3.1. Preparation of Eurotiumide A Derivatives.
3.1.1. General Procedure
All the reactions were carried out in a round-bottomed flask with an appropriate number of necks and side arms connected to a three-way stopcock and/or a rubber septum cap under an argon atmosphere. All vessels were first evacuated by rotary pump and then flushed with argon prior to use. Solutions and solvents were introduced by hypodermic syringe through a rubber septum. During the reaction, the vessel was kept under a positive pressure of argon. Dry THF was freshly prepared by distillation from benzophenone ketyl before use. Anhydrous CH
2Cl
2, DMF, ethanol, MeCN, methanol, pyridine, and toluene were purchased from Kanto Chemical Co. Inc. Infrared (IR) spectra were recorded on a JASCO FT/IR-4100 spectrophotometer using a 5 mm KBr plate. Wavelengths of maximum absorbance are quoted in cm
−1. 1H-NMR spectra were recorded on a JEOL ECA–400 (400 MHz), Bruker AV-400N (400 MHz), and Bruker AV–500 (500 MHz) in CDCl
3. Chemical shifts are reported in parts per million (ppm), and signals are expressed as singlet (s), doublet (d), triplet (t), multiplet (m), broad (br), and overlapped. 13C-NMR spectra were recorded on a JEOL ECA–400 (100 MHz), Bruker AV–400N (100 MHz), and Bruker AV–500 (125 MHz) in CDCl
3. Chemical shifts are reported in parts per million (ppm) (see
Supplementary Materials). High resolution mass (HRMS) spectra were recorded on a Thermo Scientific Exactive. All melting points were measured with a Yanaco MP-500D. Analytical thin layer chromatography (TLC) was performed using 0.25 mm E. Merck Silica gel (60F-254) plates. Reaction components were visualized phosphomolybdic acid or ninhydrin or
p-anisaldehyde in 10% sulfuric acid in ethanol. Kanto Chem. Co. Silica Gel 60N (particle size 0.040–0.050 mm) was used for column chromatography.
3.1.2. Synthesis of (3S,4S)-5,8-dihydroxy-4-methoxy-3-pentylisochroman-1-one (4)
To a solution of bromo compound 3 (10.0 mg, 30.8 µmol) in MeOH (2.3 mL) was added 6 M aqueous HCl (0.77 mL) at 0 °C. After stirring for 30 min at 40 °C, the reaction was quenched by adding saturated aqueous NaHCO3 at 0 °C. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by preparative thin layer chromatography (PTLC) (EtOAc:n-hexane = 3:7) to give non-substituted derivative 4 (6.8 mg, 79%) as a white solid. m.p. 120–121 °C; 1H-NMR (400 MHz, CDCl3) δ 10.62 (1H, s), 7.06 (1H, d, J = 9.0 Hz), 6.91 (1H, d, J = 9.0 Hz), 5.89 (1H, br-s), 4.77 (1H, d, J = 2.7 Hz), 4.50 (1H, ddd, J = 2.7, 5.4, 8.3 Hz), 3.40 (3H, s), 1.95 (1H, m), 1.85 (1H, m), 1.70–1.50 (1H, overlapped), 1.46 (1H, m), 1.40–1.25 (4H, overlapped), 0.91 (3H, t, J = 6.8 Hz); 13C-NMR (100 MHz, CDCl3) δ 169.0, 156.2, 145.7, 125.1, 121.7, 118.8, 107.6, 81.4, 69.8, 56.8, 31.6, 29.8, 24.9, 22.5, 14.0.; IR (KBr) 3219, 2955, 2924, 2860, 1661, 1586, 1471, 1293, 1204, 905 cm−1; HRMS (ESI) m/z (M + Na)+ calculated for (C15H20O5Na)+ 303.1208, found 303.1200.
3.1.3. Synthesis of (3S,4S)-5,8-dihydroxy-7-isopentyl-4-methoxy-3-pentylisochroman-1-one (6)
To a solution of eurotiumide A (1) (1.6 mg, 4.6 µmol) in MeOH (0.23 mL) was added Pd/C (1.6 mg, 100 w/w%) at room temperature. After stirring for 1.5 h under hydrogen atmosphere (balloon), the reaction mixture was passed through Celite and the organic solvent was removed under reduced pressure. The residue was purified with flash column chromatography (EtOAc:n-hexane = 2:3) to give isopentyl derivative 6 (1.4 mg, 88%) as a white wax. 1H-NMR (500 MHz, CDCl3) δ 10.91 (1H, s), 6.93 (1H, s), 5.62 (1H, br-s), 4.74 (1H, d, J = 2.5 Hz), 4.48 (1H, ddd, J = 2.6, 5.4, 8.6 Hz), 3.38 (3H, s), 2.62 (2H, m), 1.95 (1H, m), 1.85 (1H, m), 1.65–1.50 (2H, overlapped), 1.50–1.40 (3H, overlapped), 1.40–1.30 (4H, overlapped), 0.95 (6H, d, J = 6.3 Hz), 0.90 (3H, J = 6.9 Hz); 13C-NMR (125 MHz, CDCl3) δ 169.4, 154.7, 145.0, 133.6, 124.8, 118.6, 106.8, 81.4, 69.9, 56.6, 38.4, 31.6, 29.8, 29.7, 27.9, 27.5, 14.9, 22.5, 14.0.; IR (KBr) 3290, 2956, 2927, 2870, 1761, 1445, 1171, 807 cm−1; HRMS (ESI) m/z (M + H)+ calculated for (C20H31O5)+ 351.2171, found 351.2177.
3.1.4. (3S,4S)-4-methoxy-5,8-bis(methoxymethoxy)-7-methyl-3-pentylisochroman-1-one (5a)
To a solution of bromo compound 3 (40.0 mg, 89.4 µmol) and CsF (16.3 mg, 107 µmol) in degassed DMF (0.45 mL) were added Me4Sn (15 µL, 107 µmol) and PdCl2(PPh3)2 (6.3 mg, 8.94 µmol) at room temperature. After stirring for 50 min at 80 °C, the reaction was quenched by adding water. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with flash column chromatography (EtOAc:n-hexane = 3:7) to give diMOM-protected methyl derivative 5a (28.5 mg, 83%) as a yellow amorphous. 1H-NMR (400 MHz, CDCl3) δ 7.255 (1H, s), 5.21 (2H, s), 5.10 (1H, d, J = 6.8 Hz), 5.07 (1H, d, J = 6.8 Hz), 4.59 (1H, d, J = 1.5 Hz), 4.26 (1H, ddd, J = 1.5, 5.9, 7.5 Hz), 3.60 (3H, s), 3.50 (3H, s), 3.30 (3H, s), 2.39 (3H, s), 2.02 (1H, m), 1.81 (1H, m), 1.70–1.50 (1H, overlapped), 1.43 (1H, m), 1.40–1.25 (4H, overlapped), 0.91 (3H, t, J = 6.8 Hz); 13C-NMR (125 MHz, CDCl3) δ 162.4, 152.3, 149.8, 135.7, 126.3, 121.3, 118.7, 101.5, 95.0, 80.9, 68.2, 57.5, 56.7, 56.4, 31.6, 30.6, 24.9, 22.6, 17.6, 14.0.; IR (KBr) 2958, 2927, 2858, 2828, 1728, 1478, 1153 cm−1; HRMS (ESI) m/z (M + H)+ calculated for (C20H31O7)+ 383.2070, found 383.2069.
3.1.5. (3S,4S)-5,8-dihydroxy-4-methoxy-7-methyl-3-pentylisochroman-1-one (5)
To a solution of diMOM-protected methyl derivative 5a (10.0 mg, 26.0 µmol) in MeOH (2.0 mL) was added 6 M aqueous HCl (0.65 mL) at 0 °C. After stirring for 1 h at 40 °C, the reaction was quenched by adding saturated aqueous NaHCO3. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with PTLC (EtOAc:n-hexane = 3:7) to give methyl derivative 5 (5.2 mg, 68%) as a yellow solid. m.p. 113 °C; 1H-NMR (400 MHz, CDCl3) δ 10.89 (1H, s), 6.93 (1H, s), 5.59 (1H, br-s), 4.75 (1H, d, J = 2.7 Hz), 4.48 (1H, ddd, J = 2.7, 5.4, 8,3 Hz), 3.37 (3H, s), 2.25 (3H, s), 1.93 (1H, m), 1.84 (1H, m), 1.70-1.50 (1H, overlapped), 1.45 (1H, m), 1.40–1.25 (4H, overlapped), 0.91 (3H, t, J = 6.6 Hz); 13C-NMR (125 MHz, CDCl3) δ 169.4, 154.9, 144.9, 128.7, 125.8, 118.6, 106.6, 81.4, 69.8, 56.5, 31.6, 29.8, 24.9, 22.5, 15.8, 14.0.; IR (KBr) 3340, 2957, 2928, 2859, 1682, 1654, 1604, 1296, 1172 cm−1; HRMS (ESI) m/z (M + Na)+ calculated for (C16H22O5Na)+ 317.1365, found 317.1350.
3.1.6. (3S,4S)-4-methoxy-5,8-bis(methoxymethoxy)-3-pentyl-7-vinylisochroman-1-one (7a)
To a solution of bromo compound 3 (200 mg, 0.447 mmol) and CsF (135.8 mg, 0.894 mmol) in degassed DMF (2.2 mL) were added tributylvinyltin (0.26 mL, 0.894 mmol) and PdCl2(PPh3)2 (62.8 mg, 89.0 µmol) at room temperature. After stirring for 1 h at 80 °C, the reaction was quenched by adding water. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with flash column chromatography (EtOAc:n-hexane = 3:7) to give diMOM-protected vinyl derivative 7a (185.1 mg, quant) as a yellow solid. m.p. 63–64 °C; 1H-NMR (500 MHz, CDCl3) δ 7.56 (1H, s), 7.14 (1H, dd, J = 11.1, 17.7 Hz), 5.76 (1H, d, J = 17.7 Hz), 5.40 (1H, d, J = 11.1 Hz), 5.24 (2H, s), 5.08 (1H, d, J = 6.3 Hz), 5.05 (1H, d, J = 6.3 Hz), 4.60 (1H, d, J = 1.3 Hz), 4.26 (1H, ddd, J = 1.3, 5.8, 7.4 Hz), 3.58 (3H, s), 3.50 (3H, s), 3.31 (3H, s), 2.03 (1H, m), 1.81 (1H, m), 1.56 (1H, m), 1.43 (1H, m), 1.40–1.25 (4H, overlapped), 0.90 (3H, t, J = 6.9 Hz); 13C-NMR (125 MHz, CDCl3) δ 162.0, 150.7, 150.2, 134.9, 131.3, 128.5, 119.7, 116.7, 116.0, 101.5, 95.2, 80.8, 68.3, 57.9, 56.8, 56.4, 31.6, 30.6, 24.9, 22.5, 14.0.; IR (KBr) 2953, 2931, 2861, 2829, 1730, 1471, 1426, 1155, 929 cm−1; HRMS (ESI) m/z (M + H)+ calculated for (C21H31O7)+ 395.2070, found 395.2078.
3.1.7. (3S,4S)-5,8-dihydroxy-4-methoxy-3-pentyl-7-vinylisochroman-1-one (7)
To a solution of diMOM-protected methyl derivative 7a (13.7 mg, 34.7 µmol) in MeOH (2.6 mL) was added 6 M aqueous HCl (0.87 mL) at 0 °C. After stirring for 3 h at 40 °C, the reaction was quenched by adding saturated aqueous NaHCO3. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with PTLC (EtOAc:n-hexane = 3:7) to give vinyl derivative 7 (8.5 mg, 75%) as a yellow wax. 1H-NMR (500 MHz, CDCl3) δ 11.10 (1H, s), 7.23 (1H, s), 7.01 (1H, dd, J = 11.4, 17.7 Hz), 5.82 (1H, br-s), 5.80 (1H, d, J = 18.0 Hz), 5.37 (1H, d, J = 11.0 Hz), 4.77 (1H, br-s), 4.50 (1H, br-s), 3.40 (3H, s), 1.95 (1H, m), 1.85 (1H, m), 1.58 (1H, m), 1.45 (1H,m), 1.40–1.25 (4H, overlapped), 0.90 (3H, br-s); 13C-NMR (125 MHz, CDCl3) δ 169.3, 153.9, 145.4, 129.8, 128.0, 121.4, 120.9, 116.5, 107.7, 81.5, 69.7, 56.8, 31.6, 29.8, 24.9, 22.5, 14.0.; IR (KBr) 3311, 2956, 2930, 2859, 1659, 1438, 1171 cm−1; HRMS (ESI) m/z (M + Na)+ calculated for (C17H22O5Na)+ 329.1365, found 329.1368.
3.1.8. (3S,4S)-4-methoxy-5,8-bis(methoxymethoxy)-3-pentyl-7-phenylisochroman-1-one (9a)
Bromo compound 3 (10.0 mg, 22.4 µmol), Cs2CO3 (21.9 mg, 67.1 µmol), phenylboronic acid (5.5 mg, 44.7 µM), and PdCl2(PPh3)2 (3.1 mg, 44.7 µmol) were dissolved in degassed dioxane (0.22 mL) at room temperature. After stirring for 1 h under reflux condition, the reaction was quenched by adding saturated aqueous NH4Cl. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with flash column chromatography (EtOAc:n-hexane = 3:7) to give diMOM-protected phenyl derivative 9a (7.4 mg, 75%) as a white wax. 1H-NMR (500 MHz, CDCl3) δ 7.55 (1H, d, J = 7.6 Hz), 7.50-7.38 (3H, overlapped), 7.36 (1H, dd, J = 7.3 Hz), 5.25 (2H, s), 4.80 (2H, s), 4.66 (1H, s), 4.33 (1H, t, J = 7.0 Hz), 3.50 (3H, s), 3.37 (3H, s), 2.92 (3H, s), 2.06 (1H, m), 1.85 (1H, m), 1.70–1.50 (1H, overlapped), 1.50–1.25 (5H, overlapped), 0.92 (3H, br-s); 13C-NMR (125 MHz, CDCl3) δ 162.1, 150.5, 150.0, 139.5, 137.9, 129.8, 128.3, 128.1, 127.7, 121.0, 119.9, 101.0, 95.1, 80.8, 68.3, 57.1, 56.4, 31.6, 30.6, 24.9, 22.5, 14.0.; IR (KBr) 2956, 2927, 2859, 2828, 1728, 1467, 1152, 1008, 932 cm−1; HRMS (ESI) m/z (M + Na)+ calculated for (C25H32O7Na)+ 467.2046, found 467.2043.
3.1.9. (3S,4S)-5,8-dihydroxy-4-methoxy-3-pentyl-7-phenylisochroman-1-one (9)
To a solution of diMOM-protected methyl derivative 9a (7.4 mg, 16.8 µmol) in THF (1.0 mL) was added 6 M aqueous HCl (0.50 mL) at 0 °C. After stirring for 6 h at room temperature, the reaction was quenched by adding saturated aqueous NaHCO3. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with PTLC (EtOAc:n-hexane = 3:7) to give phenyl derivative 9 (6.0 mg, 90%) as a yellow solid. m.p. 173–174 °C; 1H-NMR (400 MHz, CDCl3) δ 11.21 (1H, s), 7.58 (2H, d, J = 7.3 Hz), 7.44 (2H, t, J = 7.3 Hz), 7.38 (1H, d, J = 7.6 Hz), 7.13 (1H, s), 5.76 (1H, br-s), 4.82 (1H, d, J = 2.7 Hz), 4.55 (1H, ddd, J = 2.7, 5.1, 8.3 Hz), 3.44 (3H, s), 1.98 (1H, m), 1.89 (1H, m), 1.70–1.40 (2H, overlapped), 1.40–1.25 (4H, overlapped), 0.92 (3H, t, J = 6.8 Hz); 13C-NMR (125 MHz, CDCl3) δ 169.5, 153.7, 145.4, 136.2, 131.8, 129.2, 128.3, 127.9, 125.5, 121.1, 107.8, 81.6, 69.6, 56.9, 31.6, 29.8, 24.9, 22.5, 14.0.; IR (KBr) 3307, 2955, 2928, 2859, 1650, 1425, 1295, 1194 cm−1; HRMS (ESI) m/z (M + H)+ calculated for (C21H25O5)+ 357.1702, found 357.1707.
3.1.10. (3S,4S)-7-([1,1’-biphenyl]-4-yl)-4-methoxy-5,8-bis(methoxymethoxy)-3-pentylisochroman-1-one (10a)
Bromo compound 3 (20.0 mg, 44.7 µmol), Cs2CO3 (21.9 mg, 67.1 µmol), 4-biphenylboronic acid (5.5 mg, 44.7 µmol), and PdCl2(PPh3)2 (3.2 mg, 4.47 µmol) were dissolved in degassed dioxane (0.23 mL) at room temperature. After stirring for 1 h under reflux condition, the reaction was quenched by adding saturated aqueous NH4Cl. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with PTLC (EtOAc:n-hexane = 3:7) to give diMOM-protected biphenyl derivative 10a (18.0 mg, 88%) as a white solid. 1H-NMR (500 MHz, CDCl3) δ 7.74–7.60 (6H, overlapped), 7.53–7.40 (3H, overlapped), 7.38 (1H, t, J = 7.3 Hz), 5.28 (2H, s), 4.85 (1H, d, J = 7.0 Hz), 4.84 (1H, d, J = 7.0 Hz), 4.68 (1H, d, J = 1.3 Hz), 4.35 (1H, ddd, J = 1.3, 6.0, 7.6 Hz), 3.51 (3H, s), 3.38 (3H, s), 2.99 (3H, s), 2.08 (1H, m), 1.86 (1H, m), 1.70-1.50 (1H, overlapped), 1.46 (1H, m), 1.40–1.25 (4H, overlapped), 0.92 (3H, t, J = 6.9 Hz); 13C-NMR (125 MHz, CDCl3) δ 162.1, 150.6, 150.0, 140.5, 140.4, 139.1, 136.8, 130.2, 128.9, 128.1, 127.5, 127.0, 126.9, 120.9, 120.0, 101.1, 95.1, 80.8, 68.3, 57.2, 56.9, 56.4, 31.6, 30.6, 24.9, 22.5, 14.0.; IR (KBr) 2956, 2927, 2858, 2827, 1728, 1467, 1152, 1007, 931 cm−1; HRMS (ESI) m/z (M + H)+ calculated for (C31H37O7)+ 521.2539, found 521.2539.
3.1.11. (3S,4S)-7-([1,1’-biphenyl]-4-yl)-5,8-dihydroxy-4-methoxy-3-pentylisochroman-1-one (10)
To a solution of diMOM-protected biphenyl derivative 10a (12.9 mg, 24.8 µmol) in THF (1.7 mL) was added 6 M aqueous HCl (0.83 mL) at 0 °C. After stirring for 17 h at room temperature, the reaction was quenched by adding saturated aqueous NaHCO3 at 0 °C. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with PTLC (EtOAc:n-hexane = 3:7) to give biphenyl derivative 10 (9.9 mg, 92%) as a yellow solid. m.p. 181–182 °C; 1H-NMR (400 MHz, CDCl3) δ 11.28 (1H, s), 7.67 (4H, s), 7.64 (2H, d, J = 7.3 Hz), 7.46 (2H, t, J = 7.3 Hz), 7.37 (1H, t, J = 7.3 Hz), 7.19 (1H, s), 5.75 (1H, br-s), 4.84 (1H, d, J = 2.7 Hz), 4.56 (1H, ddd, J = 2.7, 5.4, 8.3 Hz), 3.46 (3H, s), 1.98 (1H, m), 1.89 (1H, m), 1.70–1.50 (2H, overlapped), 1.45–1.25 (4H, overlapped), 0.92 (3H, t, J = 6.8 Hz); 13C-NMR (125 MHz, CDCl3) δ 169.4, 153.8, 145.5, 140.7, 135.2, 131.4, 129.6, 128.8, 127.5,127.15, 127.07, 125.3, 121.0, 107.9, 81.5, 69.8, 56.9, 31.6, 29.8, 24.9, 22.5, 14.0.; IR (KBr) 3283, 2954, 2929, 2863, 1668, 1595, 1295, 1220, 772 cm−1; HRMS (ESI) m/z (M + Na)+ calculated for (C27H28O5Na)+ 455.1834, found 455.1831.
3.1.12. (3S,4S)-7-ethynyl-4-methoxy-5,8-bis(methoxymethoxy)-3-pentylisochroman-1-one (8a)
To a solution of aldehyde 12a (5.4 mg, 13.6 µmol) in MeOH (0.14 mL) were added K2CO3 (5.7 mg, 40.9 µmol) and Ohira–Bestmann reagent (3.9 mg, 20.4 µmol) at room temperature. After stirring for 40 min at the same temperature, the mixture was concentrated under reduced pressure. The residue was purified with column chromatography (EtOAc:n-hexane = 1:4 to 1:1) to give diMOM alkyne derivative 8a (6.3 mg, quant) as a yellow oil. 1H-NMR (500 MHz, CDCl3) δ 7.52 (1H, s), 5.27 (1H, d, J = 6.0 Hz), 5.22 (2H, s), 5.17 (1H, d, J = 6.0 Hz), 4.59 (1H, d, J = 1.3 Hz), 4.27 (1H, ddd, J = 1.3, 5.8, 7.4 Hz), 3.65 (3H, s), 3.49 (3H, s), 3.32 (3H, s), 2.05 (1H, m), 1.82 (1H, m), 1.65–1.50 (1H, overlapped), 1.42 (1H, m), 1.40–1.25 (4H, overlapped), 0.91 (3H, t, J = 7.1 Hz); 13C-NMR (125 MHz, CDCl3) δ 161.2, 154.6, 149.5, 130.0, 123.5, 120.4, 120.1, 101.0, 95.2, 82.7, 80.7, 79.3, 68.3, 58.1, 57.0, 56.5, 31.6, 30.5, 24.8, 22.5, 14.0.; IR (KBr) 3260, 2954, 2932, 2861, 2830, 1730, 1155, 1012, 931 cm−1; HRMS (ESI) m/z (M + H)+ calculated for (C21H29O7)+ 393.1913, found 393.1903.
3.1.13. (3S,4S)-7-ethynyl-5,8-dihydroxy-4-methoxy-3-pentylisochroman-1-one (8)
To a solution of diMOM alkyne derivative 8a (6.3 mg, 13.6 µmol) in MeOH (1.2 mL) was added 6 M aqueous HCl (0.40 mL) at room temperature. After stirring for 24 h at the same temperature, the reaction was quenched by adding saturated aqueous NaHCO3 at 0 °C. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with column chromatography (EtOAc:n-hexane = 1:4 to 1:1) to give alkyne derivative 8 (3.3 mg, 67%) as a yellow solid. m.p. 132–133 °C; 1H-NMR (500 MHz, CDCl3) δ 11.20 (1H, s), 7.22 (1H, s), 6.03 (1H, br-s), 4.76 (1H, d, J = 2.5 Hz), 4.51 (1H, ddd, J = 2.5, 5.1, 8.2 Hz), 3.40 (3H, s), 3.39 (1H, s), 1.94 (1H, m), 1.84 (1H, m), 1.70–1.50 (1H, overlapped), 1.45 (1H, m), 1.40–1.25 (4H, overlapped), 0.91 (3H, t, J = 7.0 Hz); 13C-NMR (125 MHz, CDCl3) δ 168.6, 157.3, 145.1, 127.8, 123.4, 112.6, 108.0, 83.2, 81.4, 77.7, 69.7, 57.0, 31.5, 29.7, 24.8, 22.5, 14.0.; IR (KBr) 3294, 2956, 2930, 2859, 1679, 1434, 1172 cm−1; HRMS (ESI) m/z (M + H)+ calculated for (C17H21O5)+ 305.1389, found 305.1391.
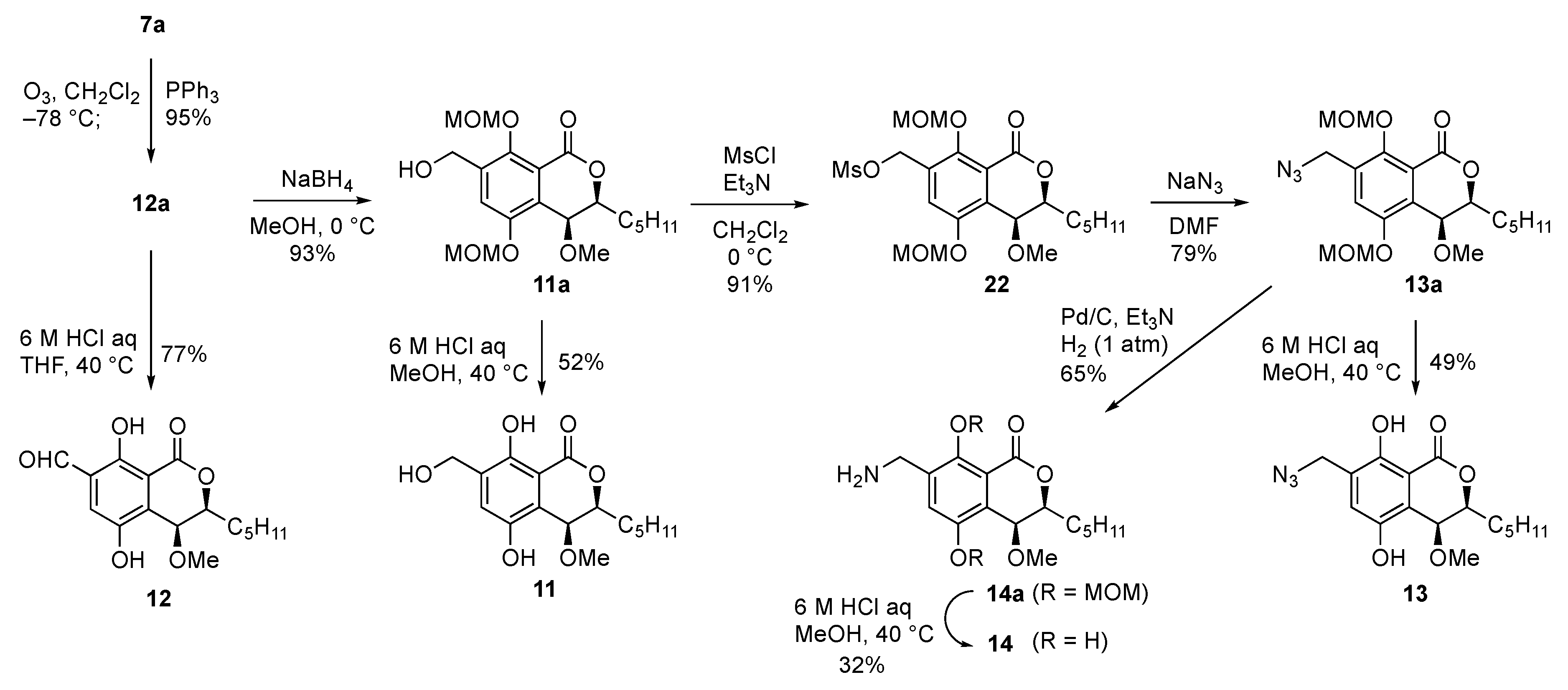
3.1.14. (3S,4S)-4-methoxy-5,8-bis(methoxymethoxy)-1-oxo-3-pentylisochromane-7-carbaldehyde (12a)
A stirred solution of 7a (185.1 mg, 0.469 mmol) in CH2Cl2 (10.0 mL) was cooled to −78 °C and a stream of ozone was passed through it for 30 min. At this time, ozone gas was bubbled into the reaction mixture until the color of the reaction mixture turned to blue. After completion of the reaction, the mixture was purged with oxygen gas for 30 min before being treated with PPh3 (246.2 mg, 0.939 mmol) and allowed to warm to room temperature. After stirring at the same temperature for 12 h, the mixture was concentrated under reduced pressure and the resultant mixture was purified with column chromatography (EtOAc:n-hexane = 1:4 to 2:3) to give diMOM benzaldehyde derivative 12a (177.4 mg, 95%) as a white solid. m.p. 38–39 °C; 1H-NMR (400 MHz, CDCl3) δ 10.42 (1H, s), 7.83 (1H, s), 5.29 (2H, s), 5.2 (2H, s), 4.65 (1H, d, J = 1.0 Hz), 4.29 (1H, J = 1.0, 5.6, 8.3 Hz), 3.59 (3H, s), 3.50 (3H, s), 3.35 (3H, s), 2.06 (1H, m), 1.83 (1H, m), 1.70-1.50 (1H, overlapped), 1.44 (1H, m), 1.40-1.30 (4H, overlapped), 0.91 (3H, t, J = 7.1 Hz); 13C-NMR (125 MHz, CDCl3) δ 189.9, 161.4, 156.6, 150.6, 135.8, 132.5, 120.8, 116.9, 103.0, 95.4, 81.0, 68.7, 58.4, 57.8, 57.0, 31.9, 30.8, 25.2, 22.8, 14.3.; IR (KBr) 2957, 2929, 2859, 2829, 1730, 1691, 1379, 1155, 930 cm−1; HRMS (ESI) m/z (M + H)+ calculated for (C20H29O8)+ 397.1862, found 397.1866.
3.1.15. (3S,4S)-5,8-dihydroxy-4-methoxy-1-oxo-3-pentylisochromane-7-carbaldehyde (12)
To a solution of diMOM aldehyde derivative 12a (10.0 mg, 25.2 µmol) in THF (1.9 mL) was added 6 M aqueous HCl (0.63 mL) at 0 °C. After stirring for 4 h at room temperature, the reaction was quenched by adding saturated aqueous NaHCO3 at 0 °C. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with PTLC (EtOAc:n-hexane = 2:3) to give benzaldehyde derivative 12 (6.0 mg, 77%) as a pale yellow solid. m.p. 170 °C (dec); 1H-NMR (400 MHz, CDCl3) δ 11.33 (1H, s), 10.47 (1H, s), 7.70 (1H, d, J = 1.5 Hz), 6.62 (1H, br-s), 4.75 (1H, d, J = 2.2 Hz), 4.49 (1H, ddd, J = 2.2, 5.6, 8.0 Hz), 3.43 (3H, s), 2.03 (1H, s), 1.88 (1H, m), 1.61 (1H, m), 1.48 (1H, m), 1.42–1.30 (4H, overlapped), 0.92 (3H, t, J = 6.8 Hz); 13C-NMR (125 MHz, CDCl3) δ 189.0, 168.8, 158.9, 146.0, 131.3, 124.9, 121.5, 110.2, 82.2, 69.2, 57.9, 31.9, 30.3, 25.1, 22.8, 14.3.; IR (KBr) 3444, 3169, 2953, 2940, 2920, 1676, 1455, 1395, 1299 cm−1; HRMS (ESI) m/z (M + H)+ calculated for (C16H21O6)+ 309.1338, found 309.1342.
3.1.16. (3S,4S)-7-(hydroxymethyl)-4-methoxy-5,8-bis(methoxymethoxy)-3-pentylisochroman-1-one (11a)
To a solution of diMOM aldehyde derivative 12a (20.0 mg, 50.5 µmol) in MeOH (0.25 mL) was added NaBH4 (2.1 mg, 55.5 µmol) at 0 °C. After stirring for 15 min at the same temperature, the reaction was quenched by adding water at 0 °C. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with PTLC (EtOAc:n-hexane = 1:1) to give diMOM hydroxymethyl derivative 11a (18.6 mg, 93%) as a white wax. 1H-NMR (400 MHz, CDCl3) δ 7.46 (1H, s), 5.25 (1H, d, J = 6.8 Hz), 5.24 (1H, d, J = 6.8 Hz), 5.15 (2H, s), 4.72 (1H, dd, J = 6.4, 12.5 Hz), 4.62 (1H, d, J = 1.2 Hz), 4.58 (1H, dd, J = 7.8, 12.5 Hz), 4.25 (1H, ddd, J = 1.2, 5.8, 8.0 Hz), 3.64 (3H, s), 3.55 (1H, t, J = 6.8 Hz), 3.50 (3H, s), 3.31 (3H, s), 2.05 (1H, m), 1.83 (1H, m), 1.65-1.50 (1H, overlapped), 1.43 (1H, m), 1.42–1.30 (4H, overlapped), 0.91 (3H, t, J = 6.8 Hz); 13C-NMR (125 MHz, CDCl3) δ 162.4, 152.7, 150.7, 138.7, 128.9, 120.8, 119.3, 102.2, 95.4, 81.2, 68.4, 61.4, 57.8, 57.2, 56.8, 31.9, 30.9, 25.2, 22.9, 14.4.; IR (KBr) 3443, 2957, 2928, 2859, 2828, 1724, 1153, 1012 cm−1; HRMS (ESI) m/z (M + H)+ calculated for (C20H31O8)+ 399.2019, found 399.2017.
3.1.17. (3S,4S)-5,8-dihydroxy-7-(hydroxymethyl)-4-methoxy-3-pentylisochroman-1-one (11)
To a solution of diMOM hydroxymethyl derivative 11a (7.2 mg, 24.1 µmol) in MeOH (1.8 mL) was added 6 M aqueous HCl (0.45 mL) at 0 °C. After stirring for 4 h at 40 °C, the reaction was quenched by adding saturated aqueous NaHCO3 at 0 °C. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with PTLC (EtOAc:n-hexane = 1:1) to give hydroxymethyl derivative 11 (3.9 mg, 52%) as a white solid. m.p. 143–145 °C; 1H-NMR (400 MHz, CDCl3) δ 10.99 (1H, s), 7.12 (1H, s), 6.03 (1H, br-s), 4.74 (1H, d, J = 2.4 Hz), 4.72 (2H, br-s), 4.48 (1H, ddd, J = 2.4, 5.2, 8.0 Hz), 3.38 (3H, s), 2.53 (1H, br-s), 1.96 (1H, m), 1.86 (1H, m), 1.70–1.50 (1H, overlapped), 1.46 (1H, m), 1.40–1.25 (4H, overlapped), 0.91 (3H, t, J = 6.8 Hz); 13C-NMR (125 MHz, CDCl3) δ 169.5, 154.2, 145.8, 130.8, 123.8, 121.4, 107.8, 82.1, 69.8, 61.2, 57.2, 31.9, 30.2, 25.2, 22.8, 14.3.; IR (KBr) 2951, 2921, 2854, 1682, 1440, 1302 cm−1; HRMS (ESI) m/z (M + H)+ calculated for (C16H23O6)+ 311.1495, found 311.1498.
3.1.18. ((3S,4S)-4-methoxy-5,8-bis(methoxymethoxy)-1-oxo-3-pentylisochroman-7-yl)methylmethanesulfonate (22)
To a solution of diMOM hydroxymethyl derivative 11a (7.2 mg, 24.1 µmol) in CH2Cl2 (0.47 mL) were added Et3N (10.8 µL, 77.5 µmol) and MsCl (6.0 µL, 77.5 µmol) at 0 °C. After stirring for 40 min at the same temperature, the reaction was quenched by adding water at 0 °C. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with PTLC (EtOAc:n-hexane = 2:3) to give diMOM mesylated derivative 22 (30.4 mg, 91%) as a white wax. 1H-NMR (400 MHz, CDCl3) δ 7.51 (1H, s), 5.45 (1H, d, J = 12.0 Hz), 5.37 (1H, d, J = 12.2 Hz), 5.25 (2H, s), 5.14 (1H, d, J = 6.6 Hz), 5.12 (1H, d, J = 6.6 Hz), 4.62 (1H, d, J = 1.4 Hz), 4.27 (1H, ddd, J = 1.2, 5.6, 7.8 Hz), 3.59 (3H, s), 3.50 (3H, s), 3.33 (3H, s), 3.07 (3H, s), 2.03 (1H, m), 1.82 (1H, m), 1.58 (1H, m), 1.44 (1H, m), 1.40–1.25 (4H, overlapped), 0.91 (3H, t, J = 6.8 Hz); 13C-NMR (100 MHz, CDCl3) δ 161.9, 152.2, 150.5, 131.3, 130.5, 120.3, 119.7, 102.8, 95.5, 81.2, 68.6, 66.9, 58.1, 57.4, 56.9, 38.2, 31.9, 30.9, 25.2, 22.8, 14.3.; IR (KBr) 2958, 2930, 2860, 1829, 1681, 1440, 1358, 1175, 933 cm−1; HRMS (ESI) m/z (M + Na)+ calculated for (C21H32O10SNa)+ 499.1614, found 499.1616.
3.1.19. (3S,4S)-7-(azidomethyl)-4-methoxy-5,8-bis(methoxymethoxy)-3-pentylisochroman-1-one (13a)
To a solution of diMOM mesylated derivative 22 (5.3 mg, 11.1 µmol) in DMF (55 µL) was added NaN3 (0.79 mg, 12.1 µmol) at room temperature. After stirring for 6 h at the same temperature, the reaction was quenched by adding water at 0 °C. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with PTLC (EtOAc:n-hexane = 3:7) to give diMOM azide derivative 13a (3.7 mg, 79%) as a pale-yellow oil. 1H-NMR (500 MHz, CDCl3) δ 7.44 (1H, s), 5.26 (1H, d, J = 6.9 Hz), 5.25 (1H, d, J = 6.9 Hz), 5.13 (1H, d, J = 6.9 Hz), 5.11 (1H, d, J = 6.9 Hz), 4.65 (1H, d, J = 14.5 Hz), 4.62 (1H, d, J = 1.3 Hz), 4.53 (1H, d, J = 14.5 Hz), 4.27 (1H, ddd, J = 1.3, 5.7, 7.3 Hz), 3.60 (3H, s), 3.51 (3H, s), 3.32 (3H, s), 2.04 (1H, m), 1.82 (1H, m), 1.65–1.50 (1H, overlapped), 1.43 (1H, m), 1.40–1.30 (4H, overlapped), 0.91 (3H, t, J = 7.0 Hz); 13C-NMR (125 MHz, CDCl3) δ 162.2, 152.1, 150.5, 133.5, 129.1, 119.7, 119.5, 102.6, 95.5, 81.2, 68.6, 57.9, 57.3, 56.8, 50.2, 31.9, 30.9, 25.2, 22.9, 14.4.; IR (KBr) 2957, 2928, 2858, 2829, 2105, 1729, 1153, 1009 cm−1; HRMS (ESI) m/z (M + H)+ calculated for (C20H30N3O7)+ 424.2084, found 424.2085.
3.1.20. (3S,4S)-7-(azidomethyl)-5,8-dihydroxy-4-methoxy-3-pentylisochroman-1-one (13)
To a solution of diMOM azide derivative 13a (8.3 mg, 19.6 µmol) in MeOH (1.5 mL) was added 6 M aqueous HCl (0.49 mL) at room temperature. After stirring for 4 h at 40 °C, the reaction was quenched by adding saturated aqueous NaHCO3 at 0 °C. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with PTLC (EtOAc:n-hexane = 3:7) to give nitro derivative 13 (3.1 mg, 49%) as a white solid. m.p. 98–99 °C; 1H-NMR (400 MHz, CDCl3) δ 10.98 (1H, s), 7.10 (1H, s), 5.81 (1H, br-s), 4.78 (1H, d, J = 2.9 Hz), 4.52 (1H, ddd, J = 2.9, 5.4, 8.5 Hz), 4.45 (1H, d, J = 14.4 Hz), 4.42 (1H, d, J = 14.4 Hz), 3.41 (3H, s), 1.93 (1H, m), 1.86 (1H, m), 1.70-1.50 (1H, overlapped), 1.47 (1H, m), 1.40–1.25 (4H, overlapped), 0.91 (3H, t, J = 7.1 Hz); 13C-NMR (125 MHz, CDCl3) δ 169.1, 154.4, 145.7, 126.2, 124.6, 121.8, 108.0, 81.7, 70.4, 57.2, 49.3, 31.9, 30.0, 25.2, 22.8, 14.3.; IR (KBr) 2959, 2924, 2857, 2108, 1654, 1441, 1293, 1170 cm−1; HRMS (ESI) m/z (M + H)+ calculated for (C16H22N3O5)+ 336.1559, found 336.1563.
3.1.21. (3S,4S)-7-(aminomethyl)-4-methoxy-5,8-bis(methoxymethoxy)-3-pentylisochroman-1-one (14a)
To a solution of diMOM azide derivative 13a (3.3 mg, 7.8 µmol) in MeOH (0.78 mL) was added Et3N (0.10 mL, 7.35 mmol) and Pd/C (1.6 mg, 1.5 µmol) at room temperature. After stirring for 1 h at the same temperature, the mixture was filtered, and the filtrate was concentrated under reduced pressure. The residue was purified with PTLC (MeOH:CH2Cl2 = 1:9) to give diMOM amine derivative 14a (2.0 mg, 65%) as brown oil. 1H-NMR (400 MHz, CDCl3) δ 7.49 (1H, s), 5.26 (2H, s), 5.16 (1H, d, J = 7.2 Hz), 5.07 (1H, d, J = 6.8 Hz), 4.61 (1H, d, J = 1.2 Hz), 4.27 (1H, ddd, J = 1.2, 6.0, 7.6 Hz), 4.00 (2H, s), 3.61 (3H, s), 3.50 (3H, s), 3.32 (3H, s), 2.59 (1H, br-s), 2.03 (1H, m), 1.82 (1H, m), 1.57 (1H, m), 1.43 (1H, m), 1.40-1.25 (1H, overlapped), 0.91 (3H, t, J = 6.8 Hz); 13C-NMR (125 MHz, CDCl3) δ 162.6, 152.6, 150.5, 128.1, 120.0, 119.0, 102.4, 95.4, 81.2, 68.5, 57.9, 57.2, 56.8, 42.5, 32.0, 30.9, 30.0, 25.2, 22.9, 14.4.; IR (KBr) 2957, 2925, 2857, 2827, 1726, 1470, 1153, 1005 cm−1; HRMS (ESI) m/z (M + H)+ calculated for (C20H32NO7)+ 398.2179, found 398.2178.
3.1.22. (3S,4S)-7-(aminomethyl)-5,8-dihydroxy-4-methoxy-3-pentylisochroman-1-one (14)
To a solution of diMOM amine derivative 14a (4.4 mg, 11.1 µmol) in MeOH (0.83 mL) was added 6 M aqueous HCl (0.28 mL) at 0 °C. After stirring for 5 h at room temperature, the reaction was quenched by adding saturated aqueous NaHCO3 at 0 °C. The mixture was extracted with the mixture of MeOH and CH2Cl2 (MeOH:CH2Cl2 = 1:4) (×4) and the combined organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified with PTLC (MeOH:CHCl3 saturated with NH3 = 1:9) to give amiomethyl derivative 14 (1.1 mg, 32%) as brown solid. m.p. 78–80 °C; 1H-NMR (400 MHz, CDCl3) δ 6.98 (1H, s), 4.59 (1H, d, J = 1.8 Hz), 4.35 (1H, ddd, J = 1.8, 6.0, 8.0 Hz), 3.97 (1H, d, J = 13.3 Hz), 3.88 (1H, d, J = 13.3 Hz), 3.19 (3H, s), 1.98 (1H, m), 1.83 (1H, m), 1.56 (1H, m), 1.43 (1H, m), 1.40–1.25 (4H, overlapped), 0.90 (3H, t, J = 7.0 Hz) ; 13C-NMR (125 MHz, CDCl3) δ 169.9, 154.2, 146.2, 130.0, 125.8, 122.9, 108.1, 82.8, 68.5, 56.9, 42.3, 31.9, 30.6, 25.1, 22.8, 14,3.; IR (KBr) 2956, 2921, 2857, 1676, 1441, 1171 cm−1; HRMS (ESI) m/z (M + Na)+ calculated for (C16H23NO5Na)+ 332.1474, found 332.1474.
3.1.23. ((3S,4S)-5,8-dihydroxy-4-methoxy-7-nitro-3-pentylisochroman-1-one (15)
To a solution of 3 (28.9 mg, 89.1 µmol) in AcOH (0.50 mL) was added the mixture of AcOH and 70% HNO3 (0.80 mL:0.20 mL) at 0 °C. After stirring for 10 min at the same temperature, the reaction was quenched by adding saturated aqueous NaHCO3 at 0 °C. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with saturated aqueous NaHCO3 and brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was pathed through SiO2 plug and the resultant mixture of monoMOM nitro derivative 15a was used for the next reaction without further purification. To a solution of 15a mixture in MeOH (7.5 mL) was added 6 M aqueous HCl (2.4 mL) at 0 °C. After stirring for 5 h at 40 °C, the reaction was quenched by adding saturated aqueous NaHCO3 at 0 °C. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with PTLC (EtOAc:n-hexane = 1:1) to give nitro derivative 15 (21.5 mg, 74%) as a yellow solid. m.p. 158-159; 1H-NMR (400 MHz, CDCl3) δ 11.89 (1H, s), 7.78 (1H, s), 6.80 (1H, br-s), 4.82 (1H, d, J = 2.6 Hz), 4.55 (1H, ddd, J = 2.6, 5.2, 8.3 Hz), 3.46 (3H, s), 1.96 (1H, m), 1.86 (1H, m), 1.59 (1H, m), 1.47 (1H, m), 1.40–1.25 (4H, overlapped), 0.91 (3H, t, J = 7.1 Hz); 13C-NMR (125 MHz, CDCl3) δ 167.5, 150.4, 144.9, 137.6, 129.4, 119.7, 110.7, 81.0, 70.3, 57.6 , 31.4, 29.4, 24.7, 22.4, 14.0; IR (KBr) 3416, 2962, 2927, 2857, 1679, 1445, 1261, 1018, 800 cm−1; HRMS (ESI) m/z (M + H)+ calculated for (C15H20NO7)+ 326.1240, found 326.1224.
3.1.24. (3S,4S)-7-amino-5,8-dihydroxy-4-methoxy-3-pentylisochroman-1-one (16)
To a solution of nitro derivative 15 (5.0 mg, 15.4 µmol) in THF (0.62 mL) and MeOH (80 µL) was added PtO2 (0.3 mg, 1.54 µmol) at room temperature. After stirring for 1.5 h at the same temperature under hydrogen atmosphere (1 atm), the mixture was passed through a membrane filter to remove PtO2. The mixture was concentrated under reduced pressure and the residue was purified with PTLC (EtOAc:n-hexane = 3:7, developed by three times) to give nitro derivative 16 (4.3 mg, 95%) as a yellow solid. m.p. 118–119 °C; 1H-NMR (500 MHz, CDCl3) δ 10.72 (1H, s), 6.45 (1H, s), 5.68 (1H, br-s), 4.67 (1H, d, J = 2.5 Hz), 4.46 (1H, ddd, J = 2.5, 5.5, 8.3 Hz), 4.05 (1H, br-s), 3.32 (3H, s), 1.94 (1H, m), 1.84 (1H, m), 1.75–1.50 (1H, overlapped), 1.45 (1H, m), 1.40–1.25 (4H, overlapped), 0.90 (3H, t, J = 7.0 Hz); 13C-NMR (125 MHz, CDCl3) δ 169.8, 145.9, 144.5, 137.2, 109.8, 108.4, 106.8, 82.4, 69.1, 56.1, 31.6, 30.1, 24.9, 22.5, 14.0; IR (KBr) 3378, 2957, 2926, 2858, 1681, 1464, 1217, 1171 cm−1; HRMS (ESI) m/z (M + Na)+ calculated for (C15H21NO5Na)+ 318.1317, found 318.1321.
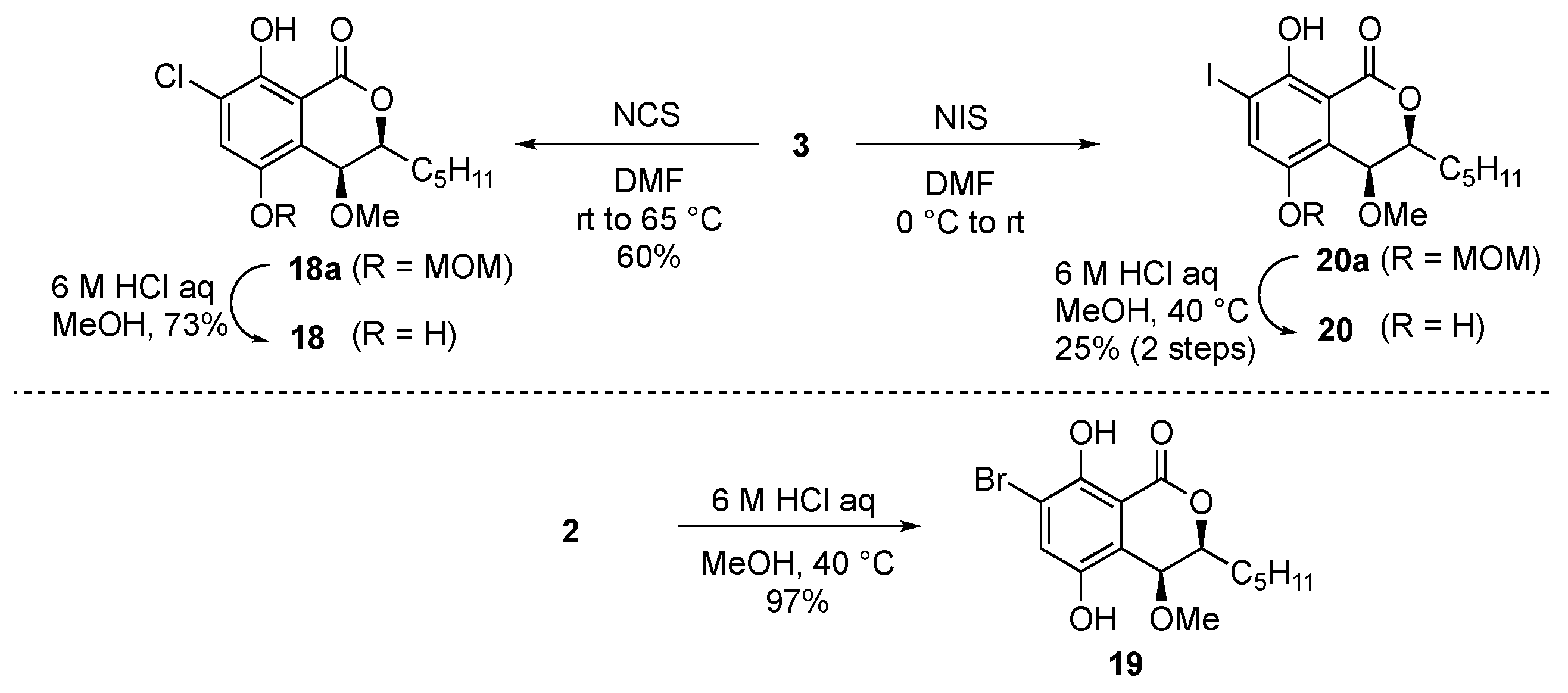
3.1.25. (3S,4S)-7-chloro-8-hydroxy-4-methoxy-5-(methoxymethoxy)-3-pentylisochroman-1-one (18a)
To a solution of 3 (5.0 mg, 15.4 µmol) in DMF (0.18 mL) was added the solution of N-chlorosuccinimide (4.1 mg, 30.8 µmol) in DMF (31 µL) at room temperature. After stirring for 5 h at 65 °C, the reaction was quenched by adding saturated aqueous NaHCO3 at 0 °C. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with PTLC (EtOAc:n-hexane = 1:9) to give monoMOM chloro derivative 18a (3.3 mg, 60%) as a brown solid. m.p. 79–81 °C; 1H-NMR (400 MHz, CDCl3) δ 11.23 (1H, s), 7.55 (1H, s), 5.18 (1H, d, J = 7.0 Hz), 5.16 (1H, d, J = 7.0 Hz), 4.59 (1H, d, J = 1.7 Hz), 4.39 (1H, ddd, J = 1.7, 6.0, 8.0 Hz), 3.50 (3H, s), 3.30 (3H, s), 2.07 (1H, m), 1.86 (1H, m), 1.70-1.50 (1H, overlapped), 1.47 (1H, m), 1.45-1.25 (4H, overlapped), 0.92 (3H, t, J = 7.1 Hz); 13C-NMR (125 MHz, CDCl3) δ 168.7, 152.8, 146.3, 125.1, 123.6, 123.0, 109.0, 95.7, 82.7, 67.4, 56.8, 56.4, 31.5, 30.4, 24.7, 22.5, 14.0.; IR (KBr) 2955, 2927, 2853, 2826, 1681, 1453, 1433, 1206 cm−1; HRMS (ESI) m/z (M + Na)+ calculated for (C17H23O6ClNa)+ 381.1081, found 381.1088.
3.1.26. (3S,4S)-7-chloro-5,8-dihydroxy-4-methoxy-3-pentylisochroman-1-one (18)
To a solution of monoMOM chloro derivative 18a (3.3 mg, 9.20 µmol) in MeOH (0.69 mL) was added 6 M aqueous HCl (0.23 mL) at 0 °C. After stirring for 2 h at 40 °C, the reaction was quenched by adding saturated NaHCO3 at 0 °C. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with PTLC (EtOAc:n-hexane = 1:9) to give chloro derivative 18 (2.1 mg, 73%) as a brown solid. m.p. 119-120 °C; 1H-NMR (400 MHz, CDCl3) δ 11.17 (1H, br-s), 7.34 (1H, s), 6.34 (1H, br-s), 4.82 (1H, br-s), 4.59 (1H, ddd, J = 2.8, 5.6, 8.4 Hz), 3.48 (3H, s), 2.03 (1H, m), 1.93 (1H, m), 1.64 (1H, m), 1.53 (1H, m), 1.51–1.35 (4H, overlapped), 0.98 (3H, t, J = 7.2 Hz); 13C-NMR (100 MHz, CDCl3) δ 168.7, 152.1, 145.6, 124.9, 122.8, 121.1, 108.5, 81.8, 69.6, 57.0, 31.5, 29.8, 24.8, 22.5, 14.0.; IR (KBr) 3282, 2958, 2929, 2860, 1681, 1437, 1198 cm−1; HRMS (ESI) m/z (M + H)+ calculated for (C15H20O5Cl)+ 315.0999, found 315.0998.
3.1.27. (3S,4S)-7-bromo-5,8-dihydroxy-4-methoxy-3-pentylisochroman-1-one (19)
To a solution of bromo derivative 2 (11.0 mg, 24.6 µmol) in MeOH (1.8 mL) was added 6 M aqueous HCl (0.62 mL) at 0 °C. After stirring for 3.5 h at 40 °C, the reaction was quenched by adding saturated aqueous NaHCO3 at 0 °C. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with PTLC (EtOAc:n-hexane = 1:9) to give bromo derivative 19 (8.6 mg, 97%) as a white solid. m.p. 132 °C; 1H-NMR (400 MHz, CDCl3) δ 11.26 (1H, s), 7.36 (1H, s), 6.00 (1H, br-s), 4.76 (1H, d, J = 2.7 Hz), 4.52 (1H, ddd, J = 2.7, 5.1, 8.3 Hz), 3.41 (3H, s), 1.95 (1H, m), 1.86 (1H, m), 1.70-1.50 (2H, overlapped), 1.40–1.25 (4H, overlapped), 0.91 (3H, t, J = 7.0 Hz); 13C-NMR (125 MHz, CDCl3) δ 168.4, 153.0, 145.8, 127.9, 121.5, 111.6, 108.2, 81.4, 70.0, 57.0, 31.5, 29.6, 24.8, 22.5, 14.0.; IR (KBr) 3296, 2955, 2930, 2859, 1679, 1432, 1197 cm−1; HRMS (ESI) m/z (M + Na)+ calculated for (C15H19O5BrNa)+ 381.0314, found 381.0322.
3.1.28. (3S,4S)-5,8-dihydroxy-7-iodo-4-methoxy-3-pentylisochroman-1-one (20)
To a solution of 3 (12.6 mg, 38.8 µmol) in DMF (0.35 mL) was added the solution of N-iodosuccinimide (17.5 mg, 77.6 µmol) in DMF (50 µL) at room temperature. After stirring for 3 h at room temperature, the reaction was quenched by adding saturated aqueous NaHCO3 at 0 °C. The mixture was extracted with CH2Cl2 (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was pathed through SiO2 plug and the resultant mixture of monoMOM iodo derivative 20a was used for the next reaction without further purification. To a solution of crude mixture of 20a in MeOH (0.83 mL) was added 6 M aqueous HCl (0.30 mL) at 0 °C. After stirring for 5 h at 40 °C, the reaction was quenched by adding saturated aqueous NaHCO3 at 0 °C. The mixture was extracted with EtOAc (×3) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified with PTLC (EtOAc:n-hexane = 1:9) to give iodo derivative 20 (4.0 mg, 87%) as a pale-yellow oil. m.p. 109–110 °C; 1H-NMR (500 MHz, CDCl3) δ 11.44 (1H, s), 7.57 (1H, s), 6.11 (1H, br-s), 4.51 (1H, ddd, J = 2.8, 5.4, 8.5 Hz), 3.40 (3H, s), 1.94 (1H, m), 1.85 (1H, m), 1.75–1.50 (4H, overlapped), 1.45 (1H, m), 1.40–1.30 (4H, overlapped), 0.91 (3H, t, J = 7.0 Hz); 13C-NMR (125 MHz, CDCl3) δ 168.3, 155.3, 146.3, 133.8, 122.6, 107.1, 85.5, 81.5, 69.8, 56.9, 31.5, 29.7, 24.8, 22.5, 14.0; IR (KBr) 3293, 2977, 298, 2857, 1674, 1427, 1197 cm−1; HRMS (ESI) m/z (M + Na)+ calculated for (C15H19O5Ina)+ 429.0175, found 429.0174.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}