Microbial Diversity and Phage–Host Interactions in the Georgian Coastal Area of the Black Sea Revealed by Whole Genome Metagenomic Sequencing

 ,
,  ,
, 

Abstract
:1. Introduction
2. Results
2.1. Physico-Chemical Parameters
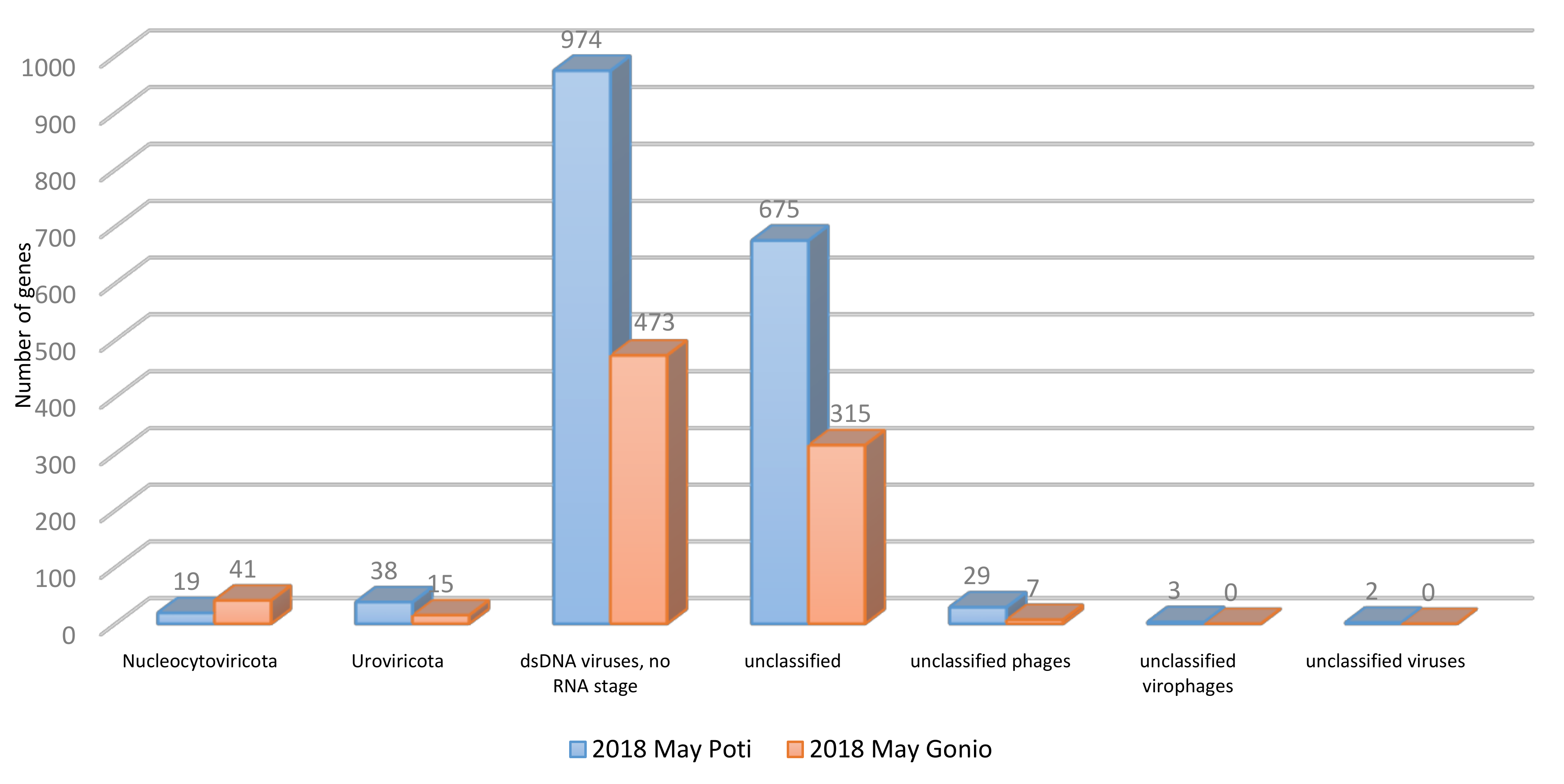
2.2. General Characteristics of Prokaryote and Viral Metagenomes
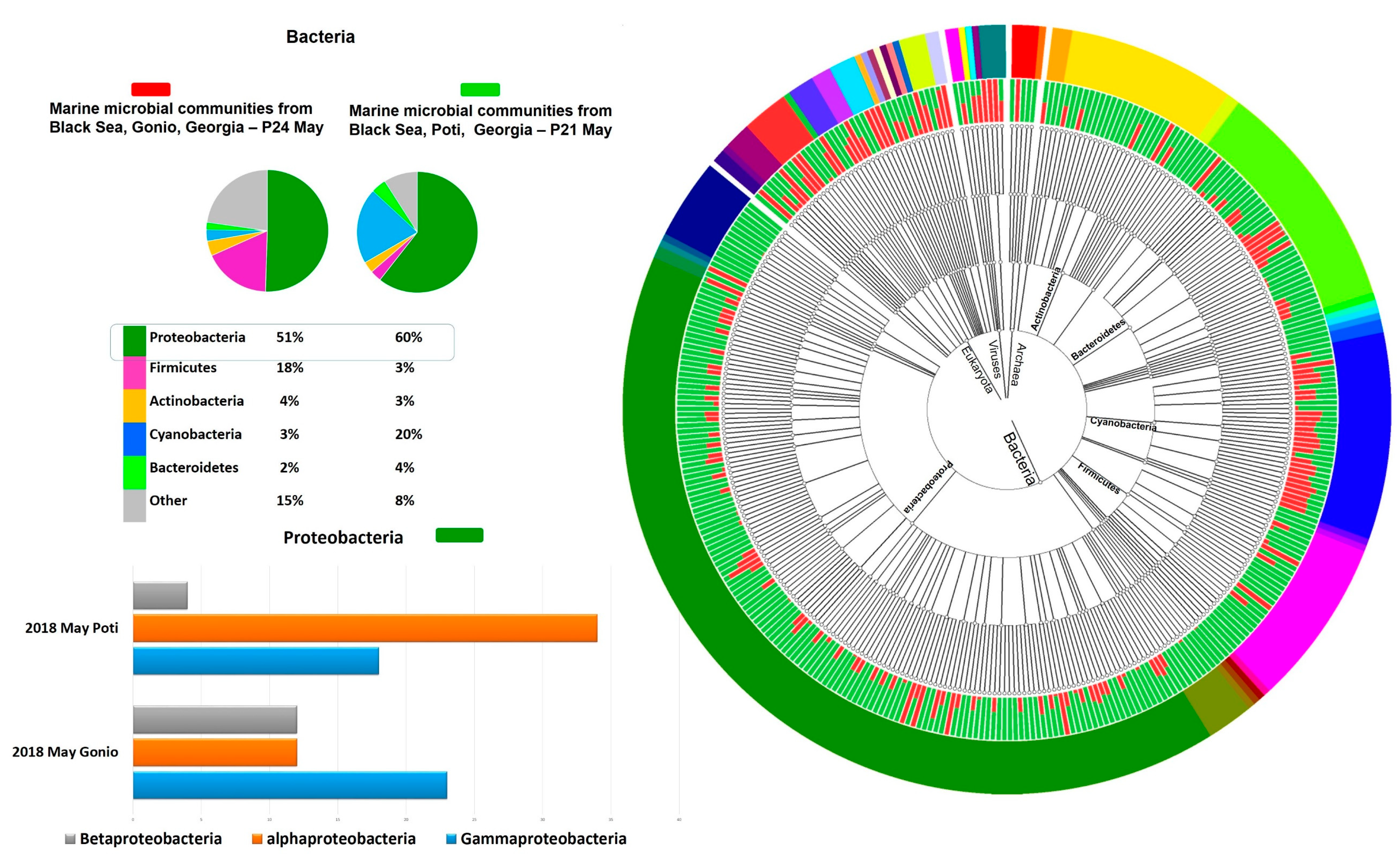
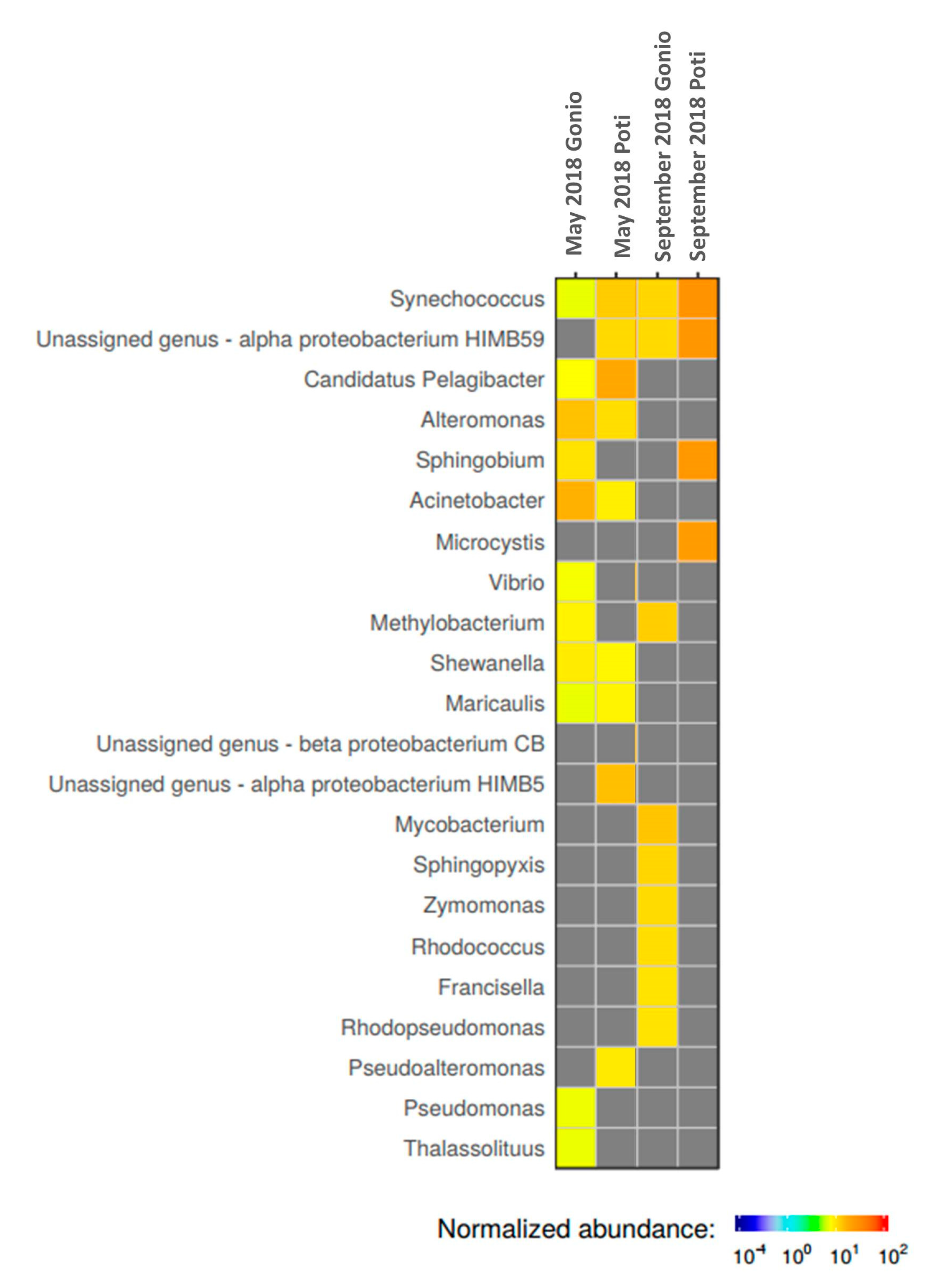
2.3. Microbial Community Structure
2.3.1. Prokaryotic Community Structure and Diversity
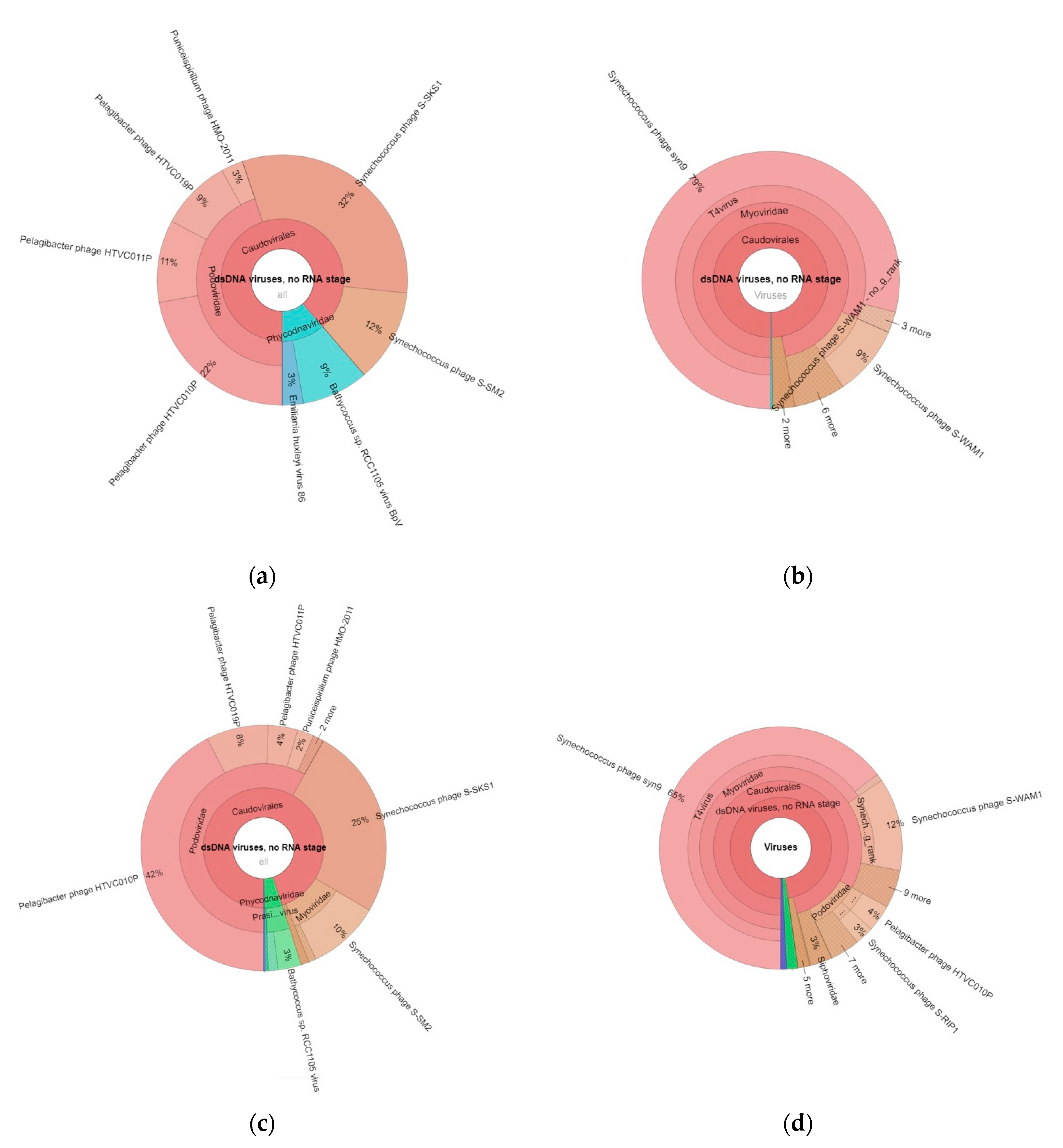
2.3.2. Viral Community Structure and Diversity
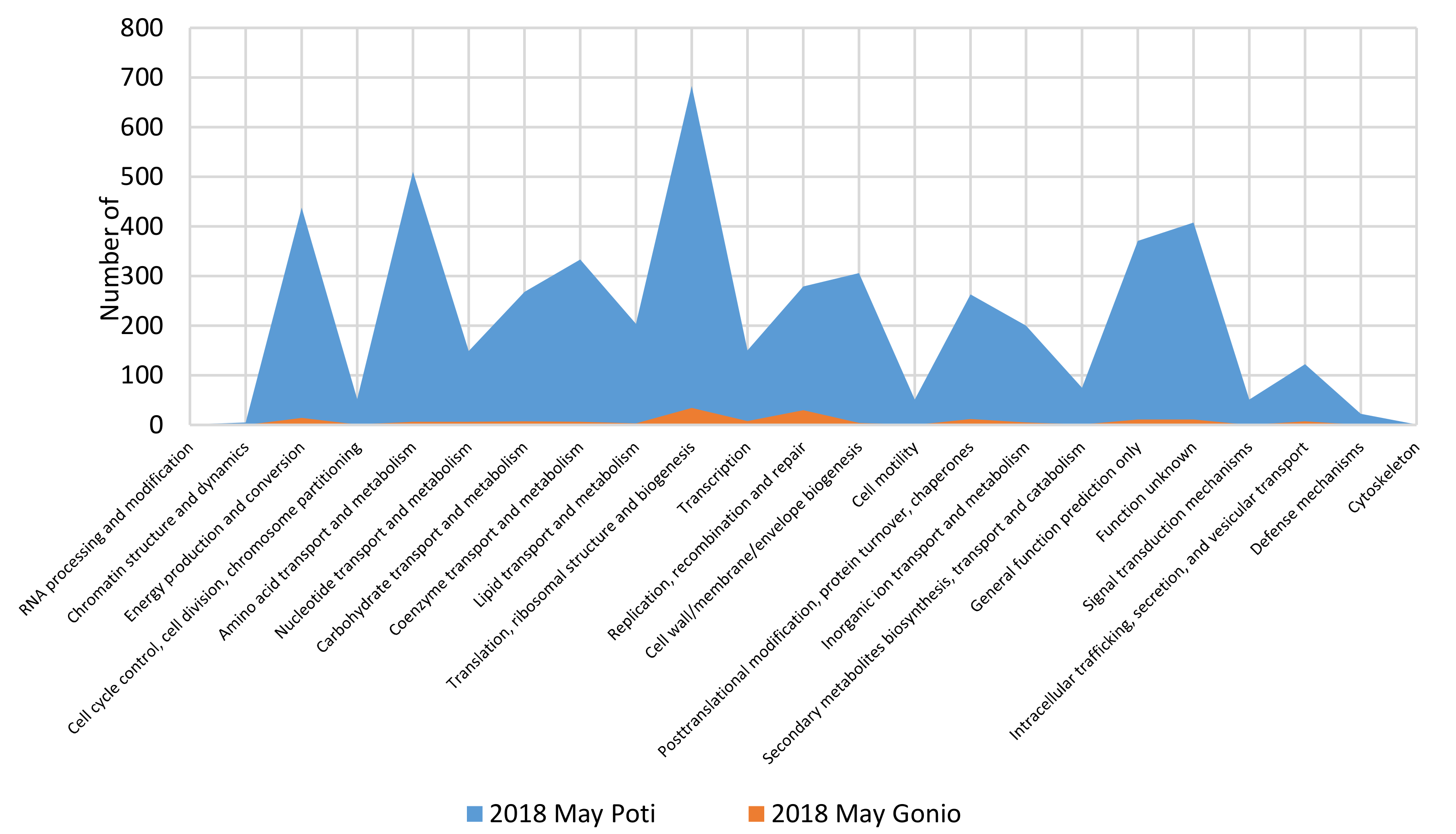
2.4. Functional Profiles of the Metagenomes
2.5. Phage–Host Interactions
2.5.1. Prophage Identification
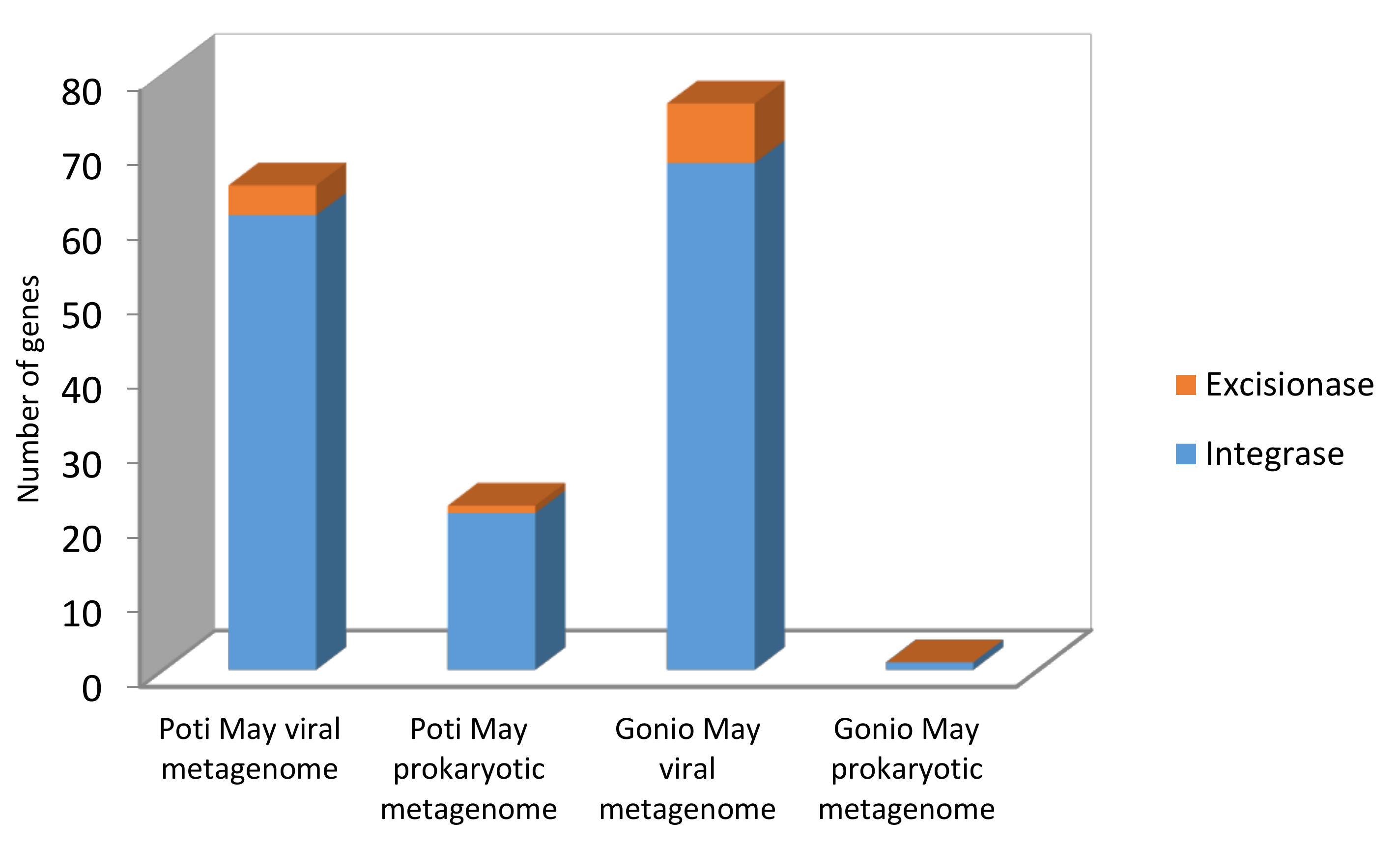
2.5.2. Integrase/Excisionase Identification
2.5.3. CRISPR Identification
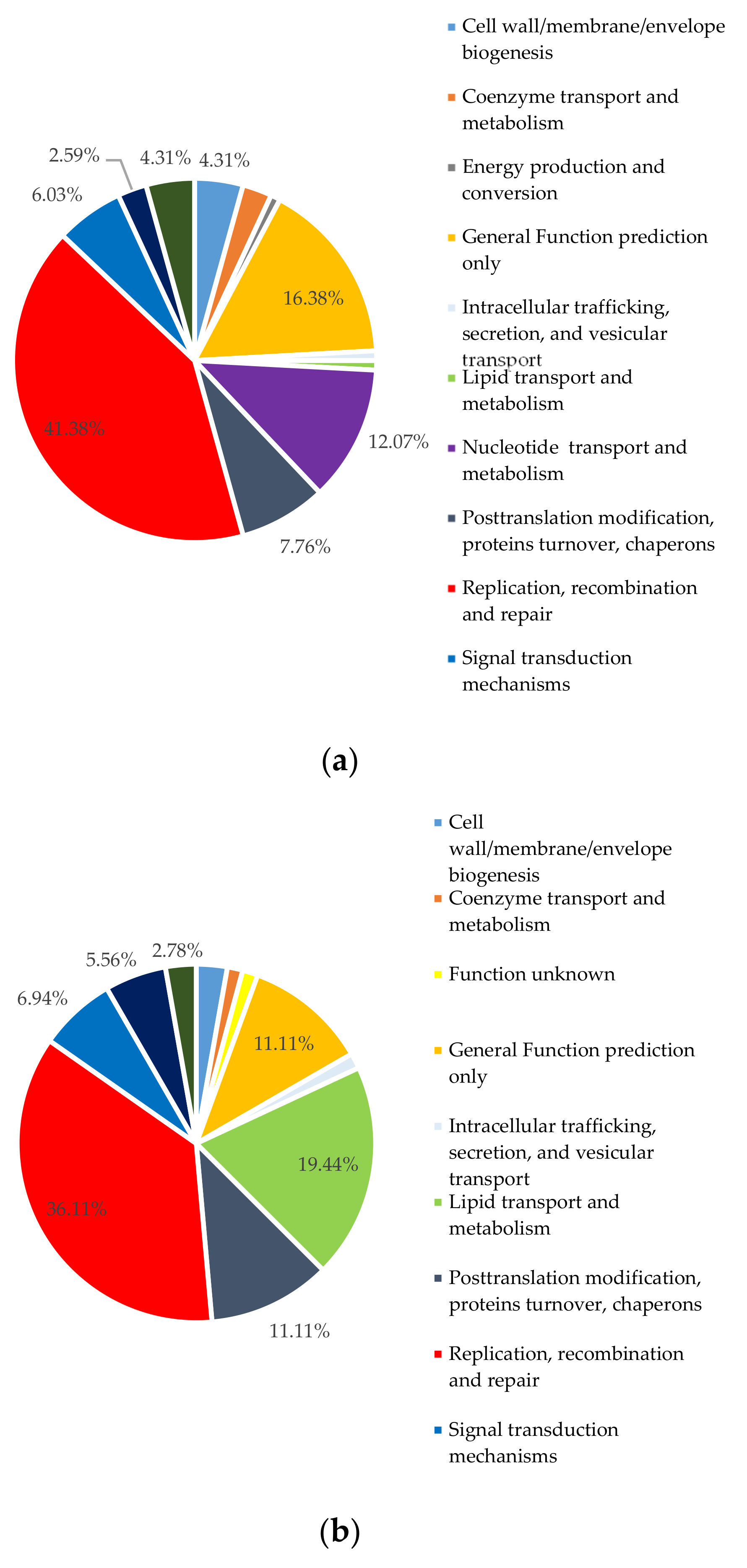
2.5.4. Putative Horizontally Transferred Genes
2.6. Identification of Peptidoglycan Hydrolase-Encoding Genes
3. Discussion
3.1. Microbial Diversity
3.2. Phage–Host Interactions
3.3. Phage-Derived Peptidoglycan Hydrolases
4. Materials and Methods
4.1. Sample Collection
4.2. Sample Processing and DNA Extraction
4.3. Bioinformatics Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Suttle, C.A. Marine viruses—Major players in the global ecosystem. Nat. Rev. Genet. 2007, 5, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Rohwer, F.; Prangishvili, D.; Lindell, D. Roles of viruses in the environment. Environ. Microbiol. 2009, 11, 2771–2774. [Google Scholar] [CrossRef]
- Middelboe, M.; Brussaard, C.P.D. Marine Viruses: Key Players in Marine Ecosystems. Viruses 2017, 9, 302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pleška, M.; Lang, M.; Refardt, D.; Levin, B.R.; Guet, C.C. Phage–host population dynamics promotes prophage acquisition in bacteria with innate immunity. Nat. Ecol. Evol. 2018, 2, 359–366. [Google Scholar] [CrossRef]
- Yosef, I.; Manor, M.; Kiro, R.; Qimron, U. Temperate and lytic bacteriophages programmed to sensitize and kill antibiotic-resistant bacteria. Proc. Natl. Acad. Sci. USA 2015, 112, 7267–7272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR Provides Acquired Resistance Against Viruses in Prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Mojica, F.J.M.; Soria, E. Intervening Sequences of Regularly Spaced Prokaryotic Repeats Derive from Foreign Genetic Elements. J. Mol. Evol. 2005, 60, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Chanishvili, N. Bacteriophages as Therapeutic and Prophylactic Means: Summary of the Soviet and Post Soviet Experiences. Curr. Drug Deliv. 2016, 13, 309–323. [Google Scholar] [CrossRef]
- Harada, L.K.; Silva, E.C.; Campos, W.F.; Del Fiol, F.S.; Vila, M.; Dąbrowska, K.; Krylov, V.N.; Balcão, V.M. Biotechnological applications of bacteriophages: State of the art. Microbiol. Res. 2018, 38–58. [Google Scholar] [CrossRef] [PubMed]
- Blondal, T.; Hjorleifsdottir, S.H.; Fridjonsson, O.F.; Aevarsson, A.; Skirnisdottir, S.; Hermannsdottir, A.G.; Hreggvidsson, G.O.; Smith, A.V.; Kristjansson, J.K. Discovery and characterization of a thermostable bacteriophage RNA ligase homologous to T4 RNA ligase 1. Nucleic Acids Res. 2003, 31, 7247–7254. [Google Scholar] [CrossRef] [Green Version]
- Makeyev, E.V.; Grimes, J.M. RNA-dependent RNA polymerases of dsRNA bacteriophages. Virus Res. 2004, 101, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Born, E.V.D.; Omelchenko, M.V.; Bekkelund, A.; Leihne, V.; Koonin, E.V.; Dolja, V.V.; Falnes, P.Ø. Viral AlkB proteins repair RNA damage by oxidative demethylation. Nucleic Acids Res. 2008, 36, 5451–5461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, K.; Kassakian, S.Z.; Krynytzky, M.; DiJulio, D.; Murray, J.W. Oxic, suboxic, and anoxic conditions in the Black Sea. In The Black Sea Flood Question: Changes in Coastline, Climate, and Human Settlement; Springer: Berlin/Heidelberg, Germany, 2006; pp. 1–21. [Google Scholar]
- Zaitsev, Y.P.; Alexandrov, B.G.; Berlinsky, N.A. The Black Sea: An Oxygen-Poor Sea. Europe’s Biodiversity-Biogeographical Regions and Seas. European Environment Agency. Available online: http://www.vliz.be/en/imis?module=ref&refid=26842&printversion=1&dropIMIStitle=1 (accessed on 9 October 2020).
- Knowles, B.; Silveira, C.B.; Bailey, B.A.; Barott, K.; Cantu, V.A.; Cobián-Güemes, A.G.; Coutinho, F.H.; Dinsdale, E.A.; Felts, B.; Furby, K.A.; et al. Erratum: Corrigendum: Lytic to temperate switching of viral communities. Nat. Cell Biol. 2016, 539, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Rubio, L.; Martínez, B.; Donovan, D.M.; Rodríguez, A.; García, P. Bacteriophage virion-associated peptidoglycan hydrolases: Potential new enzybiotics. Crit. Rev. Microbiol. 2012, 39, 427–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giovannoni, S.J. SAR11 Bacteria: The Most Abundant Plankton in the Oceans. Annu. Rev. Mar. Sci. 2017, 9, 231–255. [Google Scholar] [CrossRef] [PubMed]
- Carlson, C.; Morris, R.; Parsons, R.; Treusch, A.H.; Giovannoni, S.J.; Vergin, K. Seasonal dynamics of SAR11 populations in the euphotic and mesopelagic zones of the northwestern Sargasso Sea. ISME J. 2008, 3, 283–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, J.S.; Amaral-Zettler, L.; Rich, J.J.; Luria, C.M.; Ducklow, H.W. Bacterial community segmentation facilitates the prediction of ecosystem function along the coast of the western Antarctic Peninsula. ISME J. 2017, 11, 1460–1471. [Google Scholar] [CrossRef] [Green Version]
- Delmont, T.O.; Hammar, K.M.; Ducklow, H.W.; Yager, P.L.; Post, A.F. Phaeocystis antarctica blooms strongly influence bacterial community structures in the Amundsen Sea polynya. Front. Microbiol. 2014, 5, 646. [Google Scholar] [CrossRef]
- Kudo, T.; Kobiyama, A.; Rashid, J.; Reza, S.; Yamada, Y.; Ikeda, Y.; Ikeda, D.; Mizusawa, N.; Ikeo, K.; Sato, S.; et al. Seasonal changes in the abundance of bacterial genes related to dimethylsulfoniopropionate catabolism in seawater from Ofunato Bay revealed by metagenomic analysis. Gene 2018, 665, 174–184. [Google Scholar] [CrossRef] [Green Version]
- Dang, H.; Li, T.; Chen, M.; Huang, G. Cross-Ocean Distribution of Rhodobacterales Bacteria as Primary Surface Colonizers in Temperate Coastal Marine Waters. Appl. Environ. Microbiol. 2007, 74, 52–60. [Google Scholar] [CrossRef] [Green Version]
- Gallego, V.; García, M.T.; Ventosa, A. Methylobacterium hispanicum sp. nov. and Methylobacterium aquaticum sp. nov., isolated from drinking water. Int. J. Syst. Evol. Microbiol. 2005, 55, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-W.; Nam, J.-H.; Kim, Y.-H.; Lee, K.-H.; Lee, D.-H. Bacterial communities in the initial stage of marine biofilm formation on artificial surfaces. J. Microbiol. 2008, 46, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Biers, E.J.; Sun, S.; Howard, E.C. Prokaryotic Genomes and Diversity in Surface Ocean Waters: Interrogating the Global Ocean Sampling Metagenome. Appl. Environ. Microbiol. 2009, 75, 2221–2229. [Google Scholar] [CrossRef] [Green Version]
- Bobrova, O.; Kristoffersen, J.B.; Oulas, A.; Ivanytsia, V. Metagenomic 16s rRNA investigation of microbial communities in the Black Sea estuaries in South-West of Ukraine. Acta Biochim. Pol. 2016, 63, 315–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, C.-H.; Chiang, P.-W.; Lai, H.-C.; Shiah, F.-K.; Hsu, T.-C.; Chen, Y.-L.; Wen, L.-S.; Tseng, C.-M.; Shieh, W.Y.; Saeed, I.; et al. Prokaryotic assemblages and metagenomes in pelagic zones of the South China Sea. BMC Genom. 2015, 16, 219. [Google Scholar] [CrossRef] [Green Version]
- Math, R.K.; Jin, H.M.; Kim, J.M.; Hahn, Y.; Park, W.; Madsen, E.L.; Jeon, C.O. Comparative Genomics Reveals Adaptation by Alteromonas sp. SN2 to Marine Tidal-Flat Conditions: Cold Tolerance and Aromatic Hydrocarbon Metabolism. PLoS ONE 2012, 7, e35784. [Google Scholar] [CrossRef]
- Duhaime, M.B.; Wichels, A.; Sullivan, M.B. Six Pseudoalteromonas Strains Isolated from Surface Waters of Kabeltonne, Offshore Helgoland, North Sea. Genome Announc. 2016, 4, e01697-15. [Google Scholar] [CrossRef] [Green Version]
- Kubryakov, A.A.; Mikaelyan, A.S.; Stanichny, S.V. Summer and winter coccolithophore blooms in the Black Sea and their impact on production of dissolved organic matter from Bio-Argo data. J. Mar. Syst. 2019, 199, 103220. [Google Scholar] [CrossRef]
- Yoon, J.-H.; Kim, I.-G.; Oh, T.-K. Acinetobacter marinus sp. nov. and Acinetobacter seohaensis sp. nov., isolated from sea water of the Yellow Sea in Korea. J. Microbiol. Biotechnol. 2007, 17, 1743–1750. [Google Scholar]
- Doughari, H.J.; Ndakidemi, P.A.; Human, I.S.; Benade, S. The Ecology, Biology and Pathogenesis of Acinetobacter spp.: An Overview. Microbes Environ. 2011, 26, 101–112. [Google Scholar] [CrossRef] [Green Version]
- Nair, A.V.; Joseph, N.; Krishna, K.; Sneha, K.G.; Tom, N.; Jangid, K.; Nair, S. A comparative study of coastal and clinical isolates of Pseudomonas aeruginosa. Braz. J. Microbiol. 2015, 46, 725–734. [Google Scholar] [CrossRef] [Green Version]
- Allgaier, M.; Grossart, H.-P. Diversity and Seasonal Dynamics of Actinobacteria Populations in Four Lakes in Northeastern Germany. Appl. Environ. Microbiol. 2006, 72, 3489–3497. [Google Scholar] [CrossRef] [Green Version]
- Ibrahimi, M.; Korichi, W.; Hafidi, M.; Lemee, L.; Ouhdouch, Y.; Loqman, S. Marine Actinobacteria: Screening for Predation Leads to the Discovery of Potential New Drugs against Multidrug-Resistant Bacteria. Antibiotics 2020, 9, 91. [Google Scholar] [CrossRef] [Green Version]
- Bentley, S.D.; Chater, K.F.; Cerdeño-Tárraga, A.-M.; Challis, G.L.; Thomson, N.R.; James, K.D.; Harris, D.E.; Quail, M.; Kieser, H.M.; Harper, D.P.; et al. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nat. Cell Biol. 2002, 417, 141–147. [Google Scholar] [CrossRef]
- Hashish, E.; Merwad, A.; Elgaml, S.; Amer, A.; Kamal, H.; Elsadek, A.; Marei, A.; Sitohy, M. Mycobacterium marinum infection in fish and man: Epidemiology, pathophysiology and management; a review. Vet. Q. 2018, 38, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Flombaum, P.; Gallegos, J.L.; Gordillo, R.A.; Rincón, J.; Zabala, L.L.; Jiao, N.; Karl, D.M.; Li, W.K.W.; Lomas, M.W.; Veneziano, D.; et al. Present and future global distributions of the marine Cyanobacteria Prochlorococcus and Synechococcus. Proc. Natl. Acad. Sci. USA 2013, 110, 9824–9829. [Google Scholar] [CrossRef] [Green Version]
- Sohm, J.; Ahlgren, N.; Thomson, Z.J.; Williams, C.; Moffett, J.W.; Saito, M.; Webb, E.A.; Rocap, G. Co-occurring Synechococcus ecotypes occupy four major oceanic regimes defined by temperature, macronutrients and iron. ISME J. 2015, 10, 333–345. [Google Scholar] [CrossRef] [Green Version]
- Affe, H.M.D.J.; Rigonato, J.; Nunes, J.M.D.C.; Menezes, M. Metagenomic Analysis of Cyanobacteria in an Oligotrophic Tropical Estuary, South Atlantic. Front. Microbiol. 2018, 9, 1393. [Google Scholar] [CrossRef] [PubMed]
- Chiang, K.-P.; Kuo, M.-C.; Chang, J.; Wang, R.-H.; Gong, G.-C. Spatial and temporal variation of the Synechococcus population in the East China Sea and its contribution to phytoplankton biomass. Cont. Shelf Res. 2002, 22, 3–13. [Google Scholar] [CrossRef]
- Weinbauer, M.G. Ecology of prokaryotic viruses. FEMS Microbiol. Rev. 2004, 28, 127–181. [Google Scholar] [CrossRef] [Green Version]
- Winter, C.; Garcia, J.A.L.; Weinbauer, M.G.; Dubow, M.S.; Herndl, G.J. Comparison of Deep-Water Viromes from the Atlantic Ocean and the Mediterranean Sea. PLoS ONE 2014, 9, e100600. [Google Scholar] [CrossRef] [Green Version]
- Mann, N.H.; Clokie, M.R.J.; Millard, A.; Cook, A.; Wilson, W.H.; Wheatley, P.J.; Letarov, A.; Krisch, H.M. The Genome of S-PM2, a “Photosynthetic” T4-Type Bacteriophage That Infects Marine Synechococcus Strains. J. Bacteriol. 2005, 187, 3188–3200. [Google Scholar] [CrossRef] [Green Version]
- Weigele, P.R.; Pope, W.H.; Pedulla, M.L.; Houtz, J.M.; Smith, A.L.; Conway, J.F.; King, J.; Hatfull, G.F.; Lawrence, J.G.; Hendrix, R.W. Genomic and structural analysis of Syn9, a cyanophage infecting marine Prochlorococcus and Synechococcus. Environ. Microbiol. 2007, 9, 1675–1695. [Google Scholar] [CrossRef]
- Zhao, Y.; Temperton, B.; Thrash, J.C.; Schwalbach, M.S.; Vergin, K.L.; Landry, Z.C.; Ellisman, M.H.; Deerinck, T.J.; Sullivan, M.B.; Giovannoni, S.J. Abundant SAR11 viruses in the ocean. Nat. Cell Biol. 2013, 494, 357–360. [Google Scholar] [CrossRef]
- Antunes, A.; Alam, I.; Simões, M.F.; Daniels, C.A.; Ferreira, A.J.; Siam, R.; El-Dorry, H.; Bajic, V.B. First Insights into the Viral Communities of the Deep-sea Anoxic Brines of the Red Sea. Genom. Proteom. Bioinform. 2015, 13, 304–309. [Google Scholar] [CrossRef] [Green Version]
- Jasna, V.; Ammini, P.; Dash, A. Genetic and functional diversity of double-stranded DNA viruses in a tropical monsoonal estuary, India. Sci. Rep. 2018, 8, 16036. [Google Scholar] [CrossRef]
- Janelidze, N.; Jaiani, E.; Lashkhi, N.; Tskhvediani, A.; Kokashvili, T.; Gvarishvili, T.; Jgenti, D.; Mikashavidze, E.; Diasamidze, R.; Narodny, S.; et al. Microbial water quality of the Georgian coastal zone of the Black Sea. Mar. Pollut. Bull. 2011, 62, 573–580. [Google Scholar] [CrossRef]
- Paul, J.H. Prophages in marine bacteria: Dangerous molecular time bombs or the key to survival in the seas? ISME J. 2008, 2, 579–589. [Google Scholar] [CrossRef]
- McDaniel, L.; Houchin, L.A.; Williamson, S.J.; Paul, J.H. Lysogeny in marine Synechococcus. Nat. Cell Biol. 2002, 415, 496. [Google Scholar] [CrossRef]
- Ortmann, A.; Lawrence, J.; Suttle, C. Lysogeny and Lytic Viral Production during a Bloom of the Cyanobacterium Synechococcus spp. Microb. Ecol. 2002, 43, 225–231. [Google Scholar] [CrossRef]
- Zhao, Y.; Qin, F.; Zhang, R.; Giovannoni, S.J.; Zhang, Z.; Sun, J.; Du, S.; Rensing, C. Pelagiphages in the Podoviridae family integrate into host genomes. Environ. Microbiol. 2018, 21, 1989–2001. [Google Scholar] [CrossRef]
- Zaremba-Niedzwiedzka, K.; Viklund, J.; Zhao, W.; Ast, J.; Sczyrba, A.; Woyke, T.; McMahon, K.D.; Bertilsson, S.; Stepanauskas, R.; Andersson, S.G. Single-cell genomics reveal low recombination frequencies in freshwater bacteria of the SAR11 clade. Genome Biol. 2013, 14, R130. [Google Scholar] [CrossRef]
- Seed, K.D.; Lazinski, D.W.; Calderwood, S.B.; Camilli, A. A bacteriophage encodes its own CRISPR/Cas adaptive response to evade host innate immunity. Nat. Cell Biol. 2013, 494, 489–491. [Google Scholar] [CrossRef] [PubMed]
- Pausch, P.; Al-Shayeb, B.; Bisom-Rapp, E. Crispr-casf from huge phages is a hypercompact genome editor. Science 2020, 369, 333–337. [Google Scholar] [CrossRef]
- Jiang, S.C.; Paul, J.H. Gene Transfer by Transduction in the Marine Environment. Appl. Environ. Microbiol. 1998, 64, 2780–2787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koonin, E.V.; Makarova, K.S.; Aravind, L. Horizontal Gene Transfer in Prokaryotes: Quantification and Classification. Annu. Rev. Microbiol. 2001, 55, 709–742. [Google Scholar] [CrossRef]
- Sano, E.; Carlson, S.; Wegley, L.; Rohwer, F. Movement of Viruses between Biomes. Appl. Environ. Microbiol. 2004, 70, 5842–5846. [Google Scholar] [CrossRef] [Green Version]
- Majkowska-Skrobek, G.; Maciejewska, B. Bacteriophages and Phage-Derived Proteins–Application Approaches. Curr. Med. Chem. 2015, 22, 1757–1773. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Furuhata, K.; Fukushima, T.; Yamamoto, H.; Sekiguchi, J. Characterization of a new Bacillus subtilis peptidoglycan hydrolase gene, yvcE (named cwlO), and the enzymatic properties of its encoded protein. J. Biosci. Bioeng. 2004, 98, 174–181. [Google Scholar] [CrossRef]
- John, S.G.; Mendez, C.B.; Deng, L.; Poulos, B.; Kauffman, A.K.M.; Kern, S.; Brum, J.; Polz, M.F.; Boyle, E.A.; Sullivan, M.B. A Simple and Efficient Method for Concentration of Ocean Viruses by Chemical Flocculation. Environ. Microbiol. Rep. 2011, 3, 195–202. [Google Scholar] [CrossRef] [Green Version]
- Chen, I.-M.; Chu, K.; Palaniappan, K.; Pillay, M.; Ratner, A.; Huang, J.; Huntemann, M.; Varghese, N.; White, J.R.; Seshadri, R.; et al. IMG/M v.5.0: An integrated data management and comparative analysis system for microbial genomes and microbiomes. Nucleic Acids Res. 2019, 47, D666–D677. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [Green Version]
- Li, P.-E.; Lo, C.-C.; Anderson, J.J.; Davenport, K.W.; Bishop-Lilly, K.A.; Xu, Y.; Ahmed, S.; Feng, S.; Mokashi, V.P.; Chain, P.S. Enabling the democratization of the genomics revolution with a fully integrated web-based bioinformatics platform. Nucleic Acids Res. 2016, 45, 67–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, D.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [Green Version]
- Meyer, F.; Paarmann, D.; Souza, M.D.; Olson, R.; Glass, E.M.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A.; et al. The metagenomics RAST server—A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 2008, 9, 386. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sampling Sites and Time | T °C | pH | Salinity % | Location |
|---|---|---|---|---|
| Gonio, May 2018 | 14 | 8.0 | 17.2 | 41°36′29.9″ N 41°32′41.8″ E |
| Gonio, September 2018 | 20 | 8.1 | 17.5 | 41°36′29.9″ N 41°32′41.8″ E |
| Poti, May 2018 | 13 | 8.1 | 17.7 | 42°08′59.5″ N 41°38′16.1″ E |
| Poti, September 2018 | 20 | 7.9 | 17.2 | 42°08′59.5″ N 41°38′16.1″ E |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaiani, E.; Kusradze, I.; Kokashvili, T.; Geliashvili, N.; Janelidze, N.; Kotorashvili, A.; Kotaria, N.; Guchmanidze, A.; Tediashvili, M.; Prangishvili, D. Microbial Diversity and Phage–Host Interactions in the Georgian Coastal Area of the Black Sea Revealed by Whole Genome Metagenomic Sequencing. Mar. Drugs 2020, 18, 558. https://doi.org/10.3390/md18110558
Jaiani E, Kusradze I, Kokashvili T, Geliashvili N, Janelidze N, Kotorashvili A, Kotaria N, Guchmanidze A, Tediashvili M, Prangishvili D. Microbial Diversity and Phage–Host Interactions in the Georgian Coastal Area of the Black Sea Revealed by Whole Genome Metagenomic Sequencing. Marine Drugs. 2020; 18(11):558. https://doi.org/10.3390/md18110558
Chicago/Turabian StyleJaiani, Ekaterine, Ia Kusradze, Tamar Kokashvili, Natia Geliashvili, Nino Janelidze, Adam Kotorashvili, Nato Kotaria, Archil Guchmanidze, Marina Tediashvili, and David Prangishvili. 2020. "Microbial Diversity and Phage–Host Interactions in the Georgian Coastal Area of the Black Sea Revealed by Whole Genome Metagenomic Sequencing" Marine Drugs 18, no. 11: 558. https://doi.org/10.3390/md18110558
APA StyleJaiani, E., Kusradze, I., Kokashvili, T., Geliashvili, N., Janelidze, N., Kotorashvili, A., Kotaria, N., Guchmanidze, A., Tediashvili, M., & Prangishvili, D. (2020). Microbial Diversity and Phage–Host Interactions in the Georgian Coastal Area of the Black Sea Revealed by Whole Genome Metagenomic Sequencing. Marine Drugs, 18(11), 558. https://doi.org/10.3390/md18110558