical pharmacology of the natural marine product dolastatin 10 (NSC 376128). Drug Metab. Dispos. 1994, 22, 428–432. [Google Scholar]
- Aherne, G.W.; Hardcastle, A.; Valenti, M.; Bryant, A.; Rogers, P.; Pettit, G.R.; Srirangam, J.K.; Kelland, L.R. Antitumour evaluation of dolastatins 10 and 15 and their measurement in plasma by radioimmunoassay. Cancer Chemother. Pharmacol. 1996, 38, 225–232. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, R.; Zhao, H.; Cai, H.; Gush, K.A.; Kerr, R.G.; Pettit, G.R.; Kraft, A.S. Preclinical pharmacology of the natural product anticancer agent bryostatin 1, an activator of protein kinase C. Cancer Res. 1996, 56, 802–808. [Google Scholar]
- Wang, J.; Liu, H.; Li, N.; Zhang, Q.; Zhang, H. The protective effect of fucoidan in rats with streptozotocin-induced diabetic nephropathy. Mar. Drugs 2014, 12, 3292–3306. [Google Scholar] [CrossRef] [PubMed]
- Fitton, J.H.; Stringer, D.N.; Park, A.Y.; Karpiniec, S.S. Therapies from Fucoidan: New Developments. Mar. Drugs 2019, 17, 571. [Google Scholar] [CrossRef] [Green Version]
- Van Weelden, G.; Bobiński, M.; Okła, K.; Van Weelden, W.J.; Romano, A.; Pijnenborg, J.M.A. Fucoidan structure and activity in relation to anti-cancer mechanisms. Mar. Drugs 2019, 17, 32. [Google Scholar] [CrossRef] [Green Version]
- Krylova, N.V.; Ermakova, S.P.; Lavrov, V.F.; Leneva, I.A.; Kompanets, G.G.; Iunikhina, O.V.; Nosik, M.N.; Ebralidze, L.K.; Falynskova, I.N.; Silchenko, A.S.; et al. The comparative analysis of antiviral activity of native and modified fucoidans from brown algae Fucus evanescens in vitro and in vivo. Mar. Drugs 2020, 18, 224. [Google Scholar] [CrossRef] [Green Version]
- Pozharitskaya, O.N.; Obluchinskaya, E.D.; Shikov, A.N. Mechanisms of Bioactivities of Fucoidan from the Brown Seaweed Fucus vesiculosus L. of the Barents Sea. Mar. Drugs 2020, 18, 275. [Google Scholar] [CrossRef]
- Rosa, G.P.; Tavares, W.R.; Sousa, P.; Seca, A.M.; Pinto, D.C. Seaweed secondary metabolites with beneficial health effects: An overview of successes in in vivo studies and clinical trials. Mar. Drugs 2020, 18, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, T.; O’Keefe, B.R.; Sowder, R.C., 2nd; Bringans, S.; Gardella, R.; Berg, S.; Cochran, P.; Turpin, J.A.; Buckheit, R.W., Jr.; McMahon, J.B.; et al. Isolation and characterization of griffithsin, a novel HIV-inactivatingprotein, from the red alga Griffithsia sp. J. Biol. Chem. 2005, 280, 9345–9935. [Google Scholar] [CrossRef] [Green Version]
- Barton, C.; Kouokam, J.C.; Lasnik, A.B.; Foreman, O.; Cambon, A.; Brock, G.; Montefiori, D.C.; Vojdani, F.; McCormick, A.A.; O’Keefe, B.R.; et al. Activity of and effect of subcutaneous treatment with the broad-spectrum antiviral lectin griffithsin in two laboratory rodent models. Antimicrob. Agents Chemother. 2014, 58, 120–127. [Google Scholar] [CrossRef] [Green Version]
- Lee, C. Griffithsin, a highly potent broad-spectrum antiviral lectin from red algae: From discovery to clinical application. Mar. Drugs 2019, 17, 567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szekalska, M.; Puciłowska, A.; Szymańska, E.; Ciosek, P.; Winnicka, K. Alginate: Current use and future perspectives in pharmaceutical and biomedical applications. Int. J. Polym. Sci. 2016, 2016, 7697031. [Google Scholar] [CrossRef] [Green Version]
- Krishnaswami, V.; Kandasamy, R.; Alagarsamy, S.; Palanisamy, R.; Natesan, S. Biological macromolecules for ophthalmic drug delivery to treat ocular diseases. Int. J. Biol. Macromol. 2018, 110, 7–16. [Google Scholar] [CrossRef]
- Uyen, N.T.T.; Hamid, Z.A.A.; Tram, N.X.T.; Ahmad, N. Fabrication of alginate microspheres for drug delivery: A review. Int. J. Biol. Macromol. 2020, 153, 1035–1046. [Google Scholar] [CrossRef]
- Hagen, A.; Skjak-Braek, G.; Dornish, M. Pharmacokinetics of sodium alginate in mice. Eur. J. Pharm. Sci. 1996, 4 (Suppl. 1), S100. [Google Scholar] [CrossRef]
- Fuller, R.W.; Cardellina, J.H.; Kato, Y.; Brinen, L.S.; Clardy, J.; Snader, K.M.; Boyd, M.R. A Pentahalogenated monoterpene from the red alga Portieria hornemannii produces a novel cytotoxicity profile against a diverse panel of human tumor cell lines. J. Med. Chem. 1992, 35, 3007–3011. [Google Scholar] [CrossRef]
- Egorin, M.J.; Rosen, D.M.; Benjamin, S.E.; Callery, P.S.; Sentz, D.L.; Eiseman, J.L. In vitro metabolism by mouse and human liver preparations of halomon, an antitumor halogenated monoterpene. Cancer Chemother. Pharmacol. 1997, 41, 9–14. [Google Scholar] [CrossRef]
- Manandhar, B.; Paudel, P.; Seong, S.H.; Jung, H.A.; Choi, J.S. characterizing eckol as a therapeutic aid: A systematic review. Mar. Drugs 2019, 17, 361. [Google Scholar] [CrossRef] [Green Version]
- Paudel, P.; Seong, S.H.; Wu, S.; Park, S.; Jung, H.A.; Choi, J.S. Eckol as a potential therapeutic against neurodegenerative diseases targeting dopamine D3/D4 receptors. Mar. Drugs 2019, 17, 108. [Google Scholar] [CrossRef] [Green Version]
- Barkia, I.; Saari, N.; Manning, S.R. Microalgae for high-value products towards human health and nutrition. Mar. Drugs 2019, 17, 304. [Google Scholar] [CrossRef] [Green Version]
- Bae, M.; Kim, M.B.; Park, Y.K.; Lee, J.Y. Health benefits of fucoxanthin in the prevention of chronic diseases. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158618. [Google Scholar] [CrossRef]
- Hashimoto, T.; Ozaki, Y.; Taminato, M.; Das, S.K.; Mizuno, M.; Yoshimura, K.; Maoka, T.; Kanazawa, K. The distribution and accumulation of fucoxanthin and its metabolites after oral administration in mice. Br. J. Nutr. 2009, 102, 242–248. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.P.; Peng, J.; Yin, K.; Wang, J.H. Potential health-promoting effects of astaxanthin: A high-value carotenoid mostly from microalgae. Mol. Nutr. Food Res. 2011, 55, 150–165. [Google Scholar] [CrossRef]
- Pereira, D.M.; Valentão, P.; Andrade, P.B. Marine natural pigments: Chemistry, distribution and analysis. Dyes Pigm. 2014, 111, 124–134. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, H. Multiple mechanisms of anti-cancer effects exerted by astaxanthin. Mar. Drugs 2015, 13, 4310–4330. [Google Scholar] [CrossRef] [Green Version]
- Sorrenti, V.; Davinelli, S.; Scapagnini, G.; Willcox, B.J.; Allsopp, R.C.; Willcox, D.C. Astaxanthin as a putative geroprotector: Molecular basis and focus on brain aging. Mar. Drugs 2020, 18, 351. [Google Scholar] [CrossRef] [PubMed]
- Galasso, C.; Orefice, I.; Pellone, P.; Cirino, P.; Miele, R.; Ianora, A.; Brunet, C.; Sansone, C. On the neuroprotective role of astaxanthin: New perspectives? Mar. Drugs 2018, 16, 247. [Google Scholar] [CrossRef] [Green Version]
- Giannaccare, G.; Pellegrini, M.; Senni, C.; Bernabei, F.; Scorcia, V.; Cicero, A.F.G. Clinical applications of astaxanthin in the treatment of ocular diseases: Emerging insights. Mar. Drugs 2020, 18, 239. [Google Scholar] [CrossRef] [PubMed]
- Thanou, M.; Verhoef, J.C.; Junginger, H.E. Chitosan and its derivatives as intestinal absorption enhancers. Adv. Drug Deliv. Rev. 2001, 50, S91–S101. [Google Scholar] [CrossRef]
- D’Ayala, G.G.; Malinconico, M.; Laurienzo, P. Marine Derived Polysaccharides for Biomedical Applications: Chemical Modification Approaches. Molecules 2008, 13, 2069–2106. [Google Scholar] [CrossRef] [Green Version]
- Barbosa, A.I.; Coutinho, A.J.; Costa Lima, S.A.; Reis, S. Marine polysaccharides in pharmaceutical applications: Fucoidan and chitosan as key players in the drug delivery match field. Mar. Drugs 2019, 17, 654. [Google Scholar] [CrossRef] [Green Version]
- Hao, C.; Wang, W.; Wang, S.; Zhang, L.; Guo, Y. An Overview of the protective effects of chitosan and acetylated chitosan oligosaccharides against neuronal disorders. Mar. Drugs 2017, 15, 89. [Google Scholar] [CrossRef]
- Moraru, C.; Mincea, M.M.; Frandes, M.; Timar, B.; Ostafe, V. A Meta-Analysis on randomised controlled clinical trials evaluating the effect of the dietary supplement chitosan on weight loss, lipid parameters and blood pressure. Medicina 2018, 54, 109. [Google Scholar] [CrossRef] [Green Version]
- Chang, A.K.T.; Frias, R.R., Jr.; Alvarez, L.V.; Bigol, U.G.; Guzman, J.P.M.D. Comparative antibacterial activity of commercial chitosan and chitosan extracted from Auricularia sp. Biocatal. Agric. Biotechnol. 2019, 17, 189–195. [Google Scholar] [CrossRef]
- Richardson, S.W.; Kolbe, H.J.; Duncan, R. Potential of low molecular mass chitosan as a DNA delivery system: Biocompatibility, body distribution and ability to complex and protect DNA. Int. J. Pharm. 1999, 178, 231–243. [Google Scholar] [CrossRef]
- Pangestuti, R.; Arifin, Z. Medicinal and health benefit effects of functional sea cucumbers. J. Tradit. Complement. Med. 2018, 8, 341–351. [Google Scholar] [CrossRef]
- Zhao, Q.; Xue, Y.; Wang, J.F.; Li, H.; Long, T.T.; Li, Z.; Wang, Y.M.; Dong, P.; Xue, C.H. In vitro and in vivo anti-tumour activities of echinoside A and ds-echinoside A from Pearsonothuria graeffei. J. Sci. Food Agric. 2012, 92, 965–974. [Google Scholar] [CrossRef]
- Aminin, D.L.; Menchinskaya, E.S.; Pisliagin, E.A.; Silchenko, A.S.; Avilov, S.A.; Kalinin, V.I. Anticancer activity of sea cucumber triterpene glycosides. Mar. Drugs 2015, 13, 1202–1223. [Google Scholar] [CrossRef]
- Yun, S.-H.; Sim, E.-H.; Han, S.-H.; Han, J.-Y.; Kim, S.-H.; Silchenko, A.S.; Stonik, V.A.; Park, J.-I. Holotoxin A1 induces apoptosis by activating acid sphingomyelinase and neutral sphingomyelinase in K562 and human primary leukemia cells. Mar. Drugs 2018, 16, 123. [Google Scholar] [CrossRef] [Green Version]
- Malyarenko, O.S.; Ivanushko, L.A.; Chaikina, E.L.; Kusaykin, M.I.; Silchenko, A.S.; Avilov, S.A.; Kalinin, V.I.; Ermakova, S.P. In Vitro and in vivo effects of holotoxin a1 from the Sea Cucumber Apostichopus japonicus during ionizing radiation. Nat. Prod. Commun. 2020, 15, 1934578X20932033. [Google Scholar] [CrossRef]
- Li, X.; Roginsky, A.B.; Ding, X.Z.; Woodward, C.; Collin, P.; Newman, R.A.; Bell, R.H., Jr.; Adrian, T.E. Review of the apoptosis pathways in pancreatic cancer and the anti-apoptotic effects of the novel sea cucumber compound, Frondoside, A. Ann. N. Y. Acad. Sci. 2008, 1138, 181–198. [Google Scholar] [CrossRef]
- Jin, J.O.; Shastina, V.V.; Shin, S.W.; Xu, Q.; Park, J.I.; Rasskazov, V.A.; Avilov, S.A.; Fedorov, S.N.; Stonik, V.A.; Kwak, J.Y. Differential effects of triterpene glycosides, Frondoside A and Cucumarioside A2-2 isolated from sea cucumbers on caspase activation and apoptosis of human leukemia cells. FEBS Lett. 2009, 583, 697–702. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Kundu, N.; Collin, P.D.; Goloubeva, O.; Fulton, A.M. Frondoside A inhibits breast cancer metastasis and antagonizes prostaglandin E receptors EP4 and EP2. Breast Cancer Res. Treat. 2011, 132, 1001–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menchinskaya, E.S.; Pislyagin, E.A.; Kovalchyk, S.N.; Davydova, V.N.; Silchenko, A.S.; Avilov, S.A.; Kalinin, V.I.; Aminin, D.L. Antitumor activity of cucumarioside A2-2. Chemotherapy 2013, 59, 181–191. [Google Scholar] [CrossRef]
- Pislyagin, E.A.; Manzhulo, I.V.; Gorpenchenko, T.Y.; Dmitrenok, P.S.; Avilov, S.A.; Silchenko, A.S.; Wang, Y.-M.; Aminin, D.L. Cucumarioside A2-2 causes macrophage activation in mouse spleen. Mar. Drugs 2017, 15, 341. [Google Scholar] [CrossRef] [Green Version]
- Aminin, D.L.; Shevchenko, V.P.; Nagaev, I.Y.; Gladkikh, R.V.; Kapustina, I.I.; Likhatskaya, G.N.; Avilov, S.A.; Stonik, V.A. The use of tritium-labeled triterpene glycosides from the holothurian Cucumaria japonica in pharmacokinetic studies. Dokl. Biol. Sci. 2008, 422, 345–422. [Google Scholar] [CrossRef] [PubMed]
- Hasan, S.; Ansari, M.I.; Ahmad, A.; Mishra, M. Major bioactive metabolites from marine fungi: A Review. Bioinformation 2015, 11, 176–181. [Google Scholar] [CrossRef]
- Youssef, F.S.; Ashour, M.L.; Singab, A.N.B.; Wink, M. A comprehensive review of bioactive peptides from marine fungi and their biological significance. Mar. Drugs 2019, 17, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, W.; Deng, Z.; Hong, K. The biological activities of sesterterpenoid-type ophiobolins. Mar. Drugs 2017, 15, 229. [Google Scholar] [CrossRef] [Green Version]
- Yan, T.; Wu, W.H.; Su, T.W.; Chen, J.J.; Zhu, Q.G.; Zhang, C.Y.; Wang, X.Y.; Bao, B. Effects of a novel marine natural product: Pyrano indolone alkaloid fibrinolytic compound on thrombolysis and hemorrhagic activities In Vitro and In Vivo. Arch. Pharm. Res. 2015, 38, 1530–1540. [Google Scholar] [CrossRef]
- Guo, R.H.; Zhang, Y.T.; Duan, D.; Fu, Q.; Yu, X.W.; Wang, S.J.; Bao, B.; Wu, W.H. Fibrinolytic evaluation of compounds isolated from a marine fungus Stachybotrys longispora FG216. Chin. J. Chem. 2016, 34, 1194–1198. [Google Scholar] [CrossRef]
- Newman, D.; Cragg, G. Marine natural products and related compounds in clinical and advanced preclinical trials. J. Nat. Prod. 2004, 67, 1216–1238. [Google Scholar] [CrossRef]
- Ancheeva, E.; El-Neketi, M.; Song, W.; Lin, W.; Daletos, G.; Ebrahim, W.; Proksch, P. Structurally unprecedented metabolites from marine sponges. Curr. Org. Chem. 2017, 21, 426–449. [Google Scholar] [CrossRef]
- El-Demerdash, A.; Tammam, M.A.; Atanasov, A.G.; Hooper, J.N.A.; Al-Mourabit, A.; Kijjoa, A. Chemistry and Biological Activities of the Marine Sponges of the Genera Mycale (Arenochalina), Biemna and Clathria. Mar. Drugs 2018, 16, 214. [Google Scholar] [CrossRef] [Green Version]
- El-Demerdash, A.; Atanasov, A.G.; Horbanczuk, O.K.; Tammam, M.A.; Abdel-Mogib, M.; Hooper, J.N.A.; Sekeroglu, N.; Al-Mourabit, A.; Kijjoa, A. Chemical diversity and biological activities of marine sponges of the genus Suberea: A systematic review. Mar. Drugs 2019, 17, 115. [Google Scholar] [CrossRef] [Green Version]
- Popov, A.M.; Stekhova, S.I.; Utkina, N.K.; Rebachuk, N.M. Antimicrobial and cytotoxic activity of sesquiterpenequinones and brominated diphenyl esters isolated from marine sponges. Pharm. Chem. J. 1999, 33, 71–73. [Google Scholar] [CrossRef]
- Tziveleka, L.A.; Vagias, C.; Roussis, V. Natural products with anti-HIV activity from marine organisms. Curr. Top. Med. Chem. 2003, 3, 1512–1535. [Google Scholar] [CrossRef]
- Lee, H.Y.; Chung, K.J.; Hwang, I.H.; Gwak, J.; Park, S.; Ju, B.G.; Yun, E.; Kim, D.E.; Chung, Y.H.; Na, M.; et al. Activation of p53 with ilimaquinone and ethylsmenoquinone, marine sponge metabolites, induces apoptosis and autophagy in colon cancer cells. Mar. Drugs 2015, 13, 543–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, H.; Noh, K.; Kang, C.; Na, M.; Oh, S.; Song, I.S.; Kang, W. HPLC-MS/MS analysis of ilimaquinone and its application in a pharmacokinetic study in rats. J. Pharm. Biomed. Anal. 2019, 166, 291–294. [Google Scholar] [CrossRef]
- Sakai, R.; Higa, T.; Jefford, C.W.; Bernardinelli, G. Manzamine A, a novel antitumor alkaloid from a sponge. J. Am. Chem. Soc. 1986, 108, 6404–6405. [Google Scholar] [CrossRef]
- Ang, K.K.H.; Holmes, M.J.; Higa, T.; Hamann, M.T.; Kara, U.A.K. In vivo antimalarial activity of the beta-carboline alkaloid manzamine A. Antimicrob. Agents Chemother. 2000, 44, 1645–1649. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.-C.; Kuo, T.-T.; Chang, H.-Y.; Liu, W.-S.; Hsia, S.-M.; Huang, T.-C. Manzamine A exerts anticancer activity against human colorectal cancer cells. Mar. Drugs 2018, 16, 252. [Google Scholar] [CrossRef] [Green Version]
- Shang, X.H.; Liu, X.Y.; Zhang, J.P.; Gao, Y.; Jiao, B.H.; Zheng, H.; Lu, X.L. Traditional Chinese Medicine—Sea Urchin. Mini Rev. Med. Chem. 2014, 14, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Mishchenko, N.P.; Fedoreev, S.A.; Bagirova, V.L. Histochrome: A new original domestic drug. Pharm. Chem. J. 2003, 37, 48–52. [Google Scholar] [CrossRef]
- Kim, R.; Hur, D.; Kim, H.K.; Han, J.; Mishchenko, N.P.; Fedoreyev, S.A.; Stonik, V.A.; Chang, W. Echinochrome A attenuates cerebral ischemic injury through regulation of cell survival after middle cerebral artery occlusion in rat. Mar. Drugs 2019, 17, 501. [Google Scholar] [CrossRef] [Green Version]
- Stavitskaya, T.V.; Egorov, E.A.; Kadyrova, M.K. Features of Ocular Pharmacokinetics and Pharmacodynamics of the Drug Histochrome; All-Russian School of Ophthalmologists: Moscow, Russia, 2004; pp. 289–293. [Google Scholar]
- Katelnikova, A.E.; Kryshen, K.L.; Makarova, M.N.; Makarov, V.G.; Vorobieva, V.V.; Pozharitskaya, O.N.; Shikov, A.N. Specific pharmacological activity study of glycosylated polypeptide complex extracted from sea urchin Strongylocentrotus droebachiensis in the model of acute bronchitis induced by formalin in rats. Rus. J. Biopharm. 2016, 8, 50–57. [Google Scholar]
- Pozharitskaya, O.N.; Shikov, A.N.; Laakso, I.; Sappanen-Laakso, T.; Makarenko, I.E.; Faustova, N.M.; Makarova, M.N.; Makarov, V.G. Bioactivity and chemical characterization of gonads of green sea urchin Strongylocentrotus droebahensis from Barents Sea. J. Funct. Foods 2015, 17, 227–234. [Google Scholar] [CrossRef]
- Le Tourneau, C.; Faivre, S.; Ciruelos, E.; Domínguez, M.J.; López-Martín, J.A.; Izquierdo, M.A.; Jimeno, J.; Raymond, E. Reports of clinical benefit of plitidepsin (Aplidine), a new marine-derived anticancer agent, in patients with advanced medullary thyroid carcinoma. Am. J. Clin. Oncol. 2010, 33, 132–136. [Google Scholar] [CrossRef]
- Hamann, M.T.; Scheuer, P.J. Kahalalide F: A bioactive depsipeptide from the sacoglossan mollusk Elysia rufescens and the green alga Bryopsis sp. J. Am. Chem. Soc. 1993, 115, 5825–5826. [Google Scholar] [CrossRef]
- Suárez, Y.; González, L.; Cuadrado, A.; Berciano, M.; Lafarga, M.; Muñoz, A. Kahalalide F, a new marine-derived compound, induces oncosis in human prostate and breast cancer cells. Mol. Cancer Ther. 2003, 2, 863–872. [Google Scholar]
- Ciavatta, M.L.; Lefranc, F.; Carbone, M.; Mollo, E.; Gavagnin, M.; Betancourt, T.; Dasari, R.; Kornienko, A.; Kiss, R. Marine mollusk-derived agents with antiproliferative activity as promising anticancer agents to overcome chemotherapy resistance. Med. Res. Rev. 2017, 37, 702–801. [Google Scholar] [CrossRef] [PubMed]
- Simmons, T.L.; Andrianasolo, E.; McPhail, K.; Flatt, P.; Gerwick, W.H. Marine natural products as anticancer drugs. Mol. Cancer Ther. 2005, 4, 333–342. [Google Scholar]
- Pettit, G.R.; Herald, C.L.; Doubek, D.L.; Herald, D.L.; Arnold, E.; Clardy, J. Isolation and structure of bryostatin 1. J. Am. Chem. Soc. 1982, 104, 6846–6848. [Google Scholar] [CrossRef]
- Raghuvanshi, R.; Bharate, S.B. Preclinical and clinical studies on bryostatins, a class of marine-derived protein kinase c modulators: A mini-review. Curr. Top. Med. Chem. 2020, 20, 1124–1135. [Google Scholar] [CrossRef]
- Sun, M.-K.; Alkon, D.L. Bryostatin-1: Pharmacology and therapeutic potential as a CNS Drug. CNS Drug Rev. 2006, 12, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Nelson, T.J.; Sen, A.; Alkon, D.L.; Sun, M.K. Adduct formation in liquid chromatography-triple quadrupole mass spectrometric measurement of bryostatin 1. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2013, 944, 55–62. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, Z.; Xu, Z.; Niu, X.; Zhang, H. Effects of fucoidan on chronic renal failure in rats. Planta Med. 2003, 69, 537–541. [Google Scholar] [CrossRef]
- Gammone, M.A.; D’Orazio, N. Anti-obesity activity of the marine carotenoid fucoxanthin. Mar. Drugs 2015, 13, 2196–2214. [Google Scholar] [CrossRef]
- Hu, L.; Chen, W.; Tian, F.; Yuan, C.; Wang, H.; Yue, H. Neuroprotective role of fucoxanthin against cerebral ischemic/reperfusion injury through activation of Nrf2/HO-1 signaling. Biomed. Pharmacother. 2018, 106, 1484–1489. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Tian, X.; Zhang, W.; Zheng, P.; Huang, F.; Ding, G.; Yang, Z. Protective effects of fucoxanthin against alcoholic liver injury by activation of Nrf2-mediated antioxidant defense and inhibition of TLR4-mediated inflammation. Mar. Drugs 2019, 17, 552. [Google Scholar] [CrossRef] [Green Version]
- Petri, D.; Lundebye, A.K. Tissue distribution of astaxanthin in rats following exposure to graded levels in the feed. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2007, 145, 202–209. [Google Scholar] [CrossRef]
- Onishi, H.; Machida, Y. Biodegradation and distribution of water-soluble chitosan in mice. Biomaterials 1999, 20, 175–182. [Google Scholar] [CrossRef]
- Song, S.; Zhang, L.; Cao, J.; Xiang, G.; Cong, P.; Dong, P.; Li, Z.; Xue, C.; Xue, Y.; Wang, Y. Characterization of metabolic pathways and absorption of sea cucumber saponins, holothurin A and echinoside A, in vitro and in vivo. J. Food Sci. 2017, 82, 1961–1967. [Google Scholar] [CrossRef]
- Guseva, M.R.; Beslaneeva, M.B.; Mishchenko, N.P.; Hurai, A.R. The specific features of penetration of the antioxidant histochrome across the blood-ocular barrier. Vestn. Ophthalmol. 2007, 123, 38–40. [Google Scholar]
- Nakazato, K.; Takada, H.; Iha, M.; Nagamine, T. Attenuation of N-nitrosodiethylamine-induced liver fibrosis by high-molecular-weight fucoidan derived from Cladosiphon okamuranus. J. Gastroenterol. Hepatol. 2010, 25, 1692–1701. [Google Scholar] [CrossRef]
- Nagamine, T.; Nakazato, K.; Tomioka, S.; Iha, M.; Nakajima, K. Intestinal absorption of fucoidan extracted from the brown seaweed, Cladosiphon okamuranus. Mar. Drugs 2015, 13, 48–64. [Google Scholar] [CrossRef]
- Suda, M.; Ohno, N.; Hashimoto, T.; Koizumi, K.; Adachi, Y.; Yadomae, T. Kupffer cells play important roles in the metabolic degradation of a soluble anti-tumor (1→3)-β-d-glucan, SSG, in mice. FEMS Immunol. Med. Microbiol. 1996, 15, 93–100. [Google Scholar] [CrossRef] [Green Version]
- Ben, J.; Zhang, Y.; Zhou, R.; Zhang, H.; Zhu, X.; Li, X.; Zhang, H.; Li, N.; Zhou, X.; Bai, H.; et al. Major vault protein regulates class A scavenger receptor-mediated tumor necrosis factor-α synthesis and apoptosis in macrophages. J. Biol. Chem. 2013, 288, 20076–20084. [Google Scholar] [CrossRef] [Green Version]
- Inui, K.I.; Masuda, S.; Saito, H. Cellular and molecular aspects of drug transport in the kidney. Kidney Int. 2000, 58, 944–958. [Google Scholar] [CrossRef] [Green Version]
- Sugawara, T.; Baskaran, V.; Tsuzuki, W.; Nagao, A. Brown algae fucoxanthin is hydrolyzed to fucoxanthinol during absorption by Caco-2 human intestinal cells and mice. J. Nutr. 2002, 132, 946–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asai, A.; Sugawara, T.; Ono, H. Biotransformation of fucoxanthinol into amarouciaxanthin A in mice and HepG2cells: Formation and cytotoxicity of fucoxanthin metabolites. Drug Metab. Dispos. 2004, 32, 205–211. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Ishikawa, C.; Katano, H.; Yasumoto, T.; Mori, N. Fucoxanthin and its deacetylated product, fucoxanthinol, induce apoptosis of primary effusion lymphomas. Cancer Lett. 2011, 300, 225–234. [Google Scholar] [CrossRef]
- Talalaeva, O.S.; Mishchenko, N.P.; Briukhanov, V.M.; Zverev, I.F.; Lampatov, V.V.; Dvornikova, L.G. Identification of histochrome metabolism products in urine for studying drug pharmacokinetics. Eksp. Klin. Farmakol. 2014, 77, 29–32. [Google Scholar]
- Garteiz, D.A.; Madden, T.; Beck, D.E.; Huie, W.R.; McManus, K.T.; Abbruzzese, J.L.; Chen, W.; Newman, R.A. Quantitation of dolastatin-10 using HPLC/electrospray ionization mass spectrometry: Application in a phase I clinical trial. Cancer Chemother. Pharmacol. 1998, 41, 299–306. [Google Scholar] [CrossRef]
- Chaki, T.; Kajimoto, N.; Ogawa, H.; Baba, T.; Hiura, N. Metabolism and calcium antagonism of sodium alginate oligosaccharides. Biosci. Biotechnol. Biochem. 2007, 71, 1819–1825. [Google Scholar] [CrossRef]
- Nakamura, Y.; Tonogai, Y.; Hasegawa, Y.; Ito, Y. Metabolism of alginic acid and its salts and their effects on serum concentrations of Na, K and Ca in the rat. Food Hyg. Saf. Sci. (Shokuhin Eiseigaku Zasshi) 1988, 29, 240–248. [Google Scholar] [CrossRef]
- Humphreys, E.R.; Triffitt, J.T. Absorption by the rat of alginate labelled with carbon-14. Nature 1968, 219, 1172–1173. [Google Scholar] [CrossRef]
- Faivre, S.; Chièze, S.; Delbaldo, C.; Ady-Vago, N.; Guzman, C.; Lopez-Lazaro, L.; Lozahic, S.; Jimeno, J.; Pico, F.; Armand, J.P.; et al. Phase I and pharmacokinetic study of aplidine, a new marine cyclodepsipeptide in patients with advanced malignancies. J. Clin. Oncol. 2005, 23, 7871–7880. [Google Scholar] [CrossRef]
- Van Andel, L.; Rosing, H.; Schellens, J.H.; Beijnen, J.H. Review of chromatographic bioanalytical assays for the quantitative determination of marine-derived drugs for cancer treatment. Mar. Drugs 2018, 16, 246. [Google Scholar] [CrossRef] [Green Version]
- Nalda-Molina, R.; Valenzuela, B.; Ramon-Lopez, A.; Miguel-Lillo, B.; Soto-Matos, A.; Perez-Ruixo, J.J. Population pharmacokinetics meta-analysis of plitidepsin (Aplidin) in cancer subjects. Cancer Chemother. Pharmacol. 2009, 64, 97–108. [Google Scholar] [CrossRef]
- Rademaker-Lakhai, J.M.; Horenblas, S.; Meinhardt, W.; Stokvis, E.; de Reijke, T.M.; Jimeno, J.M.; Lopez-Lazaro, L.; Lopez Martin, J.A.; Beijnen, J.H.; Schellens, J.H. Phase I clinical and pharmacokinetic study of kahalalide F in patients with advanced androgen refractory prostate cancer. Clin. Cancer Res. 2005, 11, 1854–1862. [Google Scholar] [CrossRef] [Green Version]
- Pardo, B.; Paz-Ares, L.; Tabernero, J.; Ciruelos, E.; García, M.; Salazar, R.; López, A.; Blanco, M.; Nieto, A.; Jimeno, J.; et al. Phase i clinical and pharmacokinetic study of kahalalide f administered weekly as a 1-hour infusion to patients with advanced solid tumors. Clin. Cancer Res. 2008, 14, 1116–1123. [Google Scholar] [CrossRef] [Green Version]
- Martin-Algarra, S.; Espinosa, E.; Rubio, J.; Lopez Lopez, J.J.; Manzano, J.L.; Carrion, L.A.; Plazaola, A.; Tanovic, A.; Paz-Ares, L. Phase II study of weekly kahalalide F in patients with advanced malignant melanoma. Eur. J. Cancer 2009, 45, 732–735. [Google Scholar] [CrossRef] [PubMed]
- Van Kesteren, C.; Cvitkovic, E.; Taamma, A.; López-Lázaro, L.; Jimeno, J.M.; Guzman, C.; Hillebrand, M.J.; Mathôt, R.A.A.; Schellens, J.H.M.; Misset, J.-L.; et al. Pharmacokinetics and pharmacodynamics of the novel marine-derived anticancer agent ecteinascidin 743 in a phase I dose-finding study. Clin. Cancer Res. 2000, 6, 4725–4732. [Google Scholar]
- Zangarini, M.; Ceriani, L.; Sala, F.; Marangon, E.; Bagnati, R.; D’Incalci, M.; Grosso, F.; Zucchetti, M. Quantification of trabectedin in human plasma: Validation of a high-performance liquid chromatography–mass spectrometry method and its application in a clinical pharmacokinetic study. J. Pharm. Biomed. Anal. 2014, 95, 107–112. [Google Scholar] [CrossRef]
- Twelves, C.; Hoekman, K.; Bowman, A.; Vermorken, J.B.; Anthoney, A.; Smyth, J.; Van Kesteren, C.; Beijnen, J.H.; Uiters, J.; Wanders, J.; et al. Phase I and pharmacokinetic study of Yondelis (Ecteinascidin 743; ET-743) administered as an infusion over 1h or 3h every 21 days in patients with solid tumors. Eur. J. Cancer 2003, 39, 1842–1851. [Google Scholar] [CrossRef]
- Zakirova, A.N.; Ivanova, M.V.; Golubiatnikov, V.B.; Mishchenko, N.P.; Kol’tsova, E.A.; Fedoreev, S.A.; Krasnovid, N.J.; Lebedev, A.V. Pharmacokinetics and clinical efficacy of histochrome in patients with acute myocardial infarction. Eksp. Klin. Farmakol. 1997, 60, 21–24. [Google Scholar]
- Michel, C.; Lahaye, M.; Bonnet, C.; Mabeau, S.; Barry, J.-L. In vitro fermentation by human faecal bacteria of total and purified dietary fibres from brown seaweeds. Br. J. Nutr. 1996, 75, 263–280. [Google Scholar] [CrossRef]
- Tokita, Y.; Hirayama, M.; Nakajima, K.; Tamaki, K.; Iha, M.; Nagamine, T. Detection of Fucoidan in Urine after Oral Intake of Traditional Japanese Seaweed, Okinawa mozuku (Cladosiphon okamuranus Tokida). J. Nutr. Sci. Vitaminol. 2017, 63, 419–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadena, K.; Tomori, M.; Iha, M.; Nagamine, T. Absorption study of mozuku fucoidan in Japanese volunteers. Mar. Drugs 2018, 16, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomori, M.; Nagamine, T.; Iha, M. Are Helicobacter pylori infection and fucoidan consumption associated with fucoidan absorption? Mar. Drugs 2020, 18, 235. [Google Scholar] [CrossRef]
- Imbs, T.I.; Zvyagintseva, T.N.; Ermakova, S.P. Is the transformation of fucoidans in human body possible? Int. J. Biol. Macromol. 2020, 142, 778–781. [Google Scholar] [CrossRef]
- Osterlie, M.; Bjerkeng, B.; Liaaen-Jensen, S. Plasma appearance and distribution of astaxanthin E/Z isomers in plasma lipoproteins of after single dose administration of astaxanthin. J. Nutr. Biochem. 2000, 11, 482–492. [Google Scholar] [CrossRef]
- Odeberg, M.J.; Lignell, A.; Pettersson, A.; Hoglund, P. Oral bioavailability of the antioxidant astaxanthin in humans is enhanced by incorporation of lipid based formulations. Eur. J. Pharm. Sci. 2003, 19, 299–304. [Google Scholar] [CrossRef]
- Okada, Y.; Ishikura, M.; Maoka, T. Bioavailability of astaxanthin in Haematococcus algal extract: The effects of timing of diet and smoking habits. Biosci. Biotechnol. Biochem. 2009, 73, 1928–1932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergström, C.A.S.; Holm, R.; Jørgensen, S.A.; Andersson, S.B.E.; Artursson, P.; Beato, S.; Borde, A.; Box, K.; Brewster, M.; Dressman, J.; et al. Early pharmaceutical profiling to predict oral drug absorption: Current status and unmet needs. Eur. J. Pharm. Sci. 2014, 57, 173–199. [Google Scholar] [CrossRef]
- Cunha, L.; Grenha, A. Sulfated seaweed polysaccharides as multifunctional materials in drug delivery applications. Mar. Drugs 2016, 14, 42. [Google Scholar] [CrossRef]
- Citkowska, A.; Szekalska, M.; Winnicka, K. Possibilities of fucoidan utilization in the development of pharmaceutical dosage forms. Mar. Drugs 2019, 17, 458. [Google Scholar] [CrossRef] [Green Version]




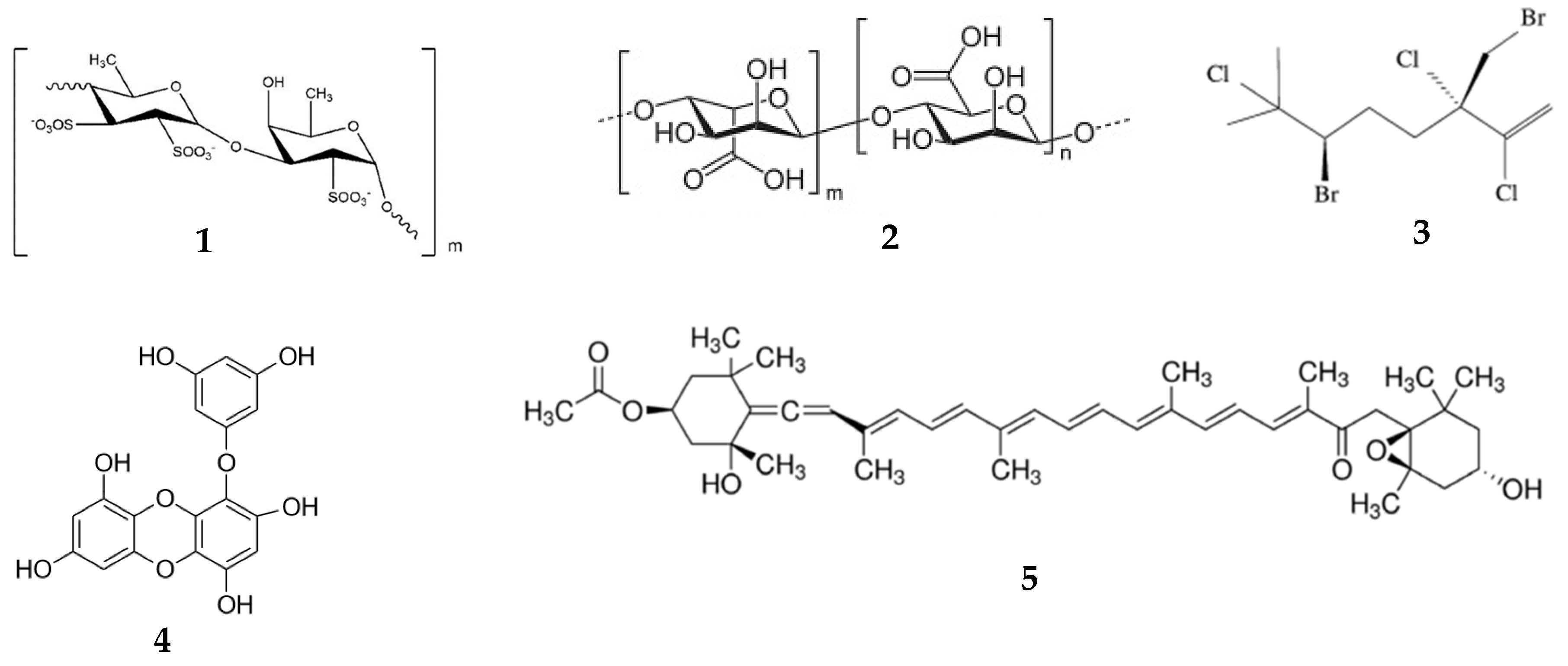{kind=link}
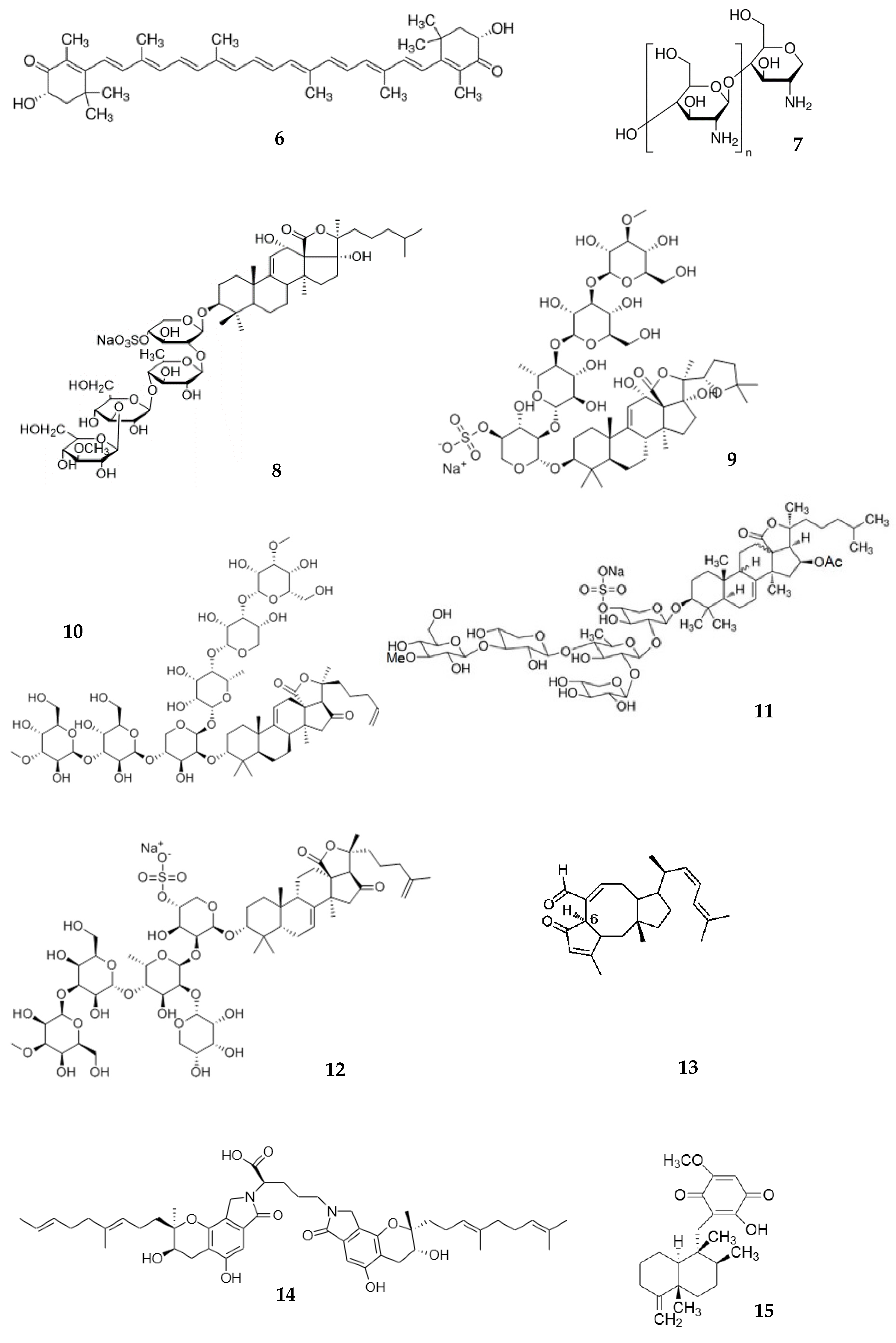{kind=link}
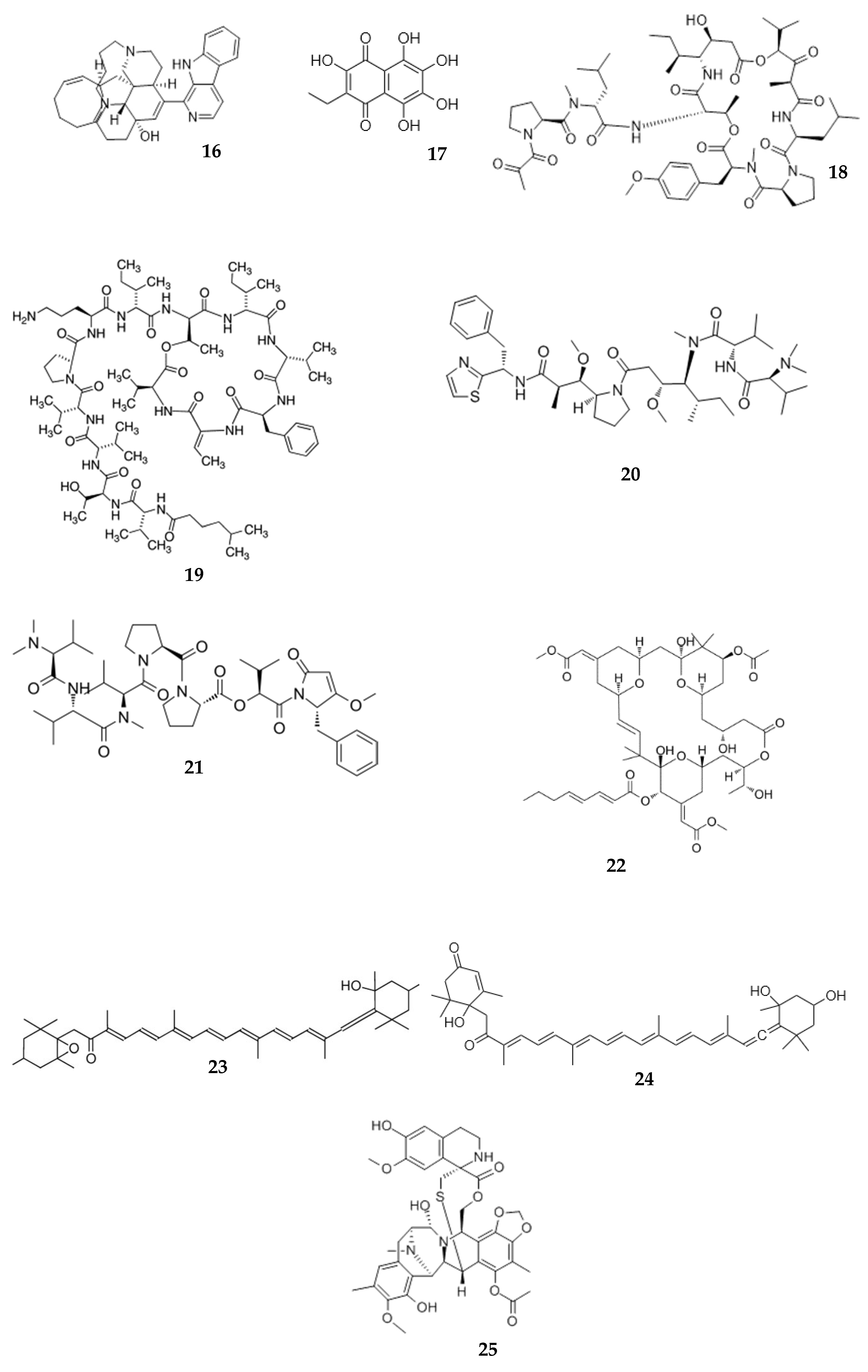{kind=link}
| Method | Recovery (%) | Linearity Range (µg/mL) | Compound | Source | Biomaterial | Reference |
|---|---|---|---|---|---|---|
| Biomarker assay Anti-Xa activity | - | 0.027–0.217 | Fucoidan | Brown algae Fucus vesiculosus | Plasma, Tissue | [27] |
| Biomarker assay Anti-Xa activity | - | 0.014–1.13 * | Fucoidan | Brown algae Fucus vesiculosus | Skin Tissue | [28] |
| Biomarker assay Dypeptydylpeptidase IV inhibition | - | 1.9–710 | Lipid extract of gonads | Sea urchins Strongylocentrotus droebachiensis | Plasma | [29] |
| Biomarker assay Lactate dehydrogenase activity | - | 0.01–7.05 | Glycopeptide | Sea urchins Strongylocentrotus droebachiensis | Plasma Tissue | [30] |
| Competitive ELISA | - | 0.078–80 | Fucoidan | Brown algae Undaria pinnatifida | Plasma | [31] |
| Fluorescent labeling | - | - | Fucoidan | Brown algae Fucus vesiculosus | Plasma Organs | [32] |
| Gas chromatography | 82–87 | 0.3–10 | Halomon | Brown algae Fucus vesiculosus | Plasma Organs | [33] |
| HPLC with derivatization | 96.6–106.4 | 0–80 *** | Fucoidan | Laminaria japonica | Plasma | [34] |
| HPLC with fluorescence detection | 93.3–96.5 | 0.5–100 | Fucoidan | Laminaria japonica | Plasma | [35] |
| HPLC with postcolumn fluorescence derivatization | 95.5–99.2 | 0.5–150 | Fucoidan | Laminaria japonica | Serum | [36] |
| HPLC with UV detection | 86.2–100.1 | 0.5–500 | Diindolinonepyrane (2,5-BHPA) | Marine fungi Stachybotrys longispora | Plasma | [37] |
| HPLC-ELSD | 90.0 | 0.1–5 0.1–20 | Echinoside A Holotoxin A1 | Sea cucumber Apostichopus japonicus | Plasma | [38] |
| HPLC-ESI-MS | 85.0 | 0.3–20 0.2–10 | Echinoside A holothurin A | Sea cucumbers Pearsonothuria graeffei | Serum, liver | [39] |
| HPLC-ESI-MS | 96.6–102.3 | 0.5–500 | 2,5-BHPA | Marine fungi Stachybotrys longispora | Plasma | [40] |
| LC-MS/MS | 88.0 | 0.025–0.25 | Frondoside A | Sea cucumber Cucumaria frondosa | Plasma | [41] |
| LC-MS/MS | 92.9–101 84.7–91.6 | 0.00153–0.72 0.00117–0.6 | Fucoxanthin Fucoxanthinol | Plasma | [42] | |
| LC-MS/MS | 80.0 | 1.0–100 | Sodium alginate | - | Plasma, urine | [43] |
| LC-MS/MS | 69 91 | 0.005–0.1 0.00125–0.125 | Aplidine (dehydrodidemnin B) | Mediterranean tunicate Aplidium albicans | Plasma, urine | [44] |
| LC-MS/MS | 90–101 90–94 | 0.01–5 ** 0.5–50 ** | Sesterterpene MHO7 (6-epi-ophiobolin G) | Mangrove fungus Aspergillus ustus | Plasma tissues | [45] |
| LC-MS/MS | - | 0.002–0.8 | Ilimaquinone Epimers | Marine Sponge Hippiospongia metachromia | Plasma | [46] |
| MALDI-MS | - | 0.001–1.000 | Cucumarioside A2-2 | Sea cucumber Cucumaria japonica | Tissue | [47] |
| RIA | - | 10 pg–10 ng | Didemnin B | Caribbean tunicate (sea squirt) Trididemnum solidum | Plasma | [48] |
| Sandwich ELISA | 97–105 86–113 97–98 | 0.001–0.1 | Fucoidan | Brown algae Cladosiphon okamuranus | Serum, plasma, urine | [49] |
| TRA | 77.4 | - | Aplidin (Plitidepsin) | - | Urine | [48] |
| Compounds | Animals/Dose (mg/kg/vehicle) | Administration | Tmax (h) | T1/2 (h) | Cmax (µg/mL) | AUC0−t (µg·h/mL) | Reference |
|---|---|---|---|---|---|---|---|
| Fucoidan (1) MW 735 kDa | Rats/100/ointment | i/v topical | N.d.1.0 | 9.5 20.7 | 9.2 0.15 | 10.8 1.9 | [28] |
| Fucoidan (1) MW 735 kDa | Rats/100/starch slime | peroral | 3.2 | 3.4 | 0.13 | 0.99 | [27] |
| Fucoidan (1) MW 107.8 kDa | Mice/50/phosphate buffer solution (pH 7.4) | i/v | 0.5 | 2.77 | 66.4 | 138.7 | [32] |
| Griffithsin | Rats/10/phosphate-buffered saline (pH 7.4) | i/v s/c | - 4.0 | 10.7 13.8 | 81.8 6.6 | 0.11 0.045 | [51] |
| Sodium alginate (2) | Mice/10 mg/500 µL saline | peroral | 0.08 | N.d. | 24.5 | N.d. | [43] |
| Halomon (3) | Mice/135/cremophor–EtOH–0.154 M NaCl (1:1:6, by vol.) | i/v | N.d. | 8.4 | N.d. | 189,960 | [33] |
| i/p | N.d. | 12.3 | N.d. | 85,620 | |||
| s/c | N.d. | 8.0 | N.d. | 89,280 | |||
| peroral | N.d. | 4.5 | N.d. | 7080 | |||
| Fucoxanthin (5) | Rats/2/mixed micelle | i/v | N.d. | 2.3 | N.d. | 9.86 | [42] |
| Fucoxanthinol (23) | 1.0 | 11.9 | 0.59 | 3.26 | |||
| Fucoxanthin (5) | Rats/65/mixed micelle | peroral | 7.7 | 1.2 | 0.03 | 0.19 | [42] |
| Fucoxanthinol (23) | 11.0 | N.d. | 0.26 | 5.0 | |||
| Astaxanthin (6) | Rats/10/polyethylene glycol 400–N,N-dimethylacetamide 50:50, v/v Rats/100/the same solution | i/v peroral | N.d. 6.5 | N.d. N.d. | 50 0.08 | 29,280 4638 | [52] |
| Chitosan (7) lactate | Rats/20/phosphate buffered saline pH 7.4 | peroral | [53] | ||||
| MW 3.8 kDa | 0.5 | N.d. | 20.23 | 24.13 | |||
| MW 7.5 kDa | 0.5 | N.d. | 9.30 | 11.55 | |||
| MW 13 kDa | 0.5 | N.d. | 5.86 | 8.71 | |||
| MW 22 kDa | 0.5 | N.d. | 4.32 | 5.59 | |||
| MW 230 kDa | 0.5 | N.d. | <0.5 | 0.97 | |||
| Chitosan (7) | Mice/500/1% (v/v) acetic acid solution | peroral | [54] | ||||
| MW 0.99 kDa | 0.5 | N.d. | 680 | N.d. | |||
| MW 39.1 kDa | 1.0 | N.d. | 190 | N.d. | |||
| MW 32.7 kDa | 1.0 | N.d. | 310 | N.d. | |||
| MW 760 kDa | 0.5 | N.d. | 60 | N.d. | |||
| Saponin extract Echinoside A (8) Holothurin A (9) | Rats/30/0.9% saline | peroral | N.d. | N.d. | [39] | ||
| 3.0 | 0.83 | ||||||
| 7.0 | 0.24 | ||||||
| 3.0 | 0.34 | ||||||
| 9.0 | 0.27 | ||||||
| Echinoside A (8) Holotoxin A1 (10) | Rats/20/water | peroral | 3.0 | 6.9 | 0.9 | 9.3 | [38] |
| i/v | 0.08 | 8.5 | 4.0 | 16.4 | |||
| i/v | 0.08 | 4.4 | 2.9 | 6.5 | |||
| Frondoside * A (11) | Mice/0.1/0.7% DMSO in saline | i/v | 0.08 | 8.5 | 0.17 | 0.73 | [41] |
| i/p | 1.0 | 14.0 | 0.024 | 0.16 | |||
| Cucumarioside * A2-2 (12) | Mice/5/water | i/p | 0.64 | 15.1 | 62.6 | 1544 | [55] |
| peroral | 0.67 | 0.35 | 74.4 | 1680 | |||
| MHO7 (6-epi-ophiobolin G) (13) | Mice/500/corn oil | peroral | 8.0 | 6.97 | 1.38 | 10.50 | [45] |
| 2,5-BHPA ** (14) | Rats/20/normal saline with NaHCO3 | i/v | N.d. | N.d. 23.2 | N.d. | 53,940 | [37] |
| 2,5-BHPA (14) | Dogs/7.5/normal saline with NaHCO3 | i/v | N.d. | 0.82 | 56.5 | 19.7 | [40] |
| Mixture Ilimaquinone (15) + epi-Ilimaquinone | Rats/2 + 1/corn oil | i/v | N.d. | 0.6 0.4 | N.d. | 1.46 0.24 | [46] |
| Mixture Ilimaquinone (15) + epi-Ilimaquinone | Rats/20 + 10/corn oil | peroral | 1.3 1.7 | 3.8 3.9 | 1.3 0.1 | 5.5 0.3 | [46] |
| Manzamine A (16) | Rats/10/EtOHRats/50/water | i/v peroral | N.d. | N.d. | 40 | N.d. | [56] |
| 10 | N.d. | 1.1 | N.d. | ||||
| Glycopeptide | Rats/0.1/water | i/v i/n | N.d. | 0.80 | 6.15 | 8.00 | [30] |
| 0.05/water | 0.67 | 0.77 | 2.90 | 3.93 | |||
| 0.1/water | 0.75 | 3.53 | 4.15 | 7.14 | |||
| 0.2/water | 0.70 | 4.03 | 6.22 | 12.6 | |||
| Lipid extract of gonads | Rabbits/15/starch slime | peroral | 3.0 | 8.8 | 107 | 313 | [29] |
| Aplidine (18) (dehydrodidemnin B) | Rats/0.7/EtOH, cremophor EL 10% in saline | i/v | N.d. | 0.15 | 0.1 | N.d. | [44] |
| Kahalalide F ** (19) | Mice/0.278/dimethylformamide/sterile saline 10:90 (v/v) | i/v | N.d. | 0.26 4.4 | 0.001 | N.d. | [57] |
| Dolastatin 10 (20) | Mice/0.24/water Mice/0.32/water | i/v | N.d. | 5.6 | 0.28 | 0.067 | [58] |
| i/p | N.d. | N.d. | 0.011 | ||||
| s/c | 3.7 | 0.011 | 0.058 | ||||
| Dolastatin 10 ** (20) Dolastatin 15 ** (21) | Mice/1/water Mice/1/water | i/v i/v | N.d. N.d. | 0.04 1.6 0.09 0.52 | N.d. N.d. | 0.33 0.21 | [59] |
| Bryostatin 1 ** (22) | Mice/0.04/phosphate buffer containing 30% DMSO | i/v i/p | N.d. N.d. | 1.05 22.96 0.81 28.76 | 0.092 0.013 | 0.37 0.62 | [60] |
| Compound | Animals/Dose (mg/kg/administration) | Tmax (h) | Cmax (µg/g) | Reference | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Heart | Stomach | Liver | Spleen | Lung | Kidney | Brain | Heart | Stomach | Liver | Spleen | Lung | Kidney | Brain | |||
| Fucoidan (1) MW 735 kDa | Rats/100/peroral | N.d. | N.d. | 2 | 3 | N.d. | 5 | N.d. | N.d. | N.d. | 0.53 | 0.78 | N.d. | 1.23 | N.d. | [27] |
| Fucoidan (1) MW 107.8 kDa | Mice/50/i/v | N.d. | N.d. | 0.5 | 6 | 4 | 0.5 | N.d. | N.d. | N.d. | 284 | 78 | 111 | 1092 | N.d. | [32] |
| Griffithsin ** | Mice/50/s/c | N.d. | N.d. | N.d. | N.d. | N.d. | N.d. | N.d. | - | - | 2.5 | 6.0 | - | 4.6 | - | [68] |
| Fucoxanthin (5) Fucoxanthinol (23) Amarouciaxanthin A (24) | Mice/0.105 mg per 200 µL/peroral | 4 4 | N.d. N.d. | 4 4 | 4 4 | 4 4 | 4 4 | N.d. N.d. | 0.15 0.069 | N.d. N.d. | 0.38 0.12 | 0.16 0.063 | 0.28 0.12 | 0.15 0.067 | N.d. N.d. | [80] |
| Astaxanthin (6) | Rats/100/peroral | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 0.12 | 7.3 | 0.14 | 0.16 | 0.15 | 0.21 | 0.26 | [52] |
| Cucumarioside * A2-2 (12) | Mice/5/i/p | 0.42 | 0.05 | 0.067 | 0.42 | N.d. | 0.067 | N.d. | 120 | 158 | 69 | 69 | N.d. | 73 | N.d. | [55] |
| Mice/5/peroral | 0.33 | 0.17 | 0.33 | 0.33 | N.d. | 0.33 | N.d. | 95 | 153 | 74 | 52 | N.d. | 70 | N.d. | ||
| MHO7 (6-epi-ophiobolin G) (13) | Mice/50 mg/kg/peroral | 4 | 8 | 1 | 8 | 12 | 12 | 1 | 0.95 | 8.4 | 4.0 | 0.65 | 2.5 | 8.0 | 1.0 | [45] |
| 2,5-BHPA (14) | Rats/20/i/v | 0.25 | 0.25 | 0.25 | 0.25 | 0.25 | 0.25 | N.d. | 2.4 | 4.1 | 235 | 16.8 | 16.0 | 18.0 | - | [37] |
| 2,5-BHPA (14) | Dogs/7.5/i/v | 1 | N.d. | 1 | 1 | 1 | 1 | 1 | 1.5 | N.d. | 52 | 5.0 | 6.0 | 7.5 | 3.5 | [40] |
| Glycopeptide | Rats/0.1/i/n | N.d. | N.d. | 1.6 | 2.4 | N.d. | 3.6 | N.d. | N.d. | N.d. | 0.73 | 2.53 | N.d. | 0.98 | N.d. | [30] |
| Bryostatin 1 (22) | Mice/0.04/i/v | 1 | 1 | 0.5 | 1 | 0.5 | 1 | 0.5 | 0.04 | 0.027 | 0.900 | 0.060 | 1.0 | 0.050 | 0.002 | [60] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shikov, A.N.; Flisyuk, E.V.; Obluchinskaya, E.D.; Pozharitskaya, O.N. Pharmacokinetics of Marine-Derived Drugs. Mar. Drugs 2020, 18, 557. https://doi.org/10.3390/md18110557
Shikov AN, Flisyuk EV, Obluchinskaya ED, Pozharitskaya ON. Pharmacokinetics of Marine-Derived Drugs. Marine Drugs. 2020; 18(11):557. https://doi.org/10.3390/md18110557
Chicago/Turabian StyleShikov, Alexander N., Elena V. Flisyuk, Ekaterina D. Obluchinskaya, and Olga N. Pozharitskaya. 2020. "Pharmacokinetics of Marine-Derived Drugs" Marine Drugs 18, no. 11: 557. https://doi.org/10.3390/md18110557
APA StyleShikov, A. N., Flisyuk, E. V., Obluchinskaya, E. D., & Pozharitskaya, O. N. (2020). Pharmacokinetics of Marine-Derived Drugs. Marine Drugs, 18(11), 557. https://doi.org/10.3390/md18110557