Marine Anticancer Agents: An Overview with a Particular Focus on Their Chemical Classes

 ,
,  ,
,  ,
,  ,
, 

 ,
,  ,
,  and
and 
Abstract
:1. Introduction
Marine Environnent
2. Natural Product Classes of Marine-Derived Anticancer Agents
2.1. Nucleoside Derivatives
2.2. Macrolide Derivatives
2.3. Peptide Derivatives
2.4. Alkaloids
2.5. β-Lactones
2.6. Polysaccharides
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Debbab, A.; Aly, A.H.; Lin, W.H.; Proksch, P. Bioactive compounds from marine bacteria and fungi. Microb. Biotechnol. 2010, 3, 544–563. [Google Scholar] [CrossRef] [Green Version]
- Hill, R.T.; Fenical, W. Pharmaceuticals from marine natural products: Surge or ebb? Curr. Opin. Biotechnol. 2010, 21, 777–779. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, P.C.; Wilke, D.V.; Branco, P.C.; Bauermeister, A.; Rezende-Teixeira, P.; Gaudencio, S.P.; Costa-Lotufo, L.V. Enriching cancer pharmacology with drugs of marine origin. Br. J. Pharmacol. 2020, 177, 3–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Proksch, P.; Putz, A.; Ortlepp, S.; Kjer, J.; Bayer, M. Bioactive natural products from marine sponges and fungal endophytes. Phytochem. Rev. 2010, 9, 475–489. [Google Scholar] [CrossRef]
- Rotter, A.; Bacu, A.; Barbier, M.; Bertoni, F.; Bones, A.M.; Cancela, M.L.; Carlsson, J.; Carvalho, M.F.; Cegłowska, M.; Dalay, M.C.; et al. A New Network for the Advancement of Marine Biotechnology in Europe and Beyond. Front. Mar. Sci. 2020, 7. [Google Scholar] [CrossRef]
- Gerwick, W.H.; Moore, B.S. Lessons from the past and charting the future of marine natural products drug discovery and chemical biology. Chem. Biol. 2012, 19, 85–98. [Google Scholar] [CrossRef] [Green Version]
- Reen, F.J.; Gutiérrez-Barranquero, J.A.; Dobson, A.D.; Adams, C.; O’Gara, F. Emerging concepts promising new horizons for marine biodiscovery and synthetic biology. Mar. Drugs 2015, 13, 2924–2954. [Google Scholar] [CrossRef] [Green Version]
- Jacobo-Velazquez, D.A.; Gonzalez-Aguero, M.; Cisneros-Zevallos, L. Cross-talk between signaling pathways: The link between plant secondary metabolite production and wounding stress response. Sci. Rep. 2015, 5, 8608. [Google Scholar] [CrossRef] [Green Version]
- Parmentier, E.; Michel, L. Boundary lines in symbiosis forms. Symbiosis 2013, 60, 1–5. [Google Scholar] [CrossRef]
- Tashiro, Y.; Yawata, Y.; Toyofuku, M.; Uchiyama, H.; Nomura, N. Interspecies interaction between Pseudomonas aeruginosa and other microorganisms. Microbes Environ. 2013, 28, 13–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abisado, R.G.; Benomar, S.; Klaus, J.R.; Dandekar, A.A.; Chandler, J.R. Bacterial Quorum Sensing and Microbial Community Interactions. MBio 2018, 9, e02331-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyc, O.; de Jager, V.C.L.; van den Berg, M.; Gerards, S.; Janssens, T.K.S.; Zaagman, N.; Kai, M.; Svatos, A.; Zweers, H.; Hordijk, C.; et al. Exploring bacterial interspecific interactions for discovery of novel antimicrobial compounds. Microb. Biotechnol. 2017, 10, 910–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, D.J.; Cragg, G.M.; Snader, K.M. The influence of natural products upon drug discovery. Nat. Prod. Rep. 2000, 17, 215–234. [Google Scholar] [CrossRef] [Green Version]
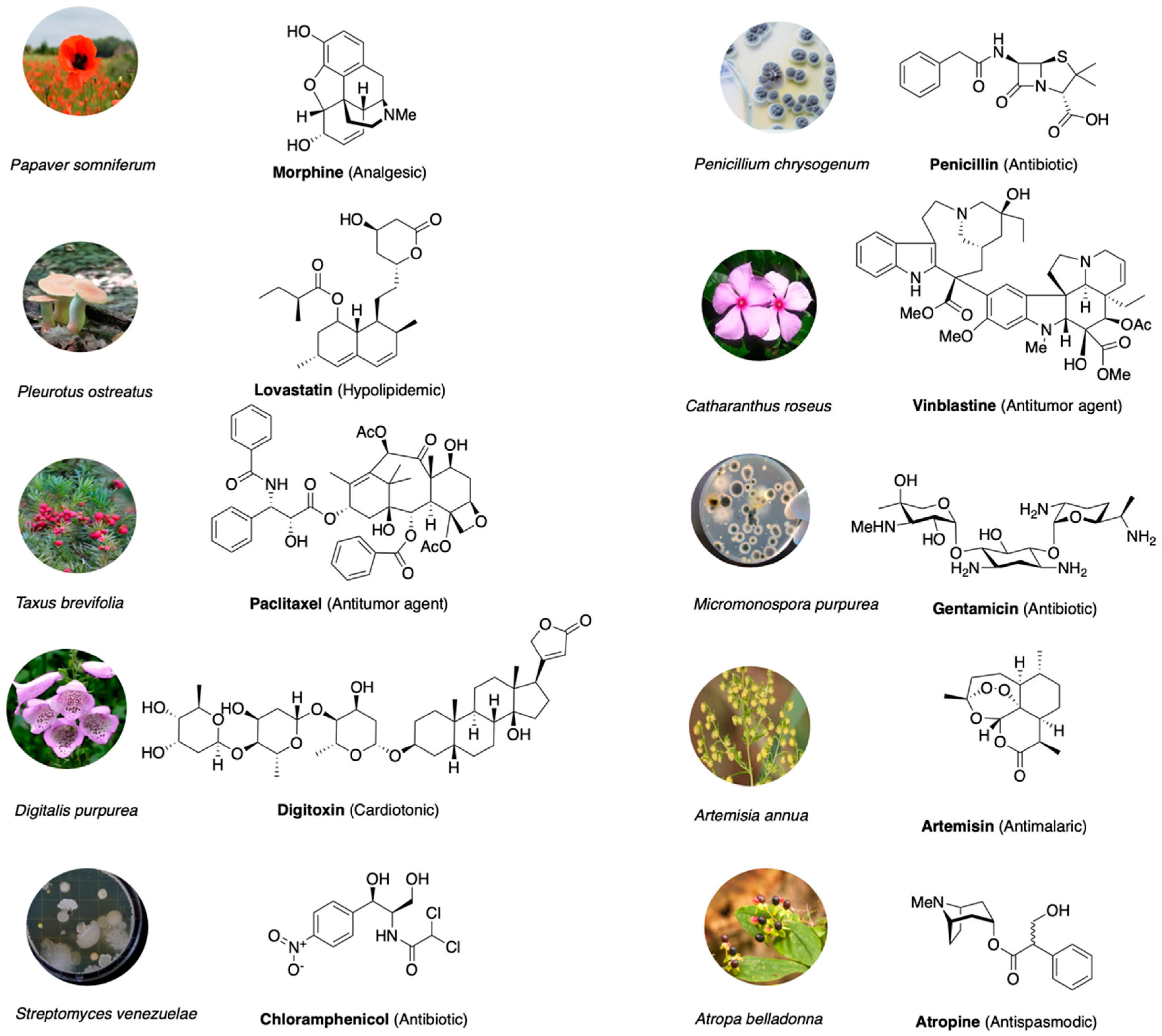
- Vane, J.R.; Botting, R.M. Mechanism of action of nonsteroidal anti-inflammatory drugs. Am. J. Med. 1998, 104, 2S–8S; discussion 21S–22S. [Google Scholar] [CrossRef]
- Krishnamurti, C.; Rao, S.C. The isolation of morphine by Serturner. Indian J. Anaesth. 2016, 60, 861–862. [Google Scholar] [CrossRef]
- Inturrisi, C.E. Clinical pharmacology of opioids for pain. Clin. J. Pain 2002, 18, S3–S13. [Google Scholar] [CrossRef]
- Elbaz, H.A.; Stueckle, T.A.; Tse, W.; Rojanasakul, Y.; Dinu, C.Z. Digitoxin and its analogs as novel cancer therapeutics. Exp. Hematol. Oncol. 2012, 1, 4. [Google Scholar] [CrossRef] [Green Version]
- Altamirano, J.; Li, Y.; DeSantiago, J.; Piacentino, V., 3rd; Houser, S.R.; Bers, D.M. The inotropic effect of cardioactive glycosides in ventricular myocytes requires Na+-Ca2+ exchanger function. J. Physiol. 2006, 575 Pt 3, 845–854. [Google Scholar] [CrossRef]
- Awan, A.R.; Shaw, W.M.; Ellis, T. Biosynthesis of therapeutic natural products using synthetic biology. Adv. Drug Deliv. Rev. 2016, 105 (Pt A), 96–106. [Google Scholar] [CrossRef]
- Lima, L.M.; Silva, B.; Barbosa, G.; Barreiro, E.J. β-lactam antibiotics: An overview from a medicinal chemistry perspective. Eur. J. Med. Chem. 2020, 208, 112829. [Google Scholar] [CrossRef] [PubMed]
- Barreca, M.; Stathis, A.; Barraja, P.; Bertoni, F. An overview on anti-tubulin agents for the treatment of lymphoma patients. Pharmacol. Ther. 2020, 211, 107552. [Google Scholar] [CrossRef] [PubMed]
- Oberlies, N.H.; Kroll, D.J. Camptothecin and taxol: Historic achievements in natural products research. J. Nat. Prod. 2004, 67, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.F.; Desai, S.D.; Li, T.K.; Mao, Y.; Sun, M.; Sim, S.P. Mechanism of action of camptothecin. Ann. N. Y. Acad. Sci. 2000, 922, 1–10. [Google Scholar] [CrossRef]
- Butler, M.S. Natural products to drugs: Natural product-derived compounds in clinical trials. Nat. Prod. Rep. 2008, 25, 475–516. [Google Scholar] [CrossRef]
- Cragg, G.M.; Grothaus, P.G.; Newman, D.J. Impact of natural products on developing new anti-cancer agents. Chem. Rev. 2009, 109, 3012–3043. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [Green Version]
- Chandra Mohana, N.; Yashavantha Rao, H.C.; Rakshith, D.; Mithun, P.R.; Nuthan, B.R.; Satish, S. Omics based approach for biodiscovery of microbial natural products in antibiotic resistance era. J. Genet. Eng. Biotechnol. 2018, 16, 1–8. [Google Scholar] [CrossRef]
- Wright, G.D. Unlocking the potential of natural products in drug discovery. Microb. Biotechnol. 2019, 12, 55–57. [Google Scholar] [CrossRef] [Green Version]
- Simmons, T.L.; Andrianasolo, E.; McPhail, K.; Flatt, P.; Gerwick, W.H. Marine natural products as anticancer drugs. Mol. Cancer Ther. 2005, 4, 333–342. [Google Scholar]
- Wagner, C.; El Omari, M.; König, G.M. Biohalogenation: Nature’s Way to Synthesize Halogenated Metabolites. J. Nat. Prod. 2009, 72, 540–553. [Google Scholar] [CrossRef] [PubMed]
- Leal, J.F.; Martínez-Díez, M.; García-Hernández, V.; Moneo, V.; Domingo, A.; Bueren-Calabuig, J.A.; Negri, A.; Gago, F.; Guillén-Navarro, M.J.; Avilés, P.; et al. PM01183, a new DNA minor groove covalent binder with potent in vitro and in vivo anti-tumour activity. Br. J. Pharmacol. 2010, 161, 1099–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, A.E.; Forleo, D.A.; Gunawardana, G.P.; Gunasekera, S.P.; Koehn, F.E.; McConnell, O.J. Antitumor tetrahydroisoquinoline alkaloids from the colonial ascidian Ecteinascidia turbinata. J. Org. Chem. 1990, 55, 4508–4512. [Google Scholar] [CrossRef]
- Fabbro, D.; Ruetz, S.; Bodis, S.; Pruschy, M.; Csermak, K.; Man, A.; Campochiaro, P.; Wood, J.; O’Reilly, T.; Meyer, T. PKC412—A protein kinase inhibitor with a broad therapeutic potential. Anticancer Drug Des. 2000, 15, 17–28. [Google Scholar]
- Urdiales, J.L.; Morata, P.; De Castro, I.N.; Sanchez-Jimenez, F. Antiproliferative effect of dehydrodidemnin B (DDB), a depsipeptide isolated from Mediterranean tunicates. Cancer Lett. 1996, 102, 31–37. [Google Scholar] [CrossRef]
- Tai, Y.T.; Mayes, P.A.; Acharya, C.; Zhong, M.Y.; Cea, M.; Cagnetta, A.; Craigen, J.; Yates, J.; Gliddon, L.; Fieles, W.; et al. Novel anti-B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood 2014, 123, 3128–3138. [Google Scholar] [CrossRef]
- Challita-Eid, P.M.; Satpayev, D.; Yang, P.; An, Z.; Morrison, K.; Shostak, Y.; Raitano, A.; Nadell, R.; Liu, W.; Lortie, D.R.; et al. Enfortumab Vedotin Antibody-Drug Conjugate Targeting Nectin-4 Is a Highly Potent Therapeutic Agent in Multiple Preclinical Cancer Models. Cancer Res. 2016, 76, 3003–3013. [Google Scholar] [CrossRef] [Green Version]
- Dornan, D.; Bennett, F.; Chen, Y.; Dennis, M.; Eaton, D.; Elkins, K.; French, D.; Go, M.A.; Jack, A.; Junutula, J.R.; et al. Therapeutic potential of an anti-CD79b antibody-drug conjugate, anti-CD79b-vc-MMAE, for the treatment of non-Hodgkin lymphoma. Blood 2009, 114, 2721–2729. [Google Scholar] [CrossRef]
- Francisco, J.A.; Cerveny, C.G.; Meyer, D.L.; Mixan, B.J.; Klussman, K.; Chace, D.F.; Rejniak, S.X.; Gordon, K.A.; DeBlanc, R.; Toki, B.E.; et al. cAC10-vcMMAE, an anti-CD30-monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood 2003, 102, 1458–1465. [Google Scholar] [CrossRef]
- Towle, M.J.; Salvato, K.A.; Budrow, J.; Wels, B.F.; Kuznetsov, G.; Aalfs, K.K.; Welsh, S.; Zheng, W.; Seletsky, B.M.; Palme, M.H.; et al. In Vitro and In Vivo Anticancer Activities of Synthetic Macrocyclic Ketone Analogues of Halichondrin B. Cancer Res. 2001, 61, 1013–1021. [Google Scholar]
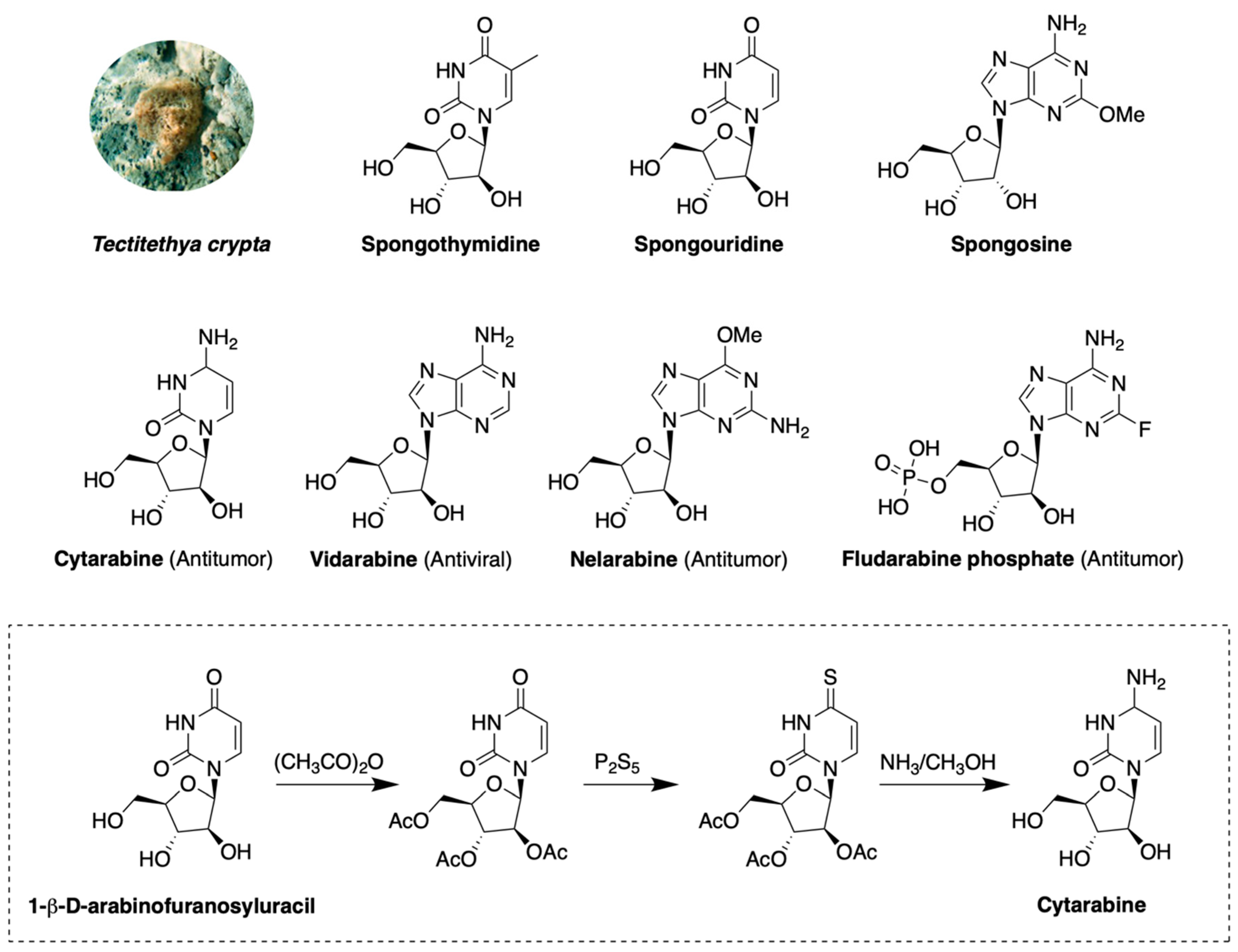
- Brockman, R.W.; Cheng, Y.-C.; Schabel, F.M.; Montgomery, J.A. Metabolism and Chemotherapeutic Activity of 9-β-D-Arabinofuranosyl-2-fluoroadenine against Murine Leukemia L1210 and Evidence for Its Phosphorylation by Deoxycytidine Kinase. Cancer Res. 1980, 40, 3610–3615. [Google Scholar] [PubMed]
- Talley, R.W.; O’Bryan, R.M.; Tucker, W.G.; Loo, R.V. Clinical pharmacology and human antitumor activity of cytosine arabinoside. Cancer 1967, 20, 809–816. [Google Scholar] [CrossRef]
- Lambe, C.U.; Averett, D.R.; Paff, M.T.; Reardon, J.E.; Wilson, J.G.; Krenitsky, T.A. 2-Amino-6-methoxypurine Arabinoside: An Agent for T-Cell Malignancies. Cancer Res. 1995, 55, 3352–3356. [Google Scholar] [PubMed]
- Park, Y.H.; Ahn, H.K.; Kim, J.-Y.; Ahn, J.S.; Im, Y.-H.; Kim, S.-H.; Lee, S.; CHUNG, H.-S.; Park, S.J. First-in-human phase I study of ALT-P7, a HER2-targeting antibody-drug conjugate in patients with HER2-positive advanced breast cancer. J. Clin. Oncol. 2020, 38, 3551. [Google Scholar] [CrossRef]
- Sandall, S.L.; Mason, M.; Olson, D.; Mazahreh, R.; Pires, T.; Sahetya, D.; Westendorf, L.; Leiske, C.; Schimpf, B.; Nguyen, L.; et al. Abstract 2688: SGN-CD228A: A novel humanized anti-CD228 antibody-drug conjugate for the treatment of solid tumors. Cancer Res. 2019, 79, 2688. [Google Scholar] [CrossRef]
- Johnson, M.L.; El-Khoueiry, A.B.; Hafez, N.; Lakhani, N.J.; Mamdani, H.; Ahnert, J.R.; Sanborn, R.E.; Ho, T.; Li, R.; Waldes, J.; et al. CX-2029, a PROBODY drug conjugate targeting CD71 (transferrin receptor): Results from a first-in-human study (PROCLAIM-CX-2029) in patients (Pts) with advanced cancer. J. Clin. Oncol. 2020, 38, 3502. [Google Scholar] [CrossRef]
- Jiang, J.; Li, S.; Shan, X.; Wang, L.; Ma, J.; Huang, M.; Dong, L.; Chen, F. Preclinical safety profile of disitamab vedotin: A novel anti-HER2 antibody conjugated with MMAE. Toxicol. Lett. 2020, 324, 30–37. [Google Scholar] [CrossRef]
- Koopman, L.A.; Terp, M.G.; Zom, G.G.; Janmaat, M.L.; Jacobsen, K.; Gresnigt-van den Heuvel, E.; Brandhorst, M.; Forssmann, U.; de Bree, F.; Pencheva, N.; et al. Enapotamab vedotin, an AXL-specific antibody-drug conjugate, shows preclinical antitumor activity in non-small cell lung cancer. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Han, H.S.; Alemany, C.A.; Brown-Glaberman, U.A.; Pluard, T.J.; Sinha, R.; Sterrenberg, D.; Albain, K.S.; Basho, R.K.; Biggs, D.; Boni, V.; et al. SGNLVA-002: Single-arm, open label phase Ib/II study of ladiratuzumab vedotin (LV) in combination with pembrolizumab for first-line treatment of patients with unresectable locally advanced or metastatic triple-negative breast cancer. J. Clin. Oncol. 2019, 37, TPS1110. [Google Scholar] [CrossRef]
- Wang, J.; Anderson, M.G.; Oleksijew, A.; Vaidya, K.S.; Boghaert, E.R.; Tucker, L.; Zhang, Q.; Han, E.K.; Palma, J.P.; Naumovski, L.; et al. ABBV-399, a c-Met Antibody-Drug Conjugate that Targets Both MET-Amplified and c-Met-Overexpressing Tumors, Irrespective of MET Pathway Dependence. Clin. Cancer Res. 2017, 23, 992–1000. [Google Scholar] [CrossRef] [Green Version]
- Theunissen, J.-W.; Cai, A.G.; Bhatti, M.M.; Cooper, A.B.; Avery, A.D.; Dorfman, R.; Guelman, S.; Levashova, Z.; Migone, T.-S. Treating Tissue Factor–Positive Cancers with Antibody–Drug Conjugates That Do Not Affect Blood Clotting. Mol. Cancer Ther. 2018, 17, 2412–2426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hingorani, D.V.; Doan, M.K.; Camargo, M.F.; Aguilera, J.; Song, S.M.; Pizzo, D.; Scanderbeg, D.J.; Cohen, E.E.W.; Lowy, A.M.; Adams, S.R.; et al. Precision Chemoradiotherapy for HER2 Tumors Using Antibody Conjugates of an Auristatin Derivative with Reduced Cell Permeability. Mol. Cancer Ther. 2020, 19, 157–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wedeh, G.; He, L.; Wittner, M.; Beghi, F.; Baral, V.; Launay, J.M.; Bibi, S.; Doñate, F.; Kouros-Mehr, H.; et al. In vitro and in vivo efficacy of an anti-CD203c conjugated antibody (AGS-16C3F) in mouse models of advanced systemic mastocytosis. Blood Adv. 2019, 3, 633–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, A.C.; Boghaert, E.R.; Vaidya, K.S.; Falls, H.D.; Mitten, M.J.; DeVries, P.J.; Benatuil, L.; Hsieh, C.M.; Meulbroek, J.A.; Panchal, S.C.; et al. Characterization of ABBV-221, a Tumor-Selective EGFR-Targeting Antibody Drug Conjugate. Mol. Cancer Ther. 2018, 17, 795–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grosso, C.; Valentao, P.; Ferreres, F.; Andrade, P.B. Bioactive marine drugs and marine biomaterials for brain diseases. Mar. Drugs 2014, 12, 2539–2589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cario, A.; Oliver, G.C.; Rogers, K.L. Exploring the Deep Marine Biosphere: Challenges, Innovations, and Opportunities. Front. Earth Sci. 2019, 7. [Google Scholar] [CrossRef]
- Malakoff, D. ECOLOGY: Extinction on the High Seas. Science 1997, 277, 486–488. [Google Scholar] [CrossRef]
- Boeuf, G. Marine biodiversity characteristics. Comptes Rendus Biol. 2011, 334, 435–440. [Google Scholar] [CrossRef]
- Peng, J.; Zhang, X.Y.; Tu, Z.C.; Xu, X.Y.; Qi, S.H. Alkaloids from the deep-sea-derived fungus Aspergillus westerdijkiae DFFSCS013. J. Nat. Prod. 2013, 76, 983–987. [Google Scholar] [CrossRef]
- Lindequist, U. Marine-Derived Pharmaceuticals—Challenges and Opportunities. Biomol. Ther. 2016, 24, 561–571. [Google Scholar] [CrossRef] [Green Version]
- Cowan, D.A. The marine biosphere: A global resource for biotechnology. Trends Biotechnol. 1997, 15, 129–131. [Google Scholar] [CrossRef]
- Tyler, P.A. Ecosystems of the Deep Oceans; Elsevier Science: Amsterdam, The Netherlands, 2003; Volume 28. [Google Scholar]
- Glud, R.N.; Wenzhöfer, F.; Middelboe, M.; Oguri, K.; Turnewitsch, R.; Canfield, D.E.; Kitazato, H. High rates of microbial carbon turnover in sediments in the deepest oceanic trench on Earth. Nat. Geosci. 2013, 6, 284–288. [Google Scholar] [CrossRef]
- Le Bris, N.; Sarradin, P.-M.; Pennec, S. A new deep-sea probe for in situ pH measurement in the environment of hydrothermal vent biological communities. Deep Sea Res. Part I Oceanogr. Res. Pap. 2001, 48, 1941–1951. [Google Scholar] [CrossRef]
- Li, G.; Lou, H.X. Strategies to diversify natural products for drug discovery. Med. Res. Rev. 2018, 38, 1255–1294. [Google Scholar] [CrossRef] [PubMed]
- Skropeta, D.; Wei, L. Recent advances in deep-sea natural products. Nat. Prod. Rep. 2014, 31, 999–1025. [Google Scholar] [CrossRef]
- Bergmann, W.; Feeney, R.J. The Isolation of a New Thymine Pentoside from Sponges1. J. Am. Chem. Soc. 1950, 72, 2809–2810. [Google Scholar] [CrossRef]
- Bergmann, W.; Feeney, R.J. Contributions to the Study of Marine Products. Xxxii. The Nucleosides of Sponges. I.1. J. Org. Chem. 1951, 16, 981–987. [Google Scholar] [CrossRef]
- Cimino, G.; De Rosa, S.; De Stefano, S. Antiviral agents from a gorgonian, Eunicella cavolini. Experientia 1984, 40, 339–340. [Google Scholar] [CrossRef]
- Nicholson, B.; Lloyd, G.K.; Miller, B.R.; Palladino, M.A.; Kiso, Y.; Hayashi, Y.; Neuteboom, S.T. NPI-2358 is a tubulin-depolymerizing agent: In-vitro evidence for activity as a tumor vascular-disrupting agent. Anticancer Drugs 2006, 17, 25–31. [Google Scholar] [CrossRef]
- Feling, R.H.; Buchanan, G.O.; Mincer, T.J.; Kauffman, C.A.; Jensen, P.R.; Fenical, W. Salinosporamide A: A Highly Cytotoxic Proteasome Inhibitor from a Novel Microbial Source, a Marine Bacterium of the New Genus Salinospora. Angew. Chem. Int. Ed. 2003, 42, 355–357. [Google Scholar] [CrossRef]
- Hwang, P.-A.; Yan, M.-D.; Kuo, K.-L.; Phan, N.N.; Lin, Y.-C. A mechanism of low molecular weight fucoidans degraded by enzymatic and acidic hydrolysis for the prevention of UVB damage. J. Appl. Phycol. 2017, 29, 521–529. [Google Scholar] [CrossRef]
- Vardanyan, R.S.; Hruby, V.J. 30—Antineoplastics. In Synthesis of Essential Drugs; Vardanyan, R.S., Hruby, V.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2006; pp. 389–418. [Google Scholar] [CrossRef]
- Lauzon, G.J.; Paterson, A.R.; Belch, A.W. Formation of 1-beta-D-arabinofuranosylcytosine diphosphate choline in neoplastic and normal cells. Cancer Res. 1978, 38, 1730–1733. [Google Scholar] [PubMed]
- Kufe, D.W.; Munroe, D.; Herrick, D.; Egan, E.; Spriggs, D. Effects of 1-beta-D-arabinofuranosylcytosine incorporation on eukaryotic DNA template function. Mol. Pharm. 1984, 26, 128–134. [Google Scholar]
- Lamba, J.K. Genetic factors influencing cytarabine therapy. Pharmacogenomics 2009, 10, 1657–1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubeek, I.; Stam, R.W.; Peters, G.J.; Broekhuizen, R.; Meijerink, J.P.; van Wering, E.R.; Gibson, B.E.; Creutzig, U.; Zwaan, C.M.; Cloos, J.; et al. The human equilibrative nucleoside transporter 1 mediates in vitro cytarabine sensitivity in childhood acute myeloid leukaemia. Br. J. Cancer 2005, 93, 1388–1394. [Google Scholar] [CrossRef] [Green Version]
- Phuphanich, S.; Maria, B.; Braeckman, R.; Chamberlain, M. A pharmacokinetic study of intra-CSF administered encapsulated cytarabine (DepoCyt) for the treatment of neoplastic meningitis in patients with leukemia, lymphoma, or solid tumors as part of a phase III study. J. Neurooncol. 2007, 81, 201–208. [Google Scholar] [CrossRef]
- Glantz, M.J.; LaFollette, S.; Jaeckle, K.A.; Shapiro, W.; Swinnen, L.; Rozental, J.R.; Phuphanich, S.; Rogers, L.R.; Gutheil, J.C.; Batchelor, T.; et al. Randomized trial of a slow-release versus a standard formulation of cytarabine for the intrathecal treatment of lymphomatous meningitis. J. Clin. Oncol. 1999, 17, 3110–3116. [Google Scholar] [CrossRef]
- Jaeckle, K.A.; Batchelor, T.; O’Day, S.J.; Phuphanich, S.; New, P.; Lesser, G.; Cohn, A.; Gilbert, M.; Aiken, R.; Heros, D.; et al. An open label trial of sustained-release cytarabine (DepoCyt) for the intrathecal treatment of solid tumor neoplastic meningitis. J. Neurooncol. 2002, 57, 231–239. [Google Scholar] [CrossRef]
- Cohen, M.H.; Johnson, J.R.; Justice, R.; Pazdur, R. FDA drug approval summary: Nelarabine (Arranon) for the treatment of T-cell lymphoblastic leukemia/lymphoma. Oncologist 2008, 13, 709–714. [Google Scholar] [CrossRef]
- Avramis, V.I.; Plunkett, W. Metabolism and Therapeutic Efficacy of 9-ß-D-Arabinofuranosyl-2-fluoroadenine against Murine Leukemia P388. Cancer Res. 1982, 42, 2587–2591. [Google Scholar]
- Huang, P.; Chubb, S.; Plunkett, W. Termination of DNA synthesis by 9-beta-D-arabinofuranosyl-2-fluoroadenine. A mechanism for cytotoxicity. J. Biol. Chem. 1990, 265, 16617–16625. [Google Scholar] [PubMed]
- Tseng, W.C.; Derse, D.; Cheng, Y.C.; Brockman, R.W.; Bennett, L.L., Jr. In vitro biological activity of 9-beta-D-arabinofuranosyl-2-fluoroadenine and the biochemical actions of its triphosphate on DNA polymerases and ribonucleotide reductase from HeLa cells. Mol. Pharm. 1982, 21, 474–477. [Google Scholar]
- Casak, S.J.; Lemery, S.J.; Shen, Y.L.; Rothmann, M.D.; Khandelwal, A.; Zhao, H.; Davis, G.; Jarral, V.; Keegan, P.; Pazdur, R.U.S. Food and drug administration approval: Rituximab in combination with fludarabine and cyclophosphamide for the treatment of patients with chronic lymphocytic leukemia. Oncologist 2011, 16, 97–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napolitano, J.G.; Daranas, A.H.; Norte, M.; Fernandez, J.J. Marine macrolides, a promising source of antitumor compounds. Anticancer Agents Med. Chem. 2009, 9, 122–137. [Google Scholar] [CrossRef] [PubMed]
- Donoghue, M.; Lemery, S.J.; Yuan, W.; He, K.; Sridhara, R.; Shord, S.; Zhao, H.; Marathe, A.; Kotch, L.; Jee, J.; et al. Eribulin mesylate for the treatment of patients with refractory metastatic breast cancer: Use of a “physician’s choice” control arm in a randomized approval trial. Clin. Cancer Res. 2012, 18, 1496–1505. [Google Scholar] [CrossRef] [Green Version]
- Cortes, J.; O’Shaughnessy, J.; Loesch, D.; Blum, J.L.; Vahdat, L.T.; Petrakova, K.; Chollet, P.; Manikas, A.; Dieras, V.; Delozier, T.; et al. Eribulin monotherapy versus treatment of physician’s choice in patients with metastatic breast cancer (EMBRACE): A phase 3 open-label randomised study. Lancet 2011, 377, 914–923. [Google Scholar] [CrossRef]
- Osgood, C.L.; Chuk, M.K.; Theoret, M.R.; Huang, L.; He, K.; Her, L.; Keegan, P.; Pazdur, R. FDA Approval Summary: Eribulin for Patients with Unresectable or Metastatic Liposarcoma Who Have Received a Prior Anthracycline-Containing Regimen. Clin. Cancer Res. 2017, 23, 6384–6389. [Google Scholar] [CrossRef] [Green Version]
- Schöffski, P.; Chawla, S.; Maki, R.G.; Italiano, A.; Gelderblom, H.; Choy, E.; Grignani, G.; Camargo, V.; Bauer, S.; Rha, S.Y.; et al. Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: A randomised, open-label, multicentre, phase 3 trial. Lancet 2016, 387, 1629–1637. [Google Scholar] [CrossRef]
- Uemura, D.; Takahashi, K.; Yamamoto, T.; Katayama, C.; Tanaka, J.; Okumura, Y.; Hirata, Y. Norhalichondrin A: An antitumor polyether macrolide from a marine sponge. J. Am. Chem. Soc. 1985, 107, 4796–4798. [Google Scholar] [CrossRef]
- Litaudon, M.; Hart, J.B.; Blunt, J.W.; Lake, R.J.; Munro, M.H. Isohomohalichondrin B, a new antitumour polyether macrolide from the New Zealand deep-water sponge Lissodendoryx sp. Tetrahedron Lett. 1994, 35, 9435–9438. [Google Scholar] [CrossRef]
- Swami, U.; Shah, U.; Goel, S. Marine Sponge Derived Eribulin in Preclinical and Clinical Studies for Cancer. In Handbook of Anticancer Drugs from Marine Origin; Kim, S.K., Ed.; Springer International Publishing: Basel, Switzerland, 2001; pp. 59–100. [Google Scholar] [CrossRef]
- Pettit, G.R.; Herald, C.L.; Boyd, M.R.; Leet, J.E.; Dufresne, C.; Doubek, D.L.; Schmidt, J.M.; Cerny, R.L.; Hooper, J.N.; Rutzler, K.C. Isolation and structure of the cell growth inhibitory constituents from the western Pacific marine sponge Axinella sp. J. Med. Chem. 1991, 34, 3339–3340. [Google Scholar] [CrossRef] [PubMed]
- Aicher, T.D.; Buszek, K.R.; Fang, F.G.; Forsyth, C.J.; Jung, S.H.; Kishi, Y.; Matelich, M.C.; Scola, P.M.; Spero, D.M.; Yoon, S.K. Total synthesis of halichondrin B and norhalichondrin B. J. Am. Chem. Soc. 1992, 114, 3162–3164. [Google Scholar] [CrossRef]
- Jordan, M.A.; Kamath, K.; Manna, T.; Okouneva, T.; Miller, H.P.; Davis, C.; Littlefield, B.A.; Wilson, L. The primary antimitotic mechanism of action of the synthetic halichondrin E7389 is suppression of microtubule growth. Mol. Cancer Ther. 2005, 4, 1086–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.A.; Wilson, L.; Azarenko, O.; Zhu, X.; Lewis, B.M.; Littlefield, B.A.; Jordan, M.A. Eribulin binds at microtubule ends to a single site on tubulin to suppress dynamic instability. Biochemistry 2010, 49, 1331–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matias, D.; Bessa, C.; Fátima Simões, M.; Reis, C.P.; Saraiva, L.; Rijo, P. Chapter 2—Natural Products as Lead Protein Kinase C Modulators for Cancer Therapy. In Studies in Natural Products Chemistry; Atta ur, R., Ed.; Elsevier: Amsterdam, The Netherlands, 2016; Volume 50, pp. 45–79. [Google Scholar]
- Mathew, M.; Bean, K.I.; Temate-Tiagueu, Y.; Caciula, A.; Mandoiu, I.I.; Zelikovsky, A.; Lopanik, N.B. Influence of symbiont-produced bioactive natural products on holobiont fitness in the marine bryozoan, Bugula neritina via protein kinase C (PKC). Mar. Biol. 2016, 163. [Google Scholar] [CrossRef]
- Lopanik, N.B.; Targett, N.M.; Lindquist, N. Isolation of two polyketide synthase gene fragments from the uncultured microbial symbiont of the marine bryozoan Bugula neritina. Appl. Environ. Microbiol. 2006, 72, 7941–7944. [Google Scholar] [CrossRef] [Green Version]
- Lopanik, N.; Lindquist, N.; Targett, N. Potent cytotoxins produced by a microbial symbiont protect host larvae from predation. Oecologia 2004, 139, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Davidson, S.K.; Allen, S.W.; Lim, G.E.; Anderson, C.M.; Haygood, M.G. Evidence for the biosynthesis of bryostatins by the bacterial symbiont “Candidatus Endobugula sertula” of the bryozoan Bugula neritina. Appl. Environ. Microbiol. 2001, 67, 4531–4537. [Google Scholar] [CrossRef] [Green Version]
- Pettit, G.R.; Day, J.F.; Hartwell, J.L.; Wood, H.B. Antineoplastic components of marine animals. Nature 1970, 227, 962–963. [Google Scholar] [CrossRef]
- Mutter, R.; Wills, M. Chemistry and clinical biology of the bryostatins. Bioorg. Med. Chem. 2000, 8, 1841–1860. [Google Scholar] [CrossRef]
- Pettit, G.R.; Herald, C.L.; Hogan, F. Biosynthetic Products for Anticancer Drug Design and Treatment: The Bryostatins. In Anticancer Drug Development; Elsevier: Amsterdam, The Netherlands, 2002; pp. 203–235. [Google Scholar] [CrossRef]
- Hale, K.J.; Hummersone, M.G.; Manaviazar, S.; Frigerio, M. The chemistry and biology of the bryostatin antitumour macrolides. Nat. Prod. Rep. 2002, 19, 413–453. [Google Scholar] [CrossRef] [PubMed]
- Hale, K.J.; Manaviazar, S. New approaches to the total synthesis of the bryostatin antitumor macrolides. Chem. Asian J. 2010, 5, 704–754. [Google Scholar] [CrossRef]
- La Barre, S.; Kornprobst, J.-M. Outstanding Marine Molecules; Wiley: Hoboken, NJ, USA, 2014. [Google Scholar] [CrossRef]
- Ruan, B.F.; Zhu, H.L. The chemistry and biology of the bryostatins: Potential PKC inhibitors in clinical development. Curr. Med. Chem. 2012, 19, 2652–2664. [Google Scholar] [CrossRef] [PubMed]
- Kraft, A.S.; Smith, J.B.; Berkow, R.L. Bryostatin, an activator of the calcium phospholipid-dependent protein kinase, blocks phorbol ester-induced differentiation of human promyelocytic leukemia cells HL-60. Proc. Natl. Acad. Sci. USA 1986, 83, 1334–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammad, R.M.; Al-Katib, A.; Pettit, G.R.; Sensenbrenner, L.L. Successful treatment of human Waldenstrom’s macroglobulinemia with combination biological and chemotherapy agents. Cancer Res. 1994, 54, 165–168. [Google Scholar] [PubMed]
- Mohammad, R.M.; Varterasian, M.L.; Almatchy, V.P.; Hannoudi, G.N.; Pettit, G.R.; Al-Katib, A. Successful treatment of human chronic lymphocytic leukemia xenografts with combination biological agents auristatin PE and bryostatin 1. Clin. Cancer Res. 1998, 4, 1337–1343. [Google Scholar] [PubMed]
- Al-Katib, A.M.; Smith, M.R.; Kamanda, W.S.; Pettit, G.R.; Hamdan, M.; Mohamed, A.N.; Chelladurai, B.; Mohammad, R.M. Bryostatin 1 down-regulates mdr1 and potentiates vincristine cytotoxicity in diffuse large cell lymphoma xenografts. Clin. Cancer Res. 1998, 4, 1305–1314. [Google Scholar]
- Biberacher, V.; Decker, T.; Oelsner, M.; Wagner, M.; Bogner, C.; Schmidt, B.; Kreitman, R.J.; Peschel, C.; Pastan, I.; Meyer Zum Büschenfelde, C.; et al. The cytotoxicity of anti-CD22 immunotoxin is enhanced by bryostatin 1 in B-cell lymphomas through CD22 upregulation and PKC-βII depletion. Haematologica 2012, 97, 771–779. [Google Scholar] [CrossRef] [Green Version]
- Dowlati, A.; Lazarus, H.M.; Hartman, P.; Jacobberger, J.W.; Whitacre, C.; Gerson, S.L.; Ksenich, P.; Cooper, B.W.; Frisa, P.S.; Gottlieb, M.; et al. Phase I and correlative study of combination bryostatin 1 and vincristine in relapsed B-cell malignancies. Clin. Cancer Res. 2003, 9 Pt 1, 5929–5935. [Google Scholar]
- Grant, S.; Roberts, J.; Poplin, E.; Tombes, M.B.; Kyle, B.; Welch, D.; Carr, M.; Bear, H.D. Phase Ib trial of bryostatin 1 in patients with refractory malignancies. Clin. Cancer Res. 1998, 4, 611–618. [Google Scholar]
- Madhusudan, S.; Protheroe, A.; Propper, D.; Han, C.; Corrie, P.; Earl, H.; Hancock, B.; Vasey, P.; Turner, A.; Balkwill, F.; et al. A multicentre phase II trial of bryostatin-1 in patients with advanced renal cancer. Br. J. Cancer 2003, 89, 1418–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prendiville, J.; Crowther, D.; Thatcher, N.; Woll, P.J.; Fox, B.W.; McGown, A.; Testa, N.; Stern, P.; McDermott, R.; Potter, M.; et al. A phase I study of intravenous bryostatin 1 in patients with advanced cancer. Br. J. Cancer 1993, 68, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.D.; Smith, M.R.; Feldman, E.J.; Cragg, L.; Millenson, M.M.; Roboz, G.J.; Honeycutt, C.; Thune, R.; Padavic-Shaller, K.; Carter, W.H.; et al. Phase I study of bryostatin 1 and fludarabine in patients with chronic lymphocytic leukemia and indolent (non-Hodgkin’s) lymphoma. Clin. Cancer Res. 2006, 12, 5809–5816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varterasian, M.L.; Mohammad, R.M.; Shurafa, M.S.; Hulburd, K.; Pemberton, P.A.; Rodriguez, D.H.; Spadoni, V.; Eilender, D.S.; Murgo, A.; Wall, N.; et al. Phase II trial of bryostatin 1 in patients with relapsed low-grade non-Hodgkin’s lymphoma and chronic lymphocytic leukemia. Clin. Cancer Res. 2000, 6, 825–828. [Google Scholar]
- Zonder, J.A.; Shields, A.F.; Zalupski, M.; Chaplen, R.; Heilbrun, L.K.; Arlauskas, P.; Philip, P.A. A phase II trial of bryostatin 1 in the treatment of metastatic colorectal cancer. Clin. Cancer Res. 2001, 7, 38–42. [Google Scholar]
- Wender, P.A.; Hardman, C.T.; Ho, S.; Jeffreys, M.S.; Maclaren, J.K.; Quiroz, R.V.; Ryckbosch, S.M.; Shimizu, A.J.; Sloane, J.L.; Stevens, M.C. Scalable synthesis of bryostatin 1 and analogs, adjuvant leads against latent HIV. Science 2017, 358, 218–223. [Google Scholar] [CrossRef] [Green Version]
- Shaala, L.A.; Youssef, D.T.A.; Ibrahim, S.R.M.; Mohamed, G.A. Callyptide A, a new cytotoxic peptide from the Red Sea marine sponge Callyspongia species. Nat. Prod. Res. 2016, 30, 2783–2790. [Google Scholar] [CrossRef]
- Suarez-Jimenez, G.M.; Burgos-Hernandez, A.; Ezquerra-Brauer, J.M. Bioactive peptides and depsipeptides with anticancer potential: Sources from marine animals. Mar. Drugs 2012, 10, 963–986. [Google Scholar] [CrossRef]
- Malaker, A.; Ahmad, S.A.I. Therapeutic potency of anticancer peptides derived from marine organism. Int. J. Eng. Appl. Sci. 2013, 2, 83–94. [Google Scholar]
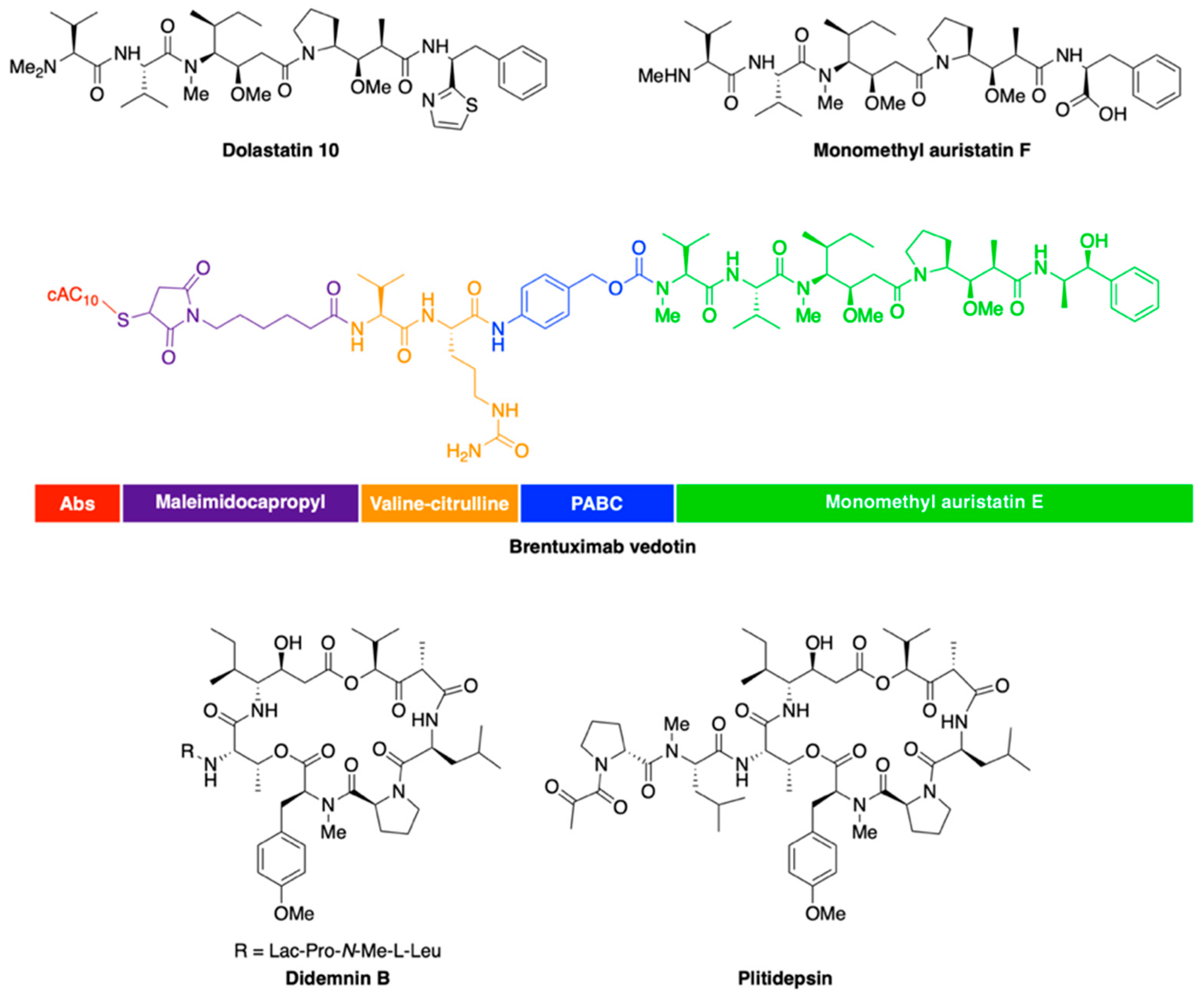
- Pettit, G.R.; Kamano, Y.; Fujii, Y.; Herald, C.L.; Inoue, M.; Brown, P.; Gust, D.; Kitahara, K.; Schmidt, J.M.; Doubek, D.L.; et al. Marine animal biosynthetic constituents for cancer chemotherapy. J. Nat. Prod. 1981, 44, 482–485. [Google Scholar] [CrossRef]
- Luesch, H.; Moore, R.E.; Paul, V.J.; Mooberry, S.L.; Corbett, T.H. Isolation of dolastatin 10 from the marine cyanobacterium Symploca species VP642 and total stereochemistry and biological evaluation of its analogue symplostatin 1. J. Nat. Prod. 2001, 64, 907–910. [Google Scholar] [CrossRef]
- Vaishampayan, U.; Glode, M.; Du, W.; Kraft, A.; Hudes, G.; Wright, J.; Hussain, M. Phase II study of dolastatin-10 in patients with hormone-refractory metastatic prostate adenocarcinoma. Clin. Cancer Res. 2000, 6, 4205–4208. [Google Scholar] [PubMed]
- Saad, E.D.; Kraut, E.H.; Hoff, P.M.; Moore, D.F., Jr.; Jones, D.; Pazdur, R.; Abbruzzese, J.L. Phase II study of dolastatin-10 as first-line treatment for advanced colorectal cancer. Am. J. Clin. Oncol. 2002, 25, 451–453. [Google Scholar] [CrossRef] [PubMed]
- Perez, E.A.; Hillman, D.W.; Fishkin, P.A.; Krook, J.E.; Tan, W.W.; Kuriakose, P.A.; Alberts, S.R.; Dakhil, S.R. Phase II trial of dolastatin-10 in patients with advanced breast cancer. Investig. New Drugs 2005, 23, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Kindler, H.L.; Tothy, P.K.; Wolff, R.; McCormack, R.A.; Abbruzzese, J.L.; Mani, S.; Wade-Oliver, K.T.; Vokes, E.E. Phase II trials of dolastatin-10 in advanced pancreaticobiliary cancers. Investig. New Drugs 2005, 23, 489–493. [Google Scholar] [CrossRef]
- Newman, D.J. The “Utility” of Highly Toxic Marine-Sourced Compounds. Mar. Drugs 2019, 17, 324. [Google Scholar] [CrossRef] [Green Version]
- Maderna, A.; Doroski, M.; Subramanyam, C.; Porte, A.; Leverett, C.A.; Vetelino, B.C.; Chen, Z.; Risley, H.; Parris, K.; Pandit, J.; et al. Discovery of cytotoxic dolastatin 10 analogues with N-terminal modifications. J. Med. Chem. 2014, 57, 10527–10543. [Google Scholar] [CrossRef]
- Ponziani, S.; Di Vittorio, G.; Pitari, G.; Cimini, A.M.; Ardini, M.; Gentile, R.; Iacobelli, S.; Sala, G.; Capone, E.; Flavell, D.J.; et al. Antibody-Drug Conjugates: The New Frontier of Chemotherapy. Int. J. Mol. Sci. 2020, 21, 5510. [Google Scholar] [CrossRef]
- Joubert, N.; Beck, A.; Dumontet, C.; Denevault-Sabourin, C. Antibody-Drug Conjugates: The Last Decade. Pharmaceuticals 2020, 13, 245. [Google Scholar] [CrossRef]
- Hafeez, U.; Parakh, S.; Gan, H.K.; Scott, A.M. Antibody-Drug Conjugates for Cancer Therapy. Molecules 2020, 25, 4764. [Google Scholar] [CrossRef]
- Dugal-Tessier, J.; Barnscher, S.D.; Kanai, A.; Mendelsohn, B.A. Synthesis and Evaluation of Dolastatin 10 Analogues Containing Heteroatoms on the Amino Acid Side Chains. J. Nat. Prod. 2017, 80, 2484–2491. [Google Scholar] [CrossRef] [PubMed]
- Doronina, S.O.; Toki, B.E.; Torgov, M.Y.; Mendelsohn, B.A.; Cerveny, C.G.; Chace, D.F.; DeBlanc, R.L.; Gearing, R.P.; Bovee, T.D.; Siegall, C.B.; et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat. Biotechnol. 2003, 21, 778–784. [Google Scholar] [CrossRef]
- Pro, B.; Advani, R.; Brice, P.; Bartlett, N.L.; Rosenblatt, J.D.; Illidge, T.; Matous, J.; Ramchandren, R.; Fanale, M.; Connors, J.M.; et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: Results of a phase II study. J. Clin. Oncol. 2012, 30, 2190–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younes, A.; Gopal, A.K.; Smith, S.E.; Ansell, S.M.; Rosenblatt, J.D.; Savage, K.J.; Ramchandren, R.; Bartlett, N.L.; Cheson, B.D.; de Vos, S.; et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin’s lymphoma. J. Clin. Oncol. 2012, 30, 2183–2189. [Google Scholar] [CrossRef] [PubMed]
- Pro, B.; Advani, R.; Brice, P.; Bartlett, N.L.; Rosenblatt, J.D.; Illidge, T.; Matous, J.; Ramchandren, R.; Fanale, M.; Connors, J.M.; et al. Five-year results of brentuximab vedotin in patients with relapsed or refractory systemic anaplastic large cell lymphoma. Blood 2017, 130, 2709–2717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connors, J.M.; Jurczak, W.; Straus, D.J.; Ansell, S.M.; Kim, W.S.; Gallamini, A.; Younes, A.; Alekseev, S.; Illes, A.; Picardi, M.; et al. Brentuximab Vedotin with Chemotherapy for Stage III or IV Hodgkin’s Lymphoma. N. Engl. J. Med. 2018, 378, 331–344. [Google Scholar] [CrossRef]
- Pfeifer, M.; Zheng, B.; Erdmann, T.; Koeppen, H.; McCord, R.; Grau, M.; Staiger, A.; Chai, A.; Sandmann, T.; Madle, H.; et al. Anti-CD22 and anti-CD79B antibody drug conjugates are active in different molecular diffuse large B-cell lymphoma subtypes. Leukemia 2015, 29, 1578–1586. [Google Scholar] [CrossRef] [PubMed]
- Tilly, H.; Morschhauser, F.; Bartlett, N.L.; Mehta, A.; Salles, G.; Haioun, C.; Munoz, J.; Chen, A.I.; Kolibaba, K.; Lu, D.; et al. Polatuzumab vedotin in combination with immunochemotherapy in patients with previously untreated diffuse large B-cell lymphoma: An open-label, non-randomised, phase 1b-2 study. Lancet Oncol. 2019, 20, 998–1010. [Google Scholar] [CrossRef]
- Sehn, L.H.; Herrera, A.F.; Flowers, C.R.; Kamdar, M.K.; McMillan, A.; Hertzberg, M.; Assouline, S.; Kim, T.M.; Kim, W.S.; Ozcan, M.; et al. Polatuzumab Vedotin in Relapsed or Refractory Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2020, 38, 155–165. [Google Scholar] [CrossRef]
- Sarfaty, M.; Rosenberg, J.E. Antibody-Drug Conjugates in Urothelial Carcinomas. Curr. Oncol. Rep. 2020, 22, 13. [Google Scholar] [CrossRef]
- Petrylak, D.P.; Balar, A.V.; O’Donnell, P.H.; McGregor, B.A.; Heath, E.I.; Yu, E.Y.; Galsky, M.D.; Hahn, N.M.; Gartner, E.M.; Pinelli, J.; et al. EV-201: Results of enfortumab vedotin monotherapy for locally advanced or metastatic urothelial cancer previously treated with platinum and immune checkpoint inhibitors. J. Clin. Oncol. 2019, 37, 4505. [Google Scholar] [CrossRef]
- Doronina, S.O.; Mendelsohn, B.A.; Bovee, T.D.; Cerveny, C.G.; Alley, S.C.; Meyer, D.L.; Oflazoglu, E.; Toki, B.E.; Sanderson, R.J.; Zabinski, R.F.; et al. Enhanced activity of monomethylauristatin F through monoclonal antibody delivery: Effects of linker technology on efficacy and toxicity. Bioconjug. Chem. 2006, 17, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Lonial, S.; Lee, H.C.; Badros, A.; Trudel, S.; Nooka, A.K.; Chari, A.; Abdallah, A.O.; Callander, N.; Lendvai, N.; Sborov, D.; et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): A two-arm, randomised, open-label, phase 2 study. Lancet Oncol. 2020, 21, 207–221. [Google Scholar] [CrossRef]
- Spicka, I.; Ocio, E.M.; Oakervee, H.E.; Greil, R.; Banh, R.H.; Huang, S.Y.; D’Rozario, J.M.; Dimopoulos, M.A.; Martinez, S.; Extremera, S.; et al. Randomized phase III study (ADMYRE) of plitidepsin in combination with dexamethasone vs. dexamethasone alone in patients with relapsed/refractory multiple myeloma. Ann. Hematol. 2019, 98, 2139–2150. [Google Scholar] [CrossRef] [Green Version]
- Leisch, M.; Egle, A.; Greil, R. Plitidepsin: A potential new treatment for relapsed/refractory multiple myeloma. Future Oncol. 2019, 15, 109–120. [Google Scholar] [CrossRef]
- Schmidt, U.; Kroner, M.; Griesser, H. Total synthesis of the didemnins—1. synthesis of the peptolide ring. Tetrahedron Lett. 1988, 29, 3057–3060. [Google Scholar] [CrossRef]
- Sakai, R.; Rinehart, K.L.; Kishore, V.; Kundu, B.; Faircloth, G.; Gloer, J.B.; Carney, J.R.; Namikoshi, M.; Sun, F.; Hughes, R.G., Jr.; et al. Structure--activity relationships of the didemnins. J. Med. Chem. 1996, 39, 2819–2834. [Google Scholar] [CrossRef]
- Andrew Dorr, F.; Kuhn, J.G.; Phillips, J.; von Hoff, D.D. Phase I clinical and pharmacokinetic investigation of didemnin B, a cyclic depsipeptide. Eur. J. Cancer Clin. Oncol. 1988, 24, 1699–1706. [Google Scholar] [CrossRef]
- Kucuk, O.; Young, M.L.; Habermann, T.M.; Wolf, B.C.; Jimeno, J.; Cassileth, P.A. Phase II trail of didemnin B in previously treated non-Hodgkin’s lymphoma: An Eastern Cooperative Oncology Group (ECOG) Study. Am. J. Clin. Oncol. 2000, 23, 273–277. [Google Scholar] [CrossRef]
- Mittelman, A.; Chun, H.G.; Puccio, C.; Coombe, N.; Lansen, T.; Ahmed, T. Phase II clinical trial of didemnin B in patients with recurrent or refractory anaplastic astrocytoma or glioblastoma multiforme (NSC 325319). Investig. New Drugs 1999, 17, 179–182. [Google Scholar] [CrossRef]
- Paniagua-Michel, J.D.J.; Olmos Soto, J.; Morales-Guerrero, E. Drugs and Leads from the Ocean Through Biotechnology. In Springer Handbook of Marine Biotechnology; Kim, S.-K., Ed.; Springer: Berlin/Heidelberg, Germany, 2015; pp. 711–729. [Google Scholar] [CrossRef]
- Losada, A.; Muñoz-Alonso, M.J.; García, C.; Sánchez-Murcia, P.A.; Martínez-Leal, J.F.; Domínguez, J.M.; Lillo, M.P.; Gago, F.; Galmarini, C.M. Translation Elongation Factor eEF1A2 is a Novel Anticancer Target for the Marine Natural Product Plitidepsin. Sci. Rep. 2016, 6, 35100. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Fernandez, L.F.; Losada, A.; Alcaide, V.; Alvarez, A.M.; Cuadrado, A.; Gonzalez, L.; Nakayama, K.; Nakayama, K.I.; Fernandez-Sousa, J.M.; Munoz, A.; et al. Aplidin induces the mitochondrial apoptotic pathway via oxidative stress-mediated JNK and p38 activation and protein kinase C delta. Oncogene 2002, 21, 7533–7544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huryn, D.M.; Wipf, P. Chapter 3—Natural Product Chemistry and Cancer Drug Discovery. In Cancer Drug Design and Discovery, 2nd ed.; Neidle, S., Ed.; Academic Press: San Diego, CA, USA, 2014; pp. 91–120. [Google Scholar] [CrossRef]
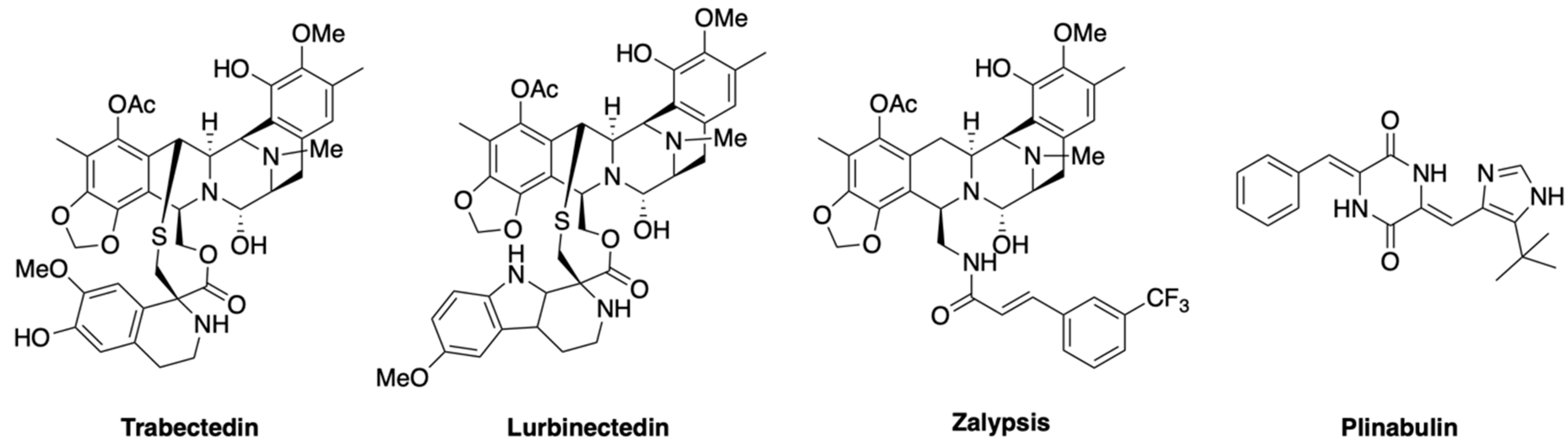
- Rath, C.M.; Janto, B.; Earl, J.; Ahmed, A.; Hu, F.Z.; Hiller, L.; Dahlgren, M.; Kreft, R.; Yu, F.; Wolff, J.J.; et al. Meta-omic characterization of the marine invertebrate microbial consortium that produces the chemotherapeutic natural product ET-743. ACS Chem. Biol. 2011, 6, 1244–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinehart, K.L. Antitumor compounds from tunicates. Med. Res. Rev. 2000, 20, 1–27. [Google Scholar] [CrossRef]
- Seaman, F.C.; Hurley, L.H. Molecular Basis for the DNA Sequence Selectivity of Ecteinascidin 736 and 743: Evidence for the Dominant Role of Direct Readout via Hydrogen Bonding. J. Am. Chem. Soc. 1998, 120, 13028–13041. [Google Scholar] [CrossRef]
- Moore, B.M.; Seaman, F.C.; Wheelhouse, R.T.; Hurley, L.H. Mechanism for the Catalytic Activation of Ecteinascidin 743 and Its Subsequent Alkylation of Guanine N2. J. Am. Chem. Soc. 1998, 120, 2490–2491. [Google Scholar] [CrossRef]
- Takebayashi, Y.; Pourquier, P.; Zimonjic, D.B.; Nakayama, K.; Emmert, S.; Ueda, T.; Urasaki, Y.; Kanzaki, A.; Akiyama, S.I.; Popescu, N.; et al. Antiproliferative activity of ecteinascidin 743 is dependent upon transcription-coupled nucleotide-excision repair. Nat. Med. 2001, 7, 961–966. [Google Scholar] [CrossRef]
- Erba, E.; Bergamaschi, D.; Bassano, L.; Damia, G.; Ronzoni, S.; Faircloth, G.T.; D’Incalci, M. Ecteinascidin-743 (ET-743), a natural marine compound, with a unique mechanism of action. Eur. J. Cancer 2001, 37, 97–105. [Google Scholar] [CrossRef]
- Cuevas, C.; Francesch, A. Development of Yondelis (trabectedin, ET-743). A semisynthetic process solves the supply problem. Nat. Prod. Rep. 2009, 26, 322–337. [Google Scholar] [CrossRef]
- Spriano, F.; Chung, E.Y.; Panini, N.; Cascione, L.; Rinaldi, A.; Erba, E.; Stathis, A.; D’Incalci, M.; Bertoni, F.; Gatta, R. Trabectedin is a novel chemotherapy agent for diffuse large B cell lymphoma. Br. J. Haematol. 2019, 184, 1022–1025. [Google Scholar] [CrossRef]
- Camorani, S.; Cerchia, L.; Fedele, M.; Erba, E.; D’Incalci, M.; Crescenzi, E. Trabectedin modulates the senescence-associated secretory phenotype and promotes cell death in senescent tumor cells by targeting NF-κB. Oncotarget 2018, 9, 19929–19944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, M.; Della Porta, M.G.; Gallì, A.; Panini, N.; Licandro, S.A.; Bello, E.; Craparotta, I.; Rosti, V.; Bonetti, E.; Tancredi, R.; et al. Antitumour activity of trabectedin in myelodysplastic/myeloproliferative neoplasms. Br. J. Cancer 2017, 116, 335–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaral, A.T.; Garofalo, C.; Frapolli, R.; Manara, M.C.; Mancarella, C.; Uboldi, S.; Di Giandomenico, S.; Ordóñez, J.L.; Sevillano, V.; Malaguarnera, R.; et al. Trabectedin efficacy in Ewing sarcoma is greatly increased by combination with anti-IGF signaling agents. Clin. Cancer Res. 2015, 21, 1373–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germano, G.; Frapolli, R.; Simone, M.; Tavecchio, M.; Erba, E.; Pesce, S.; Pasqualini, F.; Grosso, F.; Sanfilippo, R.; Casali, P.G.; et al. Antitumor and anti-inflammatory effects of trabectedin on human myxoid liposarcoma cells. Cancer Res. 2010, 70, 2235–2244. [Google Scholar] [CrossRef] [Green Version]
- Allavena, P.; Signorelli, M.; Chieppa, M.; Erba, E.; Bianchi, G.; Marchesi, F.; Olimpio, C.O.; Bonardi, C.; Garbi, A.; Lissoni, A.; et al. Anti-inflammatory properties of the novel antitumor agent yondelis (trabectedin): Inhibition of macrophage differentiation and cytokine production. Cancer Res. 2005, 65, 2964–2971. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, N.; Li, W.W.; Banerjee, D.; Scotto, K.W.; Bertino, J.R. Sequence-dependent enhancement of cytotoxicity produced by ecteinascidin 743 (ET-743) with doxorubicin or paclitaxel in soft tissue sarcoma cells. Clin. Cancer Res. 2001, 7, 3251–3257. [Google Scholar]
- Ryan, D.P.; Supko, J.G.; Eder, J.P.; Seiden, M.V.; Demetri, G.; Lynch, T.J.; Fischman, A.J.; Davis, J.; Jimeno, J.; Clark, J.W. Phase I and pharmacokinetic study of ecteinascidin 743 administered as a 72-h continuous intravenous infusion in patients with solid malignancies. Clin. Cancer Res. 2001, 7, 231–242. [Google Scholar]
- Minuzzo, M.; Marchini, S.; Broggini, M.; Faircloth, G.; D’Incalci, M.; Mantovani, R. Interference of transcriptional activation by the antineoplastic drug ecteinascidin-743. Proc. Natl. Acad. Sci. USA 2000, 97, 6780–6784. [Google Scholar] [CrossRef] [Green Version]
- García-Rocha, M.; García-Gravalos, M.D.; Avila, J. Characterisation of antimitotic products from marine organisms that disorganise the microtubule network: Ecteinascidin 743, isohomohalichondrin-B and LL-15. Br. J. Cancer 1996, 73, 875–883. [Google Scholar] [CrossRef] [Green Version]
- Izbicka, E.; Lawrence, R.; Raymond, E.; Eckhardt, G.; Faircloth, G.; Jimeno, J.; Clark, G.; Von Hoff, D.D. In vitro antitumor activity of the novel marine agent, ecteinascidin-743 (ET-743, NSC-648766) against human tumors explanted from patients. Ann. Oncol. 1998, 9, 981–987. [Google Scholar] [CrossRef]
- Demetri, G.D.; von Mehren, M.; Jones, R.L.; Hensley, M.L.; Schuetze, S.M.; Staddon, A.; Milhem, M.; Elias, A.; Ganjoo, K.; Tawbi, H.; et al. Efficacy and Safety of Trabectedin or Dacarbazine for Metastatic Liposarcoma or Leiomyosarcoma After Failure of Conventional Chemotherapy: Results of a Phase III Randomized Multicenter Clinical Trial. J. Clin. Oncol. 2016, 34, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Trigo, J.; Subbiah, V.; Besse, B.; Moreno, V.; López, R.; Sala, M.A.; Peters, S.; Ponce, S.; Fernández, C.; Alfaro, V.; et al. Lurbinectedin as second-line treatment for patients with small-cell lung cancer: A single-arm, open-label, phase 2 basket trial. Lancet Oncol. 2020, 21, 645–654. [Google Scholar] [CrossRef]
- Gaillard, S.; Oaknin, A.; Ray-Coquard, I.L.; Vergote, I.B.; Scambia, G.; Colombo, N.; Ghamande, S.A.; Soto-Matos, A.; Fernandez, C.M.; Kahatt, C.; et al. Phase III trial of lurbinectedin versus PLD or topotecan in platinum-resistant ovarian cancer patients: Results of CORAIL trial. Ann. Oncol. 2018, 29, viii332. [Google Scholar] [CrossRef]
- Santamaría Nuñez, G.; Robles, C.M.; Giraudon, C.; Martínez-Leal, J.F.; Compe, E.; Coin, F.; Aviles, P.; Galmarini, C.M.; Egly, J.M. Lurbinectedin Specifically Triggers the Degradation of Phosphorylated RNA Polymerase II and the Formation of DNA Breaks in Cancer Cells. Mol. Cancer Ther. 2016, 15, 2399–2412. [Google Scholar] [CrossRef] [Green Version]
- Galmarini, C.M.; D’Incalci, M.; Allavena, P. Trabectedin and plitidepsin: Drugs from the sea that strike the tumor microenvironment. Mar. Drugs 2014, 12, 719–733. [Google Scholar] [CrossRef] [Green Version]
- Larsen, A.K.; Galmarini, C.M.; D’Incalci, M. Unique features of trabectedin mechanism of action. Cancer Chemother. Pharm. 2016, 77, 663–671. [Google Scholar] [CrossRef]
- Belgiovine, C.; Bello, E.; Liguori, M.; Craparotta, I.; Mannarino, L.; Paracchini, L.; Beltrame, L.; Marchini, S.; Galmarini, C.M.; Mantovani, A.; et al. Lurbinectedin reduces tumour-associated macrophages and the inflammatory tumour microenvironment in preclinical models. Br. J. Cancer 2017, 117, 628–638. [Google Scholar] [CrossRef]
- Germano, G.; Frapolli, R.; Belgiovine, C.; Anselmo, A.; Pesce, S.; Liguori, M.; Erba, E.; Uboldi, S.; Zucchetti, M.; Pasqualini, F.; et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell 2013, 23, 249–262. [Google Scholar] [CrossRef] [Green Version]
- Omura, S.; Iwai, Y.; Hirano, A.; Nakagawa, A.; Awaya, J.; Tsuchya, H.; Takahashi, Y.; Masuma, R. A new alkaloid AM-2282 OF Streptomyces origin. Taxonomy, fermentation, isolation and preliminary characterization. J. Antibiot. (Tokyo) 1977, 30, 275–282. [Google Scholar] [CrossRef]
- Schupp, P.; Eder, C.; Proksch, P.; Wray, V.; Schneider, B.; Herderich, M.; Paul, V. Staurosporine Derivatives from the Ascidian Eudistoma toealensis and Its Predatory Flatworm Pseudoceros sp. J. Nat. Prod. 1999, 62, 959–962. [Google Scholar] [CrossRef]
- Schupp, P.; Proksch, P.; Wray, V. Further New Staurosporine Derivatives from the Ascidian Eudistoma toealensis and Its Predatory Flatworm Pseudoceros sp. J. Nat. Prod. 2002, 65, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S. Midostaurin: First Global Approval. Drugs 2017, 77, 1251–1259. [Google Scholar] [CrossRef] [PubMed]
- Kanoh, K.; Kohno, S.; Katada, J.; Hayashi, Y.; Muramatsu, M.; Uno, I. Antitumor activity of phenylahistin in vitro and in vivo. Biosci. Biotechnol. Biochem. 1999, 63, 1130–1133. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, A.S.; Fernandez-Rodriguez, L.; Zhao, Y.; Monaco, G.; Trefny, M.P.; Yoshida, N.; Martin, K.; Sharma, A.; Olieric, N.; Shah, P.; et al. GEF-H1 Signaling upon Microtubule Destabilization Is Required for Dendritic Cell Activation and Specific Anti-tumor Responses. Cell Rep. 2019, 28, 3367–3380.e3368. [Google Scholar] [CrossRef] [Green Version]
- Mohanlal, R.W.; LLoyd, K.; Huang, L. Plinabulin, a novel small molecule clinical stage IO agent with anti-cancer activity, to prevent chemo-induced neutropenia and immune related AEs. J. Clin. Oncol. 2018, 36, 126. [Google Scholar] [CrossRef]
- Tonra, J.R.; Lloyd, G.K.; Mohanlal, R.; Huang, L. Plinabulin ameliorates neutropenia induced by multiple chemotherapies through a mechanism distinct from G-CSF therapies. Cancer Chemother. Pharmacol. 2020, 85, 461–468. [Google Scholar] [CrossRef] [Green Version]
- López-Iglesias, A.A.; González-Méndez, L.; San-Segundo, L.; Herrero, A.B.; Hernández-García, S.; Martín-Sánchez, M.; Gutiérrez, N.C.; Paíno, T.; Avilés, P.; Mateos, M.V.; et al. Synergistic DNA-damaging effect in multiple myeloma with the combination of zalypsis, bortezomib and dexamethasone. Haematologica 2017, 102, 168–175. [Google Scholar] [CrossRef] [Green Version]
- Ocio, E.M.; Oriol, A.; Bladé, J.; Teruel, A.I.; Martín, J.; de la Rubia, J.; Gutiérrez, N.C.; Rodríguez Díaz-Pavón, J.; Martínez González, S.; Coronado, C.; et al. Phase I/II study of weekly PM00104 (Zalypsis®) in patients with relapsed/refractory multiple myeloma. Br. J. Haematol. 2016, 172, 625–628. [Google Scholar] [CrossRef]
- Capdevila, J.; Clive, S.; Casado, E.; Michie, C.; Piera, A.; Sicart, E.; Carreras, M.J.; Coronado, C.; Kahatt, C.; Soto Matos-Pita, A.; et al. A phase I pharmacokinetic study of PM00104 (Zalypsis) administered as a 24-h intravenous infusion every 3 weeks in patients with advanced solid tumors. Cancer Chemother. Pharm. 2013, 71, 1247–1254. [Google Scholar] [CrossRef]
- Yap, T.A.; Cortes-Funes, H.; Shaw, H.; Rodriguez, R.; Olmos, D.; Lal, R.; Fong, P.C.; Tan, D.S.; Harris, D.; Capdevila, J.; et al. First-in-man phase I trial of two schedules of the novel synthetic tetrahydroisoquinoline alkaloid PM00104 (Zalypsis) in patients with advanced solid tumours. Br. J. Cancer 2012, 106, 1379–1385. [Google Scholar] [CrossRef]
- Massard, C.; Margetts, J.; Amellal, N.; Drew, Y.; Bahleda, R.; Stephens, P.; Armand, J.P.; Calvert, H.; Soria, J.C.; Coronado, C.; et al. Phase I study of PM00104 (Zalypsis®) administered as a 1-h weekly infusion resting every fourth week in patients with advanced solid tumors. Investig. New Drugs 2013, 31, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Colado, E.; Paíno, T.; Maiso, P.; Ocio, E.M.; Chen, X.; Alvarez-Fernández, S.; Gutiérrez, N.C.; Martín-Sánchez, J.; Flores-Montero, J.; San Segundo, L.; et al. Zalypsis has in vitro activity in acute myeloid blasts and leukemic progenitor cells through the induction of a DNA damage response. Haematologica 2011, 96, 687–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leal, J.F.; García-Hernández, V.; Moneo, V.; Domingo, A.; Bueren-Calabuig, J.A.; Negri, A.; Gago, F.; Guillén-Navarro, M.J.; Avilés, P.; Cuevas, C.; et al. Molecular pharmacology and antitumor activity of Zalypsis in several human cancer cell lines. Biochem. Pharm. 2009, 78, 162–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ocio, E.M.; Maiso, P.; Chen, X.; Garayoa, M.; Alvarez-Fernández, S.; San-Segundo, L.; Vilanova, D.; López-Corral, L.; Montero, J.C.; Hernández-Iglesias, T.; et al. Zalypsis: A novel marine-derived compound with potent antimyeloma activity that reveals high sensitivity of malignant plasma cells to DNA double-strand breaks. Blood 2009, 113, 3781–3791. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; Frapolli, R.; Zangarini, M.; Bello, E.; Porcu, L.; Galmarini, C.M.; Garcia-Fernandez, L.F.; Cuevas, C.; Allavena, P.; Erba, E.; et al. Comparison of in vitro and in vivo biological effects of trabectedin, lurbinectedin (PM01183) and Zalypsis(R) (PM00104). Int. J. Cancer 2013, 133, 2024–2033. [Google Scholar] [CrossRef]
- Robinson, S.L.; Christenson, J.K.; Wackett, L.P. Biosynthesis and chemical diversity of β-lactone natural products. Nat. Prod. Rep. 2019, 36, 458–475. [Google Scholar] [CrossRef]
- Mincer, T.J.; Jensen, P.R.; Kauffman, C.A.; Fenical, W. Widespread and persistent populations of a major new marine actinomycete taxon in ocean sediments. Appl. Environ. Microbiol. 2002, 68, 5005–5011. [Google Scholar] [CrossRef] [Green Version]
- Potts, B.C.; Lam, K.S. Generating a generation of proteasome inhibitors: From microbial fermentation to total synthesis of salinosporamide a (marizomib) and other salinosporamides. Mar. Drugs 2010, 8, 835–880. [Google Scholar] [CrossRef] [Green Version]
- Gulder, T.A.M.; Moore, B.S. Salinosporamide natural products: Potent 20 S proteasome inhibitors as promising cancer chemotherapeutics. Angew. Chem. Int. Ed. Engl. 2010, 49, 9346–9367. [Google Scholar] [CrossRef]
- Kim, S.K.; Thomas, N.V.; Li, X. Anticancer compounds from marine macroalgae and their application as medicinal foods. Adv. Food Nutr. Res. 2011, 64, 213–224. [Google Scholar] [CrossRef]
- Venkatesan, J.; Lowe, B.; Anil, S.; Manivasagan, P.; Kheraif, A.A.A.; Kang, K.-H.; Kim, S.-K. Seaweed polysaccharides and their potential biomedical applications. Starch 2015, 67, 381–390. [Google Scholar] [CrossRef]
- Namvar, F.; Mohamad, R.; Baharara, J.; Zafar-Balanejad, S.; Fargahi, F.; Rahman, H.S. Antioxidant, antiproliferative, and antiangiogenesis effects of polyphenol-rich seaweed (Sargassum muticum). Biomed. Res. Int. 2013, 2013, 604787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, T.M.; Kim, W.J.; Moon, S.K. AKT signaling is involved in fucoidan-induced inhibition of growth and migration of human bladder cancer cells. Food Chem. Toxicol. 2014, 64, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Vishchuk, O.S.; Sun, H.; Wang, Z.; Ermakova, S.P.; Xiao, J.; Lu, T.; Xue, P.; Zvyagintseva, T.N.; Xiong, H.; Shao, C.; et al. PDZ-binding kinase/T-LAK cell-originated protein kinase is a target of the fucoidan from brown alga Fucus evanescens in the prevention of EGF-induced neoplastic cell transformation and colon cancer growth. Oncotarget 2016, 7, 18763–18773. [Google Scholar] [CrossRef] [Green Version]
- Yan, M.D.; Yao, C.J.; Chow, J.M.; Chang, C.L.; Hwang, P.A.; Chuang, S.E.; Whang-Peng, J.; Lai, G.M. Fucoidan Elevates MicroRNA-29b to Regulate DNMT3B-MTSS1 Axis and Inhibit EMT in Human Hepatocellular Carcinoma Cells. Mar. Drugs 2015, 13, 6099–6116. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.Y.; Huang, T.C.; Lin, L.C.; Shieh, T.M.; Wu, C.H.; Wang, K.L.; Hong, Y.H.; Hsia, S.M. Fucoidan Inhibits the Proliferation of Leiomyoma Cells and Decreases Extracellular Matrix-Associated Protein Expression. Cell. Physiol. Biochem. 2018, 49, 1970–1986. [Google Scholar] [CrossRef]
- Bae, H.; Lee, J.Y.; Yang, C.; Song, G.; Lim, W. Fucoidan Derived from Fucus vesiculosus Inhibits the Development of Human Ovarian Cancer via the Disturbance of Calcium Homeostasis, Endoplasmic Reticulum Stress, and Angiogenesis. Mar. Drugs 2020, 18, 45. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Qi, X.; Liu, H.; Xue, K.; Xu, S.; Tian, Z. The anti-cancer effects of fucoidan: A review of both in vivo and in vitro investigations. Cancer Cell Int. 2020, 20, 154. [Google Scholar] [CrossRef]
- Malyarenko, O.S.; Ermakova, S.P. Chapter 10—Fucoidans: Anticancer Activity and Molecular Mechanisms of Action. In Seaweed Polysaccharides; Venkatesan, J., Anil, S., Kim, S.-K., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 175–203. [Google Scholar] [CrossRef]
- van Weelden, G.; Bobiński, M.; Okła, K.; van Weelden, W.J.; Romano, A.; Pijnenborg, J.M.A. Fucoidan Structure and Activity in Relation to Anti-Cancer Mechanisms. Mar. Drugs 2019, 17, 32. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, M.; Yamori, T.; Naitoh, M.; Okutani, K. Structural revision of sulfated polysaccharide B-1 isolated from a marine Pseudomonas species and its cytotoxic activity against human cancer cell lines. Mar. Biotechnol. 2003, 5, 13–19. [Google Scholar] [CrossRef]
- Wang, H.; Chiu, L.C.M.; Ooi, V.E.C.; Ang, P.O. A potent antitumor polysaccharide from the edible brown seaweed Hydroclathrus clathratus. Bot. Mar. 2010, 53. [Google Scholar] [CrossRef]
- Jiao, L.; Li, X.; Li, T.; Jiang, P.; Zhang, L.; Wu, M.; Zhang, L. Characterization and anti-tumor activity of alkali-extracted polysaccharide from Enteromorpha intestinalis. Int. Immunopharmacol. 2009, 9, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Dias, P.F.; Siqueira, J.M., Jr.; Vendruscolo, L.F.; de Jesus Neiva, T.; Gagliardi, A.R.; Maraschin, M.; Ribeiro-do-Valle, R.M. Antiangiogenic and antitumoral properties of a polysaccharide isolated from the seaweed Sargassum stenophyllum. Cancer Chemother. Pharm. 2005, 56, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Mishima, T.; Murata, J.; Toyoshima, M.; Fujii, H.; Nakajima, M.; Hayashi, T.; Kato, T.; Saiki, I. Inhibition of tumor invasion and metastasis by calcium spirulan (Ca-SP), a novel sulfated polysaccharide derived from a blue-green alga, Spirulina platensis. Clin. Exp. Metastasis 1998, 16, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.; Lynn, D.M.; Sasisekharan, R.; Langer, R. Poly(beta-amino ester)s promote cellular uptake of heparin and cancer cell death. Chem. Biol. 2004, 11, 487–498. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Marine Organism | Chemical Classes | Therapeutic Use | Ref. |
|---|---|---|---|---|
| Lurbinectedin | Tunicate | Alkaloid | Solid tumors | [32] |
| Trabectedin | Tunicate | Alkaloid | Solid tumors | [33] |
| Midostaurin | Tunicate/Actinobacteria | Indolocarbazole | Leukemias | [34] |
| Plitidepsin ^ | Tunicate | Peptide | Multiple myeloma | [35] |
| Belantamab mafodotin | Mollusk/Cyanobacteria | ADC/Peptide ** | Multiple myeloma | [36] |
| Enfortumab vedotin | Mollusk/Cyanobacteria | ADC/Peptide * | Solid tumors | [37] |
| Polatuzumab vedotin | Mollusk/Cyanobacteria | ADC/Peptide * | Lymphomas | [38] |
| Brentuximab vedotin | Mollusk/Cyanobacteria | ADC/Peptide * | Lymphomas | [39] |
| Eribulin mesylate | Sponge | Macrolide polyketide | Solid tumors | [40] |
| Fludarabine phosphate | Sponge | Nucleoside | Leukemias, lymphomas | [41] |
| Cytarabine | Sponge | Nucleoside | Leukemias, lymphomas | [42] |
| Nelarabine | Sponge | Nucleoside | Leukemias, lymphomas | [43] |
| Clinical Status | Compound | Target | Payload | Marine Organism | Therapeutic Use | FDA Orphan Drug Designation | Ref. |
|---|---|---|---|---|---|---|---|
| Phase I | ALT-P7 | HER2 | MMAE | Mollusk/Cyanobacteria | Solid tumors | Gastric cancer | [44] |
| Phase I | RC88 | Mesothelin | MMAE | Mollusk/Cyanobacteria | Solid tumors | - | * |
| Phase I | SGN-CD228A | CD228 | MMAE | Mollusk/Cyanobacteria | Solid tumors | - | [45] |
| Phase II | CX-2029 | CD71 | MMAE | Mollusk/Cyanobacteria | Solid tumors, lymphomas | - | [46] |
| Phase II | Disitamab vedotin | HER2 | MMAE | Mollusk/Cyanobacteria | Solid tumors | Gastric cancer | [47] |
| Phase II | Enapotamab vedotin | AXL | MMAE | Mollusk/Cyanobacteria | Solid tumors | - | [48] |
| Phase II | Ladiratuzumab vedotin | LIV-1 A | MMAE | Mollusk/Cyanobacteria | Solid tumors | - | [49] |
| Phase II | Telisotuzumab vedotin | MET | MMAE | Mollusk/Cyanobacteria | Solid tumors | - | [50] |
| Phase II | Tisotumab vedotin | TF | MMAE | Mollusk/Cyanobacteria | Solid tumors | - | [51] |
| Phase I | FS-1502 | HER2 | MMAF | Mollusk/Cyanobacteria | Solid tumors | [52] | |
| Phase II | AGS 16C3F | ENPP3 | MMAF | Mollusk/Cyanobacteria | Solid tumors | - | [53] |
| Phase III | Depatuxizumab mafodotin | EGFR | MMAF | Mollusk/Cyanobacteria | Solid tumors | Glioblastoma | [54] |
| Clinical Status | Compound | Marine Organism | Chemical Classes | Therapeutic Use | FDA Orphan Drug Designation | Ref. |
|---|---|---|---|---|---|---|
| Phase III | Plinabulin | Fungus | Alkaloid | Solid tumors | - | [70] |
| Phase III | Marizomib | Actinobacteria | Beta-lactone | Solid tumors, lymphomas, multiple myeloma | Multiple myeloma, glioblastoma | [71] |
| Phase II | Oligo-fucoidan | Brown seaweed | Polysaccharide | Solid tumors | - | [72] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barreca, M.; Spanò, V.; Montalbano, A.; Cueto, M.; Díaz Marrero, A.R.; Deniz, I.; Erdoğan, A.; Lukić Bilela, L.; Moulin, C.; Taffin-de-Givenchy, E.; et al. Marine Anticancer Agents: An Overview with a Particular Focus on Their Chemical Classes. Mar. Drugs 2020, 18, 619. https://doi.org/10.3390/md18120619
Barreca M, Spanò V, Montalbano A, Cueto M, Díaz Marrero AR, Deniz I, Erdoğan A, Lukić Bilela L, Moulin C, Taffin-de-Givenchy E, et al. Marine Anticancer Agents: An Overview with a Particular Focus on Their Chemical Classes. Marine Drugs. 2020; 18(12):619. https://doi.org/10.3390/md18120619
Chicago/Turabian StyleBarreca, Marilia, Virginia Spanò, Alessandra Montalbano, Mercedes Cueto, Ana R. Díaz Marrero, Irem Deniz, Ayşegül Erdoğan, Lada Lukić Bilela, Corentin Moulin, Elisabeth Taffin-de-Givenchy, and et al. 2020. "Marine Anticancer Agents: An Overview with a Particular Focus on Their Chemical Classes" Marine Drugs 18, no. 12: 619. https://doi.org/10.3390/md18120619
APA StyleBarreca, M., Spanò, V., Montalbano, A., Cueto, M., Díaz Marrero, A. R., Deniz, I., Erdoğan, A., Lukić Bilela, L., Moulin, C., Taffin-de-Givenchy, E., Spriano, F., Perale, G., Mehiri, M., Rotter, A., P. Thomas, O., Barraja, P., Gaudêncio, S. P., & Bertoni, F. (2020). Marine Anticancer Agents: An Overview with a Particular Focus on Their Chemical Classes. Marine Drugs, 18(12), 619. https://doi.org/10.3390/md18120619