Natural Bioactive Thiazole-Based Peptides from Marine Resources: Structural and Pharmacological Aspects

 ,
,  ,
,
Abstract
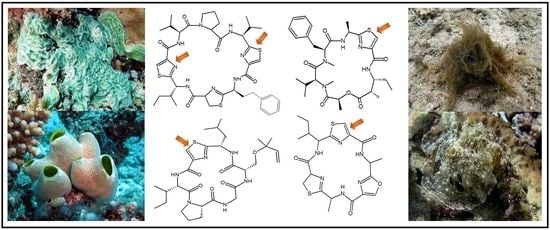
:
1. Introduction
1.1. Resources
1.2. Linear vs. Cyclic Peptides
2. Chemistry
2.1. Structural Features of Thiazole (Tzl)-Containing Cyclooligopeptides
2.2. Structural Features of Tzl-Containing Linear Peptides
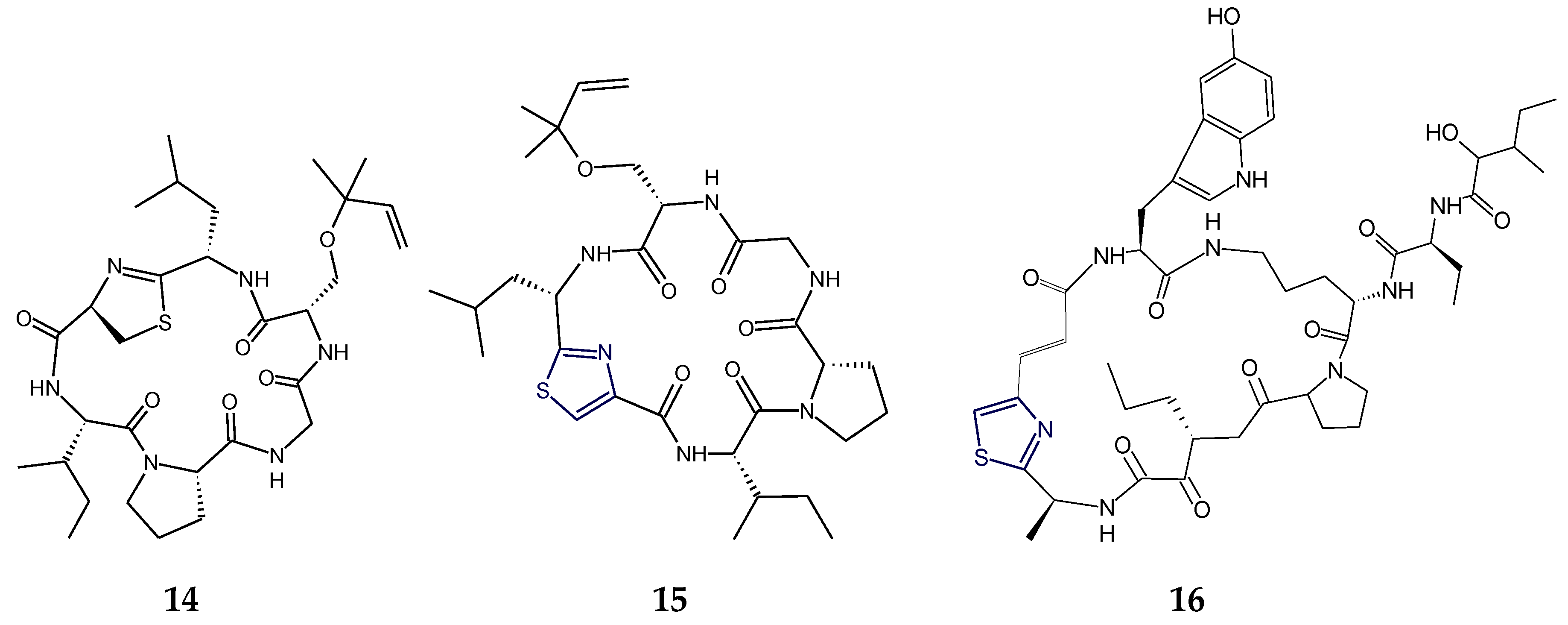
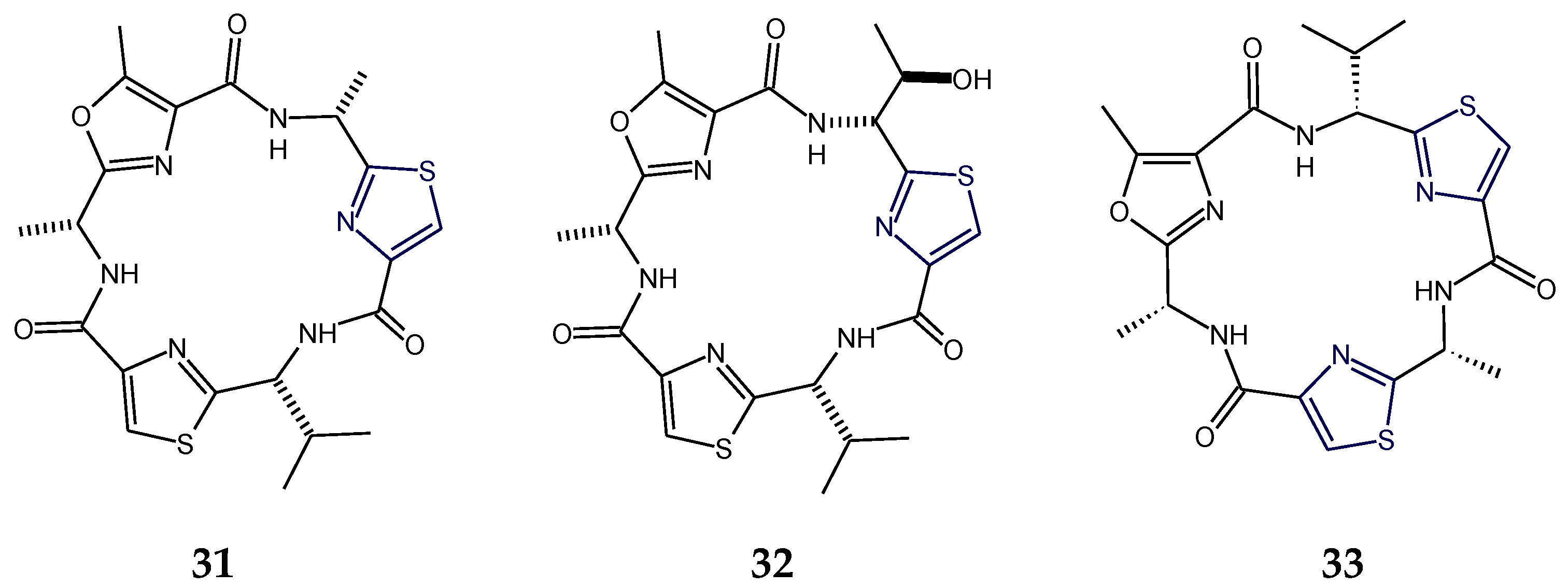
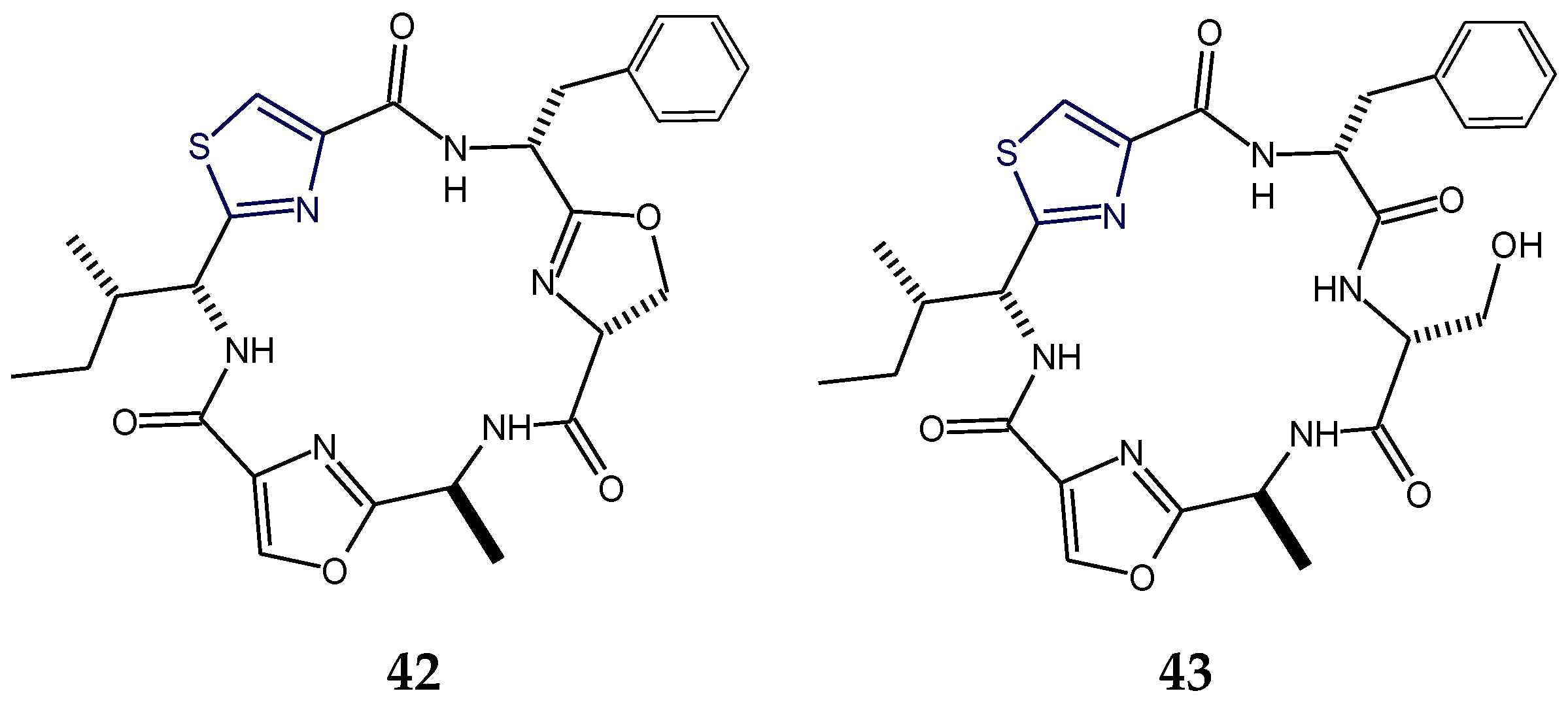
2.3. Structural Features of Thiazole (Tzl)- and Oxazole (Ozl)-Containing Cyclopeptides
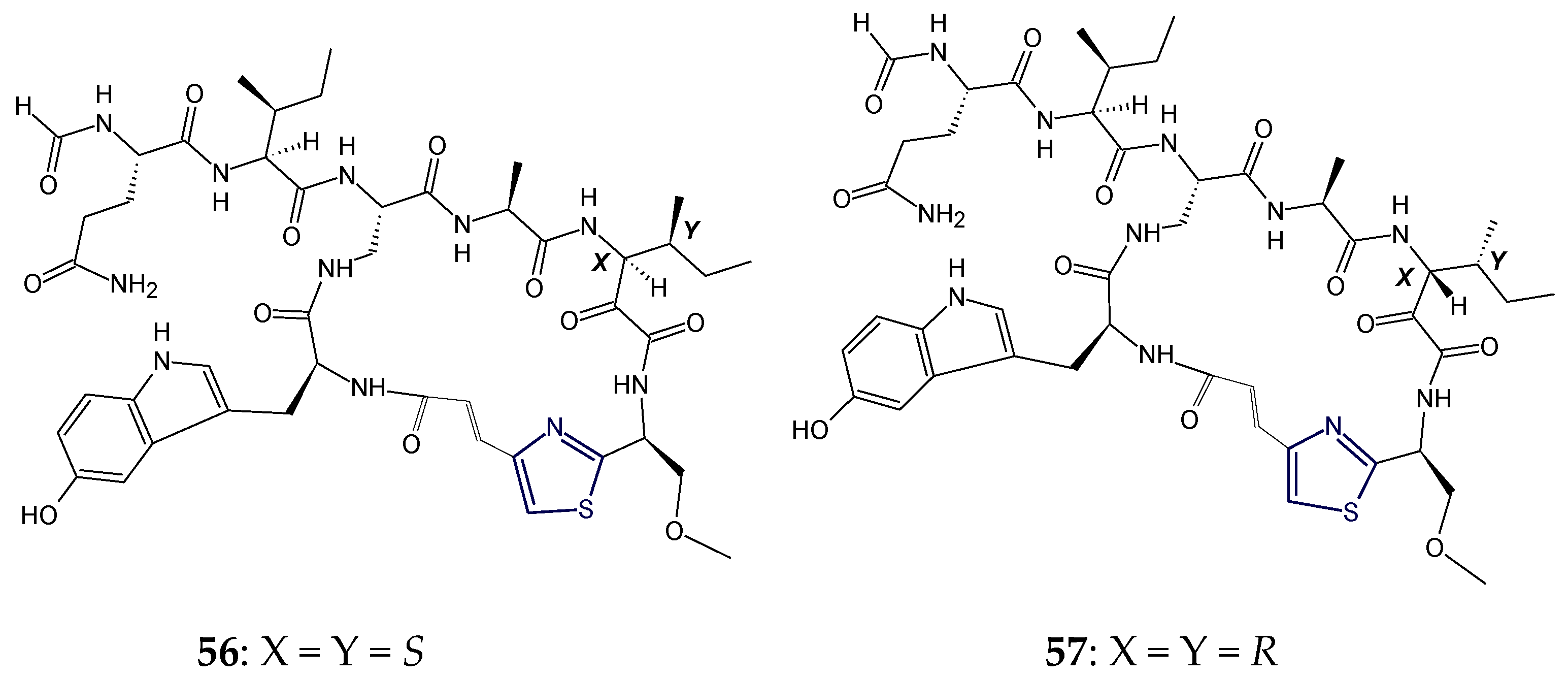
2.4. Structural Features of Thiopeptide Antibiotics
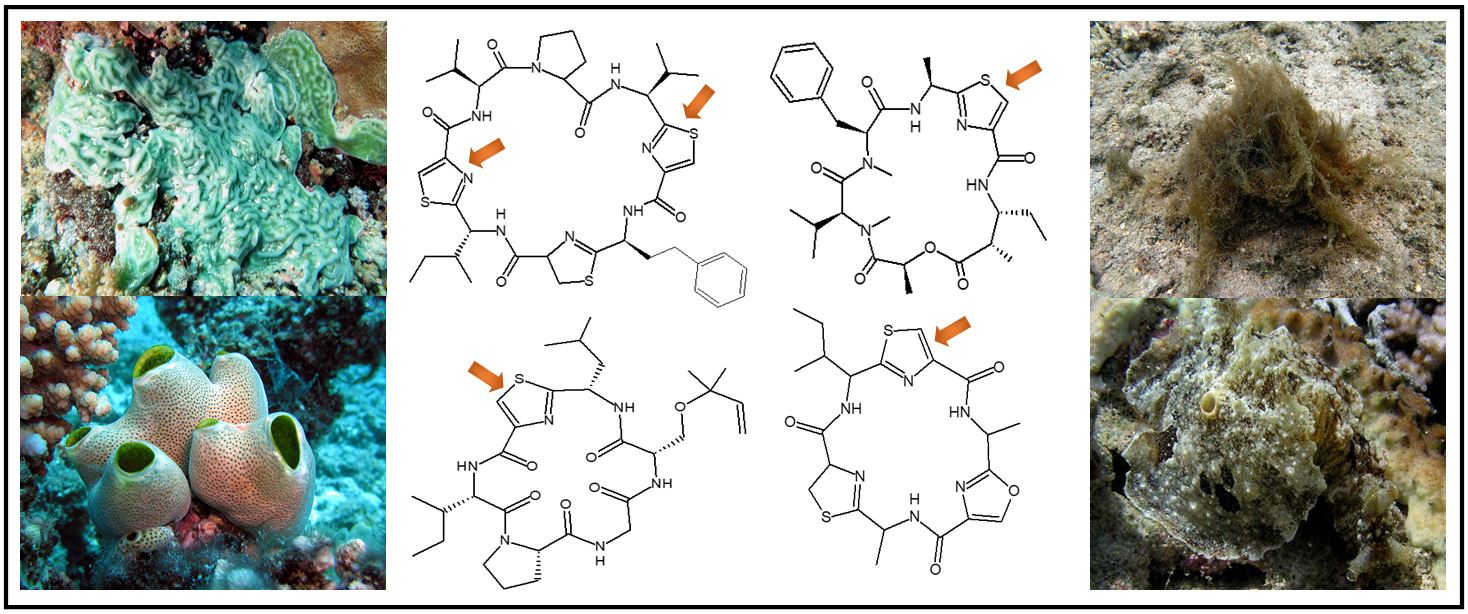
2.5. Structural Features of Bridged Heterocyclic Peptide Bicycles
2.6. Structural Features of Other Heterocyclic Peptides from Marine Resources
3. Stereochemical Aspects
4. Synthesis of Heterocyclic Peptides
5. Structural Activity Relationships
6. Biological Activity
7. Mechanism of Action
8. Issues Associated with Marine Peptides in Drug Development
9. Peptide Market and Clinical Trials
10. Conclusions and Future Prospects
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gomtsyan, A. Heterocycles in drugs and drug discovery. Chem. Heterocyl. Compd. 2012, 48, 7–10. [Google Scholar] [CrossRef]
- Kumawat, M.K. Thiazole containing heterocycles with antimalarial activity. Curr. Drug Discov. Technol. 2018, 15, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Pathak, D.; Dahiya, R.; Pathak, K.; Dahiya, S. New generation antipsychotics: A review. Indian. J. Pharm. Educ. Res. 2006, 40, 77–83. [Google Scholar]
- Fang, W.Y.; Dahiya, R.; Qin, H.L.; Mourya, R.; Maharaj, S. Natural proline-rich cyclopolypeptides from marine organisms: Chemistry, synthetic methodologies and biological status. Mar. Drugs 2016, 14, 194. [Google Scholar] [CrossRef] [Green Version]
- Dahiya, R.; Pathak, D. Cyclic peptides: New hope for antifungal therapy. Egypt. Pharm. J. 2006, 5, 189–199. [Google Scholar]
- Tiwari, J.; Gupta, G.; Dahiya, R.; Pabreja, K.; Kumar Sharma, R.; Mishra, A.; Dua, K. Recent update on biological activities and pharmacological actions of liraglutide. Excli J. 2017, 16, 742–747. [Google Scholar]
- Singh, Y.; Gupta, G.; Shrivastava, B.; Dahiya, R.; Tiwari, J.; Ashwathanarayana, M.; Sharma, R.K.; Agrawal, M.; Mishra, A.; Dua, K. Calcitonin gene-related peptide (CGRP): A novel target for Alzheimer’s disease. CNS Neurosci. Ther. 2017, 23, 457–461. [Google Scholar] [CrossRef]
- Adiv, S.; Ahronov-Nadborny, R.; Carmeli, S. New aeruginazoles, a group of thiazole-containing cyclic peptides from Microcystis aeruginosa blooms. Tetrahedron 2012, 68, 1376–1383. [Google Scholar] [CrossRef]
- Mitchell, S.S.; Faulkner, D.J.; Rubins, K.; Bushman, F.D. Dolastatin 3 and two novel cyclic peptides from a Palauan collection of Lyngbya majuscula. J. Nat. Prod. 2000, 63, 279–282. [Google Scholar] [CrossRef] [Green Version]
- McIntosh, J.A.; Lin, Z.; Tianero, M.D.; Schmidt, E.W. Aestuaramides, a natural library of cyanobactin cyclic peptides resulting from isoprene-derived Claisen rearrangements. ACS Chem. Biol. 2013, 8, 877–883. [Google Scholar] [CrossRef] [Green Version]
- Ploutno, A.; Carmeli, S. Modified peptides from a water bloom of the cyanobacterium Nostoc sp. Tetrahedron 2002, 58, 9949–9957. [Google Scholar] [CrossRef]
- Williams, P.G.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. Isolation and structure determination of obyanamide, a novel cytotoxic cyclic depsipeptide from the marine cyanobacterium Lyngbya confervoides. J. Nat. Prod. 2002, 65, 29–31. [Google Scholar] [CrossRef] [PubMed]
- Luesch, H.; Williams, P.G.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. Ulongamides A-F, new beta-amino acid-containing cyclodepsipeptides from Palauan collections of the marine cyanobacterium Lyngbya sp. J. Nat. Prod. 2002, 65, 996–1000. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.T.; Sitachitta, N.; Gerwick, W.H. The guineamides, novel cyclic depsipeptides from a Papua New Guinea collection of the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2003, 66, 764–771. [Google Scholar] [CrossRef] [PubMed]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. New apratoxins of marine cyanobacterial origin from Guam and Palau. Bioorg. Med. Chem. 2002, 10, 1973–1978. [Google Scholar] [CrossRef]
- Hong, J.; Luesch, H. Largazole: From discovery to broad-spectrum therapy. Nat. Prod. Rep. 2012, 29, 449–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudek, S.; Haygood, M.G.; Youssef, D.T.A.; Schmidt, E.W. Structure of Trichamide, a cyclic peptide from the bloom-forming cyanobacterium Trichodesmium erythraeum, predicted from the genome sequence. Appl. Environ. Microbiol. 2006, 72, 4382–4387. [Google Scholar] [CrossRef] [Green Version]
- Zafrir-Ilan, E.; Carmeli, S. Two new microcyclamides from a water bloom of the cyanobacterium Microcystis sp. Tetrahedron Lett. 2010, 51, 6602–6604. [Google Scholar] [CrossRef]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J.; Corbett, T.H. Total structure determination of apratoxin A, a potent novel cytotoxin from the marine cyanobacterium Lyngbya majuscula. J. Am. Chem. Soc. 2001, 123, 5418–5423. [Google Scholar] [CrossRef]
- Gutiérrez, M.; Suyama, T.L.; Engene, N.; Wingerd, J.S.; Matainaho, T.; Gerwick, W.H. Apratoxin D, a potent cytotoxic cyclodepsipeptide from papua new guinea collections of the marine cyanobacteria Lyngbya majuscule and Lyngbya sordida. J. Nat. Prod. 2008, 71, 1099–1103. [Google Scholar] [CrossRef]
- Matthew, S.; Schupp, P.J.; Luesch, H. Apratoxin E, a cytotoxic peptolide from a Guamanian collection of the marine cyanobacterium Lyngbya bouillonii. J. Nat. Prod. 2008, 71, 1113–1116. [Google Scholar] [CrossRef]
- Tidgewell, K.; Engene, N.; Byrum, T.; Media, J.; Doi, T.; Valeriote, F.A.; Gerwick, W.H. Evolved diversification of a modular natural product pathway: Apratoxins F and G, two cytotoxic cyclic depsipeptides from a Palmyra collection of Lyngbya bouillonii. ChemBioChem 2010, 11, 1458–1466. [Google Scholar] [CrossRef] [PubMed]
- Thornburg, C.C.; Cowley, E.S.; Sikorska, J.; Shaala, L.A.; Ishmael, J.E.; Youssef, D.T.A.; McPhail, K.L. Apratoxin H and apratoxin A sulfoxide from the Red sea cyanobacterium Moorea producens. J. Nat. Prod. 2013, 76, 1781–1788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwan, J.C.; Ratnayake, R.; Abboud, K.A.; Paul, V.J.; Luesch, H. Grassypeptolides A−C, cytotoxic bis-thiazoline containing marine cyclodepsipeptides. J. Org. Chem. 2010, 75, 8012–8023. [Google Scholar] [CrossRef] [PubMed]
- Thornburg, C.C.; Thimmaiah, M.; Shaala, L.A.; Hau, A.M.; Malmo, J.M.; Ishmael, J.E.; Youssef, D.T.A.; McPhail, K.L. Cyclic depsipeptides, grassypeptolides D, E and Ibu epidemethoxylyngbyastatin 3, from a Red sea Leptolyngbya cyanobacterium. J. Nat. Prod. 2011, 74, 1677–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popplewell, W.L.; Ratnayake, R.; Wilson, J.A.; Beutler, J.A.; Colburn, N.H.; Henrich, C.J.; McMahon, J.B.; McKee, T.C. Grassypeptolides F and G, cyanobacterial peptides from Lyngbya majuscula. J. Nat. Prod. 2011, 74, 1686–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J.; Mooberry, S.L. Isolation, Structure determination, and biological activity of lyngbyabellin A from the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2000, 63, 611–615. [Google Scholar] [CrossRef]
- Han, B.; McPhail, K.L.; Gross, H.; Goeger, D.E.; Mooberry, S.L.; Gerwick, W.H. Isolation and structure of five lyngbyabellin derivatives from a Papua New Guinea collection of the marine cyanobacterium Lyngbya majuscula. Tetrahedron 2005, 61, 11723–11729. [Google Scholar] [CrossRef]
- Choi, H.; Mevers, E.; Byrum, T.; Valeriote, F.A.; Gerwick, W.H. Lyngbyabellins K-N from two Palmyra Atoll collections of the marine cyanobacterium Moorea bouilloni. Eur. J. Org. Chem. 2012, 2012(27), 5141–5150. [Google Scholar] [CrossRef]
- Matthew, S.; Salvador, L.A.; Schupp, P.J.; Paul, V.J.; Luesch, H. Cytotoxic halogenated macrolides and modified peptides from the apratoxin-producing marine cyanobacterium Lyngbya bouillonii from Guam. J. Nat. Prod. 2010, 73, 1544–1552. [Google Scholar] [CrossRef] [Green Version]
- Soria-Mercado, I.E.; Pereira, A.; Cao, Z.; Murray, T.F.; Gerwick, W.H. Alotamide A, a novel neuropharmacological agent from the marine cyanobacterium Lyngbya bouillonii. Org. Lett. 2009, 11, 4704–4707. [Google Scholar] [CrossRef] [Green Version]
- Admi, V.; Afek, U.; Carmeli, S. Raocyclamides A and B, novel cyclic hexapeptides isolated from the cyanobacterium Oscillatoria raoi. J. Nat. Prod. 1996, 59, 396–399. [Google Scholar] [CrossRef]
- Portmann, C.; Sieber, S.; Wirthensohn, S.; Blom, J.F.; Da Silva, L.; Baudat, E.; Kaiser, M.; Brun, R.; Gademann, K. Balgacyclamides, antiplasmodial heterocyclic peptides from Microcystis aeruguinosa EAWAG 251. J. Nat. Prod. 2014, 77, 557–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linington, R.G.; González, J.; Ureña, L.-D.; Romero, L.I.; Ortega-Barría, E.; Gerwick, W.H. Venturamides A and B: Antimalarial constituents of the Panamanian marine cyanobacterium Oscillatoria sp. J. Nat. Prod. 2007, 70, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Ishida, K.; Nakagawa, H.; Murakami, M. Microcyclamide, a cytotoxic cyclic hexapeptide from the cyanobacterium Microcystis aeruginosa. J. Nat. Prod. 2000, 63, 1315–1317. [Google Scholar] [CrossRef]
- Jüttner, F.; Todorova, A.K.; Walch, N.; von Philipsborn, W. Nostocyclamide M: A cyanobacterial cyclic peptide with allelopathic activity from Nostoc 31. Phytochemistry 2001, 57, 613–619. [Google Scholar] [CrossRef]
- Portmann, C.; Blom, J.F.; Gademann, K.; Jüttner, F. Aerucyclamides A and B: Isolation and synthesis of toxic ribosomal heterocyclic peptides from the cyanobacterium Microcystis aeruginosa PCC 7806. J. Nat. Prod. 2008, 71, 1193–1196. [Google Scholar] [CrossRef]
- Portmann, C.; Blom, J.F.; Kaiser, M.; Brun, R.; Jüttner, F.; Gademann, K. Isolation of aerucyclamides C and D and structure revision of microcyclamide 7806A: Heterocyclic ribosomal peptides from Microcystis aeruginosa PCC 7806 and their antiparasite evaluation. J. Nat. Prod. 2008, 71, 1891–1896. [Google Scholar] [CrossRef]
- Chuang, P.-H.; Hsieh, P.-W.; Yang, Y.-L.; Hua, K.-F.; Chang, F.-R.; Shiea, J.; Wu, S.-H.; Wu, Y.-C. Cyclopeptides with anti-inflammatory activity from seeds of Annona montana. J. Nat. Prod. 2008, 71, 1365–1370. [Google Scholar] [CrossRef]
- Ogino, J.; Moore, R.E.; Patterson, G.M.L.; Smith, C.D. Dendroamides, new cyclic hexapeptides from a blue-green alga. Multidrug-resistance reversing activity of dendroamide A. J. Nat. Prod. 1996, 59, 581–586. [Google Scholar] [CrossRef]
- McDonald, L.A.; Foster, M.P.; Phillips, D.R.; Ireland, C.M.; Lee, A.Y.; Clardy, J. Tawicyclamides A and B, new cyclic peptides from the ascidian Lissoclinum patella: Studies on the solution- and solid-state conformations. J. Org. Chem. 1992, 57, 4616–4624. [Google Scholar] [CrossRef]
- Arrault, A.; Witczak-Legrand, A.; Gonzalez, P.; Bontemps-Subielos, N.; Banaigs, B. Structure and total synthesis of cyclodidemnamide B, a cycloheptapeptide from the ascidian Didemnum molle. Tetrahedron Lett. 2002, 43, 4041–4044. [Google Scholar] [CrossRef]
- Carroll, A.R.; Bowden, B.F.; Coll, J.C.; Hockless, D.C.R.; Skelton, B.W.; White, A.H. Studies of Australian ascidians. IV. Mollamide, a cytotoxic cyclic heptapeptide from the compound ascidian Didemnum molle. Aust. J. Chem. 1994, 47, 61–69. [Google Scholar] [CrossRef]
- Carroll, A.R.; Coll, J.C.; Bourne, J.C.; MacLeod, J.K.; Zanriskie, T.M.; Ireland, C.M.; Bowden, B.F. Patellins 1-6 and trunkamide A: Novel cyclic hexa-, hepta- and octa-peptides from colonial ascidians, Lissoclinum sp. Aust. J. Chem. 1996, 49, 659–667. [Google Scholar] [CrossRef]
- Rudi, A.; Aknin, M.; Gaydou, E.M.; Kashman, Y. Four new cytotoxic cyclic hexa- and heptapeptides from the marine ascidian Didemnum molle. Tetrahedron 1998, 54, 13203–13210. [Google Scholar] [CrossRef]
- Donia, M.S.; Wang, B.; Dunbar, D.C.; Desai, P.V.; Patny, A.; Avery, M.; Hamann, M.T. Mollamides B and C, cyclic hexapeptides from the Indonesian tunicate Didemnum molle. J. Nat. Prod. 2008, 71, 941–945. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Harper, M.K.; Pond, C.D.; Barrows, L.R.; Ireland, C.M.; Van Wagoner, R.M. Thiazoline peptides and a tris-phenethyl urea from Didemnum molle with anti-HIV activity. J. Nat. Prod. 2012, 75, 1436–1440. [Google Scholar] [CrossRef] [Green Version]
- Rudi, A.; Chill, L.; Aknin, M.; Kashman, Y. Didmolamide A and B, two new cyclic hexapeptides from the marine ascidian Didemnum molle. J. Nat. Prod. 2003, 66, 575–577. [Google Scholar] [CrossRef]
- Teruya, T.; Sasaki, H.; Suenaga, K. Hexamollamide, a hexapeptide from an Okinawan ascidian Didemnum molle. Tetrahedron Lett. 2008, 49, 5297–5299. [Google Scholar] [CrossRef]
- Perez, L.J.; Faulkner, D.J. Bistratamides E-J, modified cyclic hexapeptides from the Philippines ascidian Lissoclinum bistratum. J. Nat. Prod. 2003, 66, 247–250. [Google Scholar] [CrossRef]
- Fu, X.; Do, T.; Schmitz, F.J.; Andrusevich, V.; Engel, M.H. New cyclic peptides from the ascidian Lissoclinum patella. J. Nat. Prod. 1998, 61, 1547–1551. [Google Scholar] [CrossRef] [PubMed]
- Morris, L.A.; Jantina Kettenes van den Bosch, J.; Versluis, K.; Thompson, G.S.; Jaspars, M. Structure determination and MSn analysis of two new lissoclinamides isolated from the Indo-Pacific ascidian Lissoclinum patella: NOE restrained molecular dynamics confirms the absolute stereochemistry derived by degradative methods. Tetrahedron 2000, 56, 8345–8353. [Google Scholar] [CrossRef]
- Ireland, C.; Scheuer, P.J. Ulicyclamide and ulithiacyclamide, two new small peptides from a marine tunicate. J. Am. Chem. Soc. 1980, 102, 5688–5691. [Google Scholar] [CrossRef]
- Rashid, M.A.; Gustafson, K.R.; Cardellina II, J.H.; Boyd, M.R. Patellamide F, a new cytotoxic cyclic peptide from the colonial ascidian Lissoclinum patella. J. Nat. Prod. 1995, 58, 594–597. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, C.J.; Lavin, M.F.; Marshall, K.A.; Van den Brenk, A.L.; Watters, D.J. Structure-activity relationships of the lissoclinamides: Cytotoxic cyclic peptides from the ascidian Lissoclinum patella. J. Med. Chem. 1990, 33, 1634–1638. [Google Scholar] [CrossRef] [PubMed]
- Degnan, B.M.; Hawkins, C.J.; Lavin, M.F.; McCaffrey, E.J.; Parry, D.L.; Van den Brenk, A.L.; Watters, D.J. New cyclic peptides with cytotoxic activity from the ascidian Lissoclinum patella. J. Med. Chem. 1989, 32, 1349–1354. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.E.; Moore, R.E. The structure of ulithiacyclamide B. Antitumor evaluation of cyclic peptides and macrolides from Lissoclinum patella. J. Nat. Prod. 1989, 52, 732–739. [Google Scholar] [CrossRef]
- McDonald, L.A.; Ireland, C.M. Patellamide E: A new cyclic peptide from the ascidian Lissoclinum patella. J. Nat. Prod. 1992, 55, 376–379. [Google Scholar] [CrossRef]
- Foster, M.P.; Concepcion, G.P.; Caraan, G.B.; Ireland, C.M. Bistratamides C and D. two new oxazole-containing cyclic hexapeptides isolated from a Philippine Lissoclinum bistratum ascidian. J. Org. Chem. 1992, 57, 6671–6675. [Google Scholar] [CrossRef]
- Degnan, B.M.; Hawkins, C.J.; Lavin, M.F.; McCaffrey, E.J.; Parry, D.L.; Watters, D.J. Novel cytotoxic compounds from the ascidian Lissoclinum bistratum. J. Med. Chem. 1989, 32, 1354–1359. [Google Scholar] [CrossRef]
- Urda, C.; Fernández, R.; Rodríguez, J.; Pérez, M.; Jiménez, C.; Cuevas, C. Bistratamides M and N, oxazole-thiazole containing cyclic hexapeptides isolated from Lissoclinum bistratum interaction of zinc (II) with bistratamide K. Mar. Drugs 2017, 15, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toske, S.G.; Fenical, W. Cyclodidemnamide: A new cyclic heptapeptide from the marine ascidian Didemnum molle. Tetrahedron Lett. 1995, 36, 8355–8358. [Google Scholar] [CrossRef]
- Rashid, M.A.; Gustafson, K.R.; Boswell, J.L.; Boyd, M.R. Haligramides A and B, two new cytotoxic hexapeptides from the marine sponge Haliclona nigra. J. Nat. Prod. 2000, 63, 956–959. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.W.; Raventos-Suarez, C.; Bifano, M.; Menendez, A.T.; Fairchild, C.R.; Faulkner, D.J. Scleritodermin A, a cytotoxic cyclic peptide from the Lithistid sponge Scleritoderma nodosum. J. Nat. Prod. 2004, 67, 475–478. [Google Scholar] [CrossRef]
- Chill, L.; Kashman, Y.; Schleyer, M. Oriamide, a new cytotoxic cyclic peptide containing a novel amino acid from the marine sponge Theonella sp. Tetrahedron 1997, 53, 16147–16152. [Google Scholar] [CrossRef]
- Mau, C.M.S.; Nakao, Y.; Yoshida, W.Y.; Scheuer, P.J. Waiakeamide, a cyclic hexapeptide from the sponge Ircinia dendroides. J. Org. Chem. 1996, 61, 6302–6304. [Google Scholar] [CrossRef]
- Kobayashi, J.; Itagaki, F.; Shigemori, I.; Takao, T.; Shimonishi, Y. Keramamides E, G, H, and J, new cyclic peptides containing an oxazole or a thiazole ring from a Theonella sponge. Tetrahedron 1995, 51, 2525–2532. [Google Scholar] [CrossRef]
- Issac, M.; Aknin, M.; Gauvin-Bialecki, A.; De Voogd, N.; Ledoux, A.; Frederich, M.; Kashman, Y.; Carmeli, S. Cyclotheonellazoles A–C, potent protease inhibitors from the marine sponge Theonella aff. swinhoei. J. Nat. Prod. 2017, 80, 1110–1116. [Google Scholar] [CrossRef]
- Erickson, K.L.; Gustafson, K.R.; Milanowski, D.J.; Pannell, L.K.; Klose, J.R.; Boyd, M.R. Myriastramides A–C, new modified cyclic peptides from the Philippines marine sponge Myriastra clavosa. Tetrahedron 2003, 59, 10231–10238. [Google Scholar] [CrossRef]
- Kehraus, S.; König, G.M.; Wright, A.D.; Woerheide, G. Leucamide A: A new cytotoxic heptapeptide from the Australian sponge Leucetta microraphis. J. Org. Chem. 2002, 67, 4989–4992. [Google Scholar] [CrossRef]
- Tan, K.O.; Wakimoto, T.; Takada, K.; Ohtsuki, T.; Uchiyama, N.; Goda, Y.; Abe, I. Cycloforskamide, a cytotoxic macrocyclic peptide from the sea slug Pleurobranchus forskalii. J. Nat. Prod. 2013, 76, 1388–1391. [Google Scholar] [CrossRef] [PubMed]
- Wesson, K.J.; Hamann, M.T. Keenamide A, a bioactive cyclic peptide from the marine mollusk Pleurobranchus forskalii. J. Nat. Prod. 1996, 59, 629–631. [Google Scholar] [CrossRef] [PubMed]
- Dalisay, D.S.; Rogers, E.W.; Edison, A.S.; Molinski, T.F. Structure elucidation at the nanomole scale. 1. Trisoxazole macrolides and thiazole-containing cyclic peptides from the nudibranch Hexabranchus sanguineus. J. Nat. Prod. 2009, 72, 732–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Huang, H.; Chen, Y.; Tan, J.; Song, Y.; Zou, J.; Tian, X.; Hua, Y.; Ju, J. Marthiapeptide A, an anti-infective and cytotoxic polythiazole cyclopeptide from a 60 L scale fermentation of the deep sea-derived Marinactinospora thermotolerans SCSIO 00652. J. Nat. Prod. 2012, 75, 2251–2255. [Google Scholar] [CrossRef]
- Sone, H.; Kigoshi, H.; Yamada, K. Isolation and stereostructure of dolastatin I, a cytotoxic cyclic hexapeptide from the Japanese sea hare Dolabella auricularia. Tetrahedron 1997, 53, 8149–8154. [Google Scholar] [CrossRef]
- Ojika, M.; Nemoto, T.; Nakamura, M.; Yamada, K. Dolastatin E, a new cyclic hexapeptide isolated from the sea hare Dolabella auricularia. Tetrahedron Lett. 1995, 36, 5057–5058. [Google Scholar] [CrossRef]
- Tan, L.T.; Williamson, R.T.; Gerwick, W.H.; Watts, K.S.; McGough, K.; Jacobs, R. cis, cis- and trans, trans-Ceratospongamide, new bioactive cyclic heptapeptides from the Indonesian red alga Ceratodictyon spongiosum and symbiotic sponge Sigmadocia symbiotica. J. Org. Chem. 2000, 65, 419–425. [Google Scholar] [CrossRef]
- Matsuo, Y.; Kanoh, K.; Yamori, T.; Kasai, H.; Katsuta, A.; Adachi, K.; Shin-ya, K.; Shizuri, Y. Urukthapelstatin A, a novel cytotoxic substance from marine-derived Mechercharimyces asporophorigenens YM11-542. J. Antibiot. 2007, 60, 251–255. [Google Scholar] [CrossRef] [Green Version]
- Kanoh, K.; Matsuo, Y.; Adachi, K.; Imagawa, H.; Nishizawa, M.; Shizuri, Y. Mechercharmycins A and B, cytotoxic substances from marine-derived Thermoactinomyces sp. YM3-251. J. Antibiot. 2005, 58, 289–292. [Google Scholar] [CrossRef] [Green Version]
- Itokawa, H.; Yun, Y.; Morita, H.; Takeya, K.; Yamada, K. Estrogen-like activity of cyclic peptides from Vaccaria segetalis extracts. Planta Med. 1995, 61, 561–562. [Google Scholar] [CrossRef]
- Joo, S.H. Cyclic Peptides as therapeutic agents and biochemical tools. Biomol. Ther. (Seoul) 2012, 20, 19–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodwin, D.; Simerska, P.; Toth, I. Peptides as therapeutics with enhanced bioactivity. Curr. Med. Chem. 2012, 19, 4451–4461. [Google Scholar] [CrossRef]
- Pathak, D.; Dahiya, R. Cyclic peptides as novel antineoplastic agents: A review. J. Sci. Pharm. 2003, 4, 125–131. [Google Scholar]
- Clark, W.D.; Corbett, T.; Valeriote, F.; Crews, P. Cyclocinamide A. An unusual cytotoxic halogenated hexapeptide from the marine sponge Psammocinia. J. Am. Chem. Soc. 1997, 119, 9285–9286. [Google Scholar] [CrossRef]
- Laird, D.W.; LaBarbera, D.V.; Feng, X.; Bugni, T.S.; Harper, M.K.; Ireland, C.M. Halogenated cyclic peptides isolated from the sponge Corticium sp. J. Nat. Prod. 2007, 70, 741–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. Isolation and structure of the cytotoxin Lyngbyabellin B and absolute configuration of Lyngbyapeptin A from the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2000, 63, 1437–1439. [Google Scholar] [CrossRef]
- Kobayashi, L.; Sato, M.; Murayama, T.; Ishibashi, M.; Walchi, M.R.; Kanai, M.; Shoji, J.; Ohizumie, Y. Konbamide, a novel peptide with calmodulin antagonistic activity from the okinawan marine sponge Theonella sp. J. Chem. Soc. Chem. Commun. 1991, 1050–1052. [Google Scholar] [CrossRef]
- Gulavita, N.K.; Gunasekela, S.P.; Pomponi, S.A.; Robinson, E.V. Polydiscamide A: A new bioactive depsipeptide from the marine sponge Discodermia sp. J. Org. Chem. 1992, 57, 1767–1772. [Google Scholar] [CrossRef]
- Jamison, M.T.; Molinski, T.F. Jamaicensamide A, a peptide containing β-amino-α-keto and thiazole-homologated η-amino acid residues from the sponge Plakina jamaicensis. J. Nat. Prod. 2016, 79, 2243–2249. [Google Scholar] [CrossRef]
- Pettit, G.R.; Kamano, Y.; Holzapfel, C.W.; van Zyl, W.J.; Tuinman, A.A.; Herald, C.L.; Baczynskyj, L.; Schmidt, J.M. Antineoplastic agents. 150. The structure and synthesis of dolastatin 3. J. Am. Chem. Soc. 1987, 109, 7581–7582. [Google Scholar] [CrossRef]
- Raveh, A.; Carmeli, S. Aeruginazole A, a novel thiazole-containing cyclopeptide from the cyanobacterium Microcystis sp. Org. Lett. 2010, 12, 3536–3539. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.G.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. Micromide and guamamide: Cytotoxic alkaloids from a species of the marine cyanobacterium Symploca. J. Nat. Prod. 2004, 67, 49–53. [Google Scholar] [CrossRef]
- Poncet, J. The dolastatins, a family of promising antineoplastic agents. Curr. Pharm. Des. 1999, 5, 139–162. [Google Scholar] [PubMed]
- Luesch, H.; Moore, R.E.; Paul, V.J.; Mooberry, S.L.; Corbett, T.H. Isolation of dolastatin 10 from the marine cyanobacterium Symploca species VP642 and total stereochemistry and biological evaluation of its analogue symplostatin 1. J. Nat. Prod. 2001, 64, 907–910. [Google Scholar] [CrossRef] [PubMed]
- Harrigan, G.G.; Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Nagle, D.G.; Paul, V.J.; Mooberry, S.L.; Corbett, T.H.; Valeriote, F.A. Symplostatin 1: A dolastatin 10 analogue from the marine cyanobacterium Symploca hydnoides. J. Nat. Prod. 1998, 61, 1075–1077. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Xu, J.; Williams, M.D.; Hogan, F.; Schmidt, J.M.; Cerny, R.L. Antineoplastic agents 370. Isolation and structure of dolastatin 18. Bioorg. Med. Cem. Lett. 1997, 7, 827–832. [Google Scholar] [CrossRef]
- Klein, D.; Braekman, J.-C.; Daloze, D.; Hoffmann, L.; Castillo, G.; Demoulin, V. Lyngbyapeptin A, a modified tetrapeptide from Lyngbya bouillonii (Cyanophyceae). Tetrahedron Lett. 1999, 40, 695–696. [Google Scholar] [CrossRef]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. Structurally diverse new alkaloids from Palauan collections of the apratoxin-producing marine cyanobacterium Lyngbya sp. Tetrahedron 2002, 58, 7959–7966. [Google Scholar] [CrossRef]
- Tan, L.T. Marine Cyanobacteria: A Treasure Trove of Bioactive Secondary Metabolites for Drug Discovery. In Studies in Natural Product Chemistry, 1st ed.; Atta-ur-Rahman, Ed.; Elsevier: Amsterdam, The Netherlands, 2012; Volume 36, p. 80, Chapter 4. [Google Scholar]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. Apramides A−G, novel lipopeptides from the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2000, 63, 1106–1112. [Google Scholar] [CrossRef]
- Sorek, H. Isolation, structure elucidation and biological activity of natural products from marine organisms. Ph.D. Thesis, Tel Aviv University, Tel Aviv, Israel, 2010. [Google Scholar]
- Boden, C.; Pattenden, G. Total synthesis of lissoclinamide 5, a cytotoxic cyclic peptide from the tunicate Lissoclinum patella. Tetrahedron Lett. 1994, 35, 8271–8274. [Google Scholar] [CrossRef]
- Wipf, P.; Fritch, P.C. Total synthesis and assignment of configuration of lissoclinamide 7. J. Am. Chem. Soc. 1996, 118, 12358–12367. [Google Scholar] [CrossRef]
- Boden, C.D.J.; Pattenden, G. Total syntheses and re-assignment of configurations of the cyclopeptides lissoclinamide 4 and lissoclinamide 5 from Lissoclinum patella. J. Chem. Soc. Perkin Trans. 1 2000, 6, 875–882. [Google Scholar] [CrossRef]
- Banker, R.; Carmeli, S. Tenuecyclamides A−D, cyclic hexapeptides from the cyanobacterium Nostoc spongiaeforme var. tenue. J. Nat. Prod. 1998, 61, 1248–1251. [Google Scholar] [CrossRef] [PubMed]
- Hamamoto, Y.; Endo, M.; Nakagawa, M.; Nakanishi, T.; Mizukawa, K. A new cyclic peptide, ascidiacyclamide, isolated from ascidian. J. Chem. Soc. Chem. Commun. 1983, 6, 323–324. [Google Scholar] [CrossRef]
- Todorova, A.K.; Juettner, F.; Linden, A.; Pluess, T.; von Philipsborn, W. Nostocyclamide: A new macrocyclic, thiazole-containing allelochemical from Nostoc sp. 31 (cyanobacteria). J. Org. Chem. 1995, 60, 7891–7895. [Google Scholar] [CrossRef]
- Sera, Y.; Adachi, K.; Fujii, K.; Shizuri, Y. A new antifouling hexapeptide from a palauan sponge, Haliclona sp. J. Nat. Prod. 2003, 66, 719–721. [Google Scholar] [CrossRef]
- Uemoto, H.; Yahiro, Y.; Shigemori, H.; Tsuda, M.; Takao, T.; Shimonishi, Y.; Kobayashi, J. Keramamides K and L, new cyclic peptides containing unusual tryptophan residue from Theonella sponge. Tetrahedron 1998, 54, 6719–6724. [Google Scholar] [CrossRef]
- Kimura, M.; Wakimoto, T.; Egami, Y.; Tan, K.C.; Ise, Y.; Abe, I. Calyxamides A and B, cytotoxic cyclic peptides from the marine sponge Discodermia calyx. J. Nat. Prod. 2012, 75, 290–294. [Google Scholar] [CrossRef]
- Just-Baringo, X.; Albericio, F.; Alvarez, M. Thiopeptide antibiotics: Retrospective and recent advances. Mar. Drugs 2014, 12, 317–351. [Google Scholar] [CrossRef] [Green Version]
- Engelhardt, K.; Degnes, K.F.; Kemmler, M.; Bredholt, H.; Fjaervik, E.; Klinkenberg, G.; Sletta, H.; Ellingsen, T.E.; Zotchev, S.B. Production of a new thiopeptide antibiotic, TP-1161, by a marine Nocardiopsis species. Appl. Environ. Microbiol. 2010, 76, 4969–4976. [Google Scholar] [CrossRef] [Green Version]
- Suzumura, K.; Yokoi, T.; Funatsu, M.; Nagai, K.; Tanaka, K.; Zhang, H.; Suzuki, K. YM-266183 and YM-266184, novel thiopeptide antibiotics produced by Bacillus cereus isolated from a marine sponge II. Structure elucidation. J. Antibiot (Tokyo) 2003, 56, 129–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palomo, S.; González, I.; de la Cruz, M.; Martín, J.; Tormo, J.R.; Anderson, M.; Hill, R.T.; Vicente, F.; Reyes, F.; Genilloud, O. Sponge-derived Kocuria and Micrococcus spp. as sources of the new thiazolyl peptide antibiotic kocurin. Mar. Drugs 2013, 11, 1071–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Just-Baringo, X.; Bruno, P.; Ottesen, L.K.; Cañedo, L.M.; Albericio, F.; Álvarez, M. Total synthesis and stereochemical assignment of baringolin. Angew. Chem. Int. Ed. Engl. 2013, 52, 7818–7821. [Google Scholar] [CrossRef] [PubMed]
- Iniyan, A.M.; Sudarman, E.; Wink, J.; Kannan, R.R.; Vincent, S.G.P. Ala-geninthiocin, a new broad spectrum thiopeptide antibiotic, produced by a marine Streptomyces sp. ICN19. J. Antibiot. 2019, 72, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Ireland, C.M.; Durso, A.R.; Newman, R.A.; Hacker, M.P. Antineoplastic cyclic peptides from the marine tunicate Lissoclinum patella. J. Org. Chem. 1982, 47, 1807–1811. [Google Scholar] [CrossRef]
- Bewley, C.A.; He, H.; Williams, D.H.; Faulkner, D.J. Aciculitins A−C: Cytotoxic and antifungal cyclic peptides from the lithistid sponge Aciculites orientalis. J. Am. Chem. Soc. 1996, 118, 4314–4321. [Google Scholar] [CrossRef]
- Bewley, C.A.; Faulkner, D.J. Theonegramide, an antifungal glycopeptide from the Philippine lithistid sponge Theonella swinhoei. J. Org. Chem. 1994, 59, 4849–4852. [Google Scholar] [CrossRef]
- Matsunaga, S.; Fusetani, N. Theonellamides A-E, cytotoxic bicyclic peptides, from a marine sponge Theonella sp. J. Org. Chem. 1995, 60, 1177–1181. [Google Scholar] [CrossRef]
- Matsunaga, S.; Fusetani, N.; Hashimoto, K.; Walchli, M. Theonellamide, F. A novel antifungal bicyclic peptide from a marine sponge Theonella sp. J. Am. Chem. Soc. 1989, 111, 2582–2588. [Google Scholar] [CrossRef]
- Morita, H.; Shimbo, K.; Shigemori, H.; Kobayashi, J. Antimitotic activity of moroidin, a bicyclic peptide from the seeds of Celosia argentea. Bioorg. Med. Chem. Lett. 2000, 10, 469–471. [Google Scholar] [CrossRef]
- Kobayashi, J.; Suzuki, H.; Shimbo, K.; Takeya, K.; Morita, H. Celogentins A−C, new antimitotic bicyclic peptides from the seeds of Celosia argentea. J. Org. Chem. 2001, 66, 6626–6633. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, C.A.; Pei, D. Bicyclic Peptides as next-generation therapeutics. Chemistry 2017, 23, 12690–12703. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.-C.; Yu, S.-M.; Liu, Y.; Yao, Z.-J. Biomimetic synthesis of ent-(−)-Azonazine and stereochemical reassignment of natural product. Org. Lett. 2013, 15, 4300–4303. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Maloney, K.N.; Nam, S.-J.; Haste, N.M.; Raju, R.; Aalbersberg, W.; Jensen, P.R.; Nizet, V.; Hensler, M.E.; Fenical, W. Fijimycins A–C, three antibacterial etamycin-class depsipeptides from a marine-derived Streptomyces sp. Bioorg. Med. Chem. 2011, 19, 6557–6562. [Google Scholar] [CrossRef] [Green Version]
- Gala, F.; D’Auria, M.V.; De Marino, S.; Sepe, V.; Zollo, F.; Smith, C.D.; Keller, S.N.; Zampella, A. Jaspamides M–P: New tryptophan modified jaspamide derivatives from the sponge Jaspis splendans. Tetrahedron 2009, 65, 51–56. [Google Scholar] [CrossRef]
- Tabudravu, J.; Morris, L.A.; Kettenes-van den Bosch, J.J.; Jaspars, M. Wainunuamide, a histidine-containing proline-rich cyclic heptapeptide isolated from the Fijian marine sponge Stylotella aurantium. Tetrahedron Lett. 2001, 42, 9273–9276. [Google Scholar] [CrossRef]
- Um, S.; Kim, Y.-J.; Kwon, H.; Wen, H.; Kim, S.-H.; Kwon, H.C.; Park, S.; Shin, J.; Oh, D.-C. Sungsanpin, a lasso peptide from a deep-sea Streptomycete. J. Nat. Prod. 2013, 76, 873–879. [Google Scholar] [CrossRef]
- Song, Y.; Li, Q.; Liu, X.; Chen, Y.; Zhang, Y.; Sun, A.; Zhang, W.; Zhang, J.; Ju, J. Cyclic hexapeptides from the deep South China sea-derived Streptomyces scopuliridis SCSIO ZJ46 active against pathogenic gram-positive bacteria. J. Nat. Prod. 2014, 77, 1937–1941. [Google Scholar] [CrossRef]
- Zheng, J.; Zhu, H.; Hong, K.; Wang, Y.; Liu, P.; Wang, X.; Peng, X.; Zhu, W. Novel cyclic hexapeptides from marine-derived fungus, Aspergillus sclerotiorum PT06-1. Org. Lett. 2009, 11, 5262–5265. [Google Scholar] [CrossRef]
- Mueller, L.K.; Baumruck, A.C.; Zhdanova, H.; Tietze, A.A. Challenges and perspectives in chemical synthesis of highly hydrophobic peptides. Front. Bioeng. Biotechnol. 2020, 8, 162. [Google Scholar] [CrossRef] [Green Version]
- Dahiya, R.; Pathak, D. First total synthesis and biological evaluation of halolitoralin A. J. Serb. Chem. Soc. 2007, 72, 101–107. [Google Scholar] [CrossRef]
- Dahiya, R.; Pathak, D. Synthesis, characterization and biological evaluation of halolitoralin B-A natural cyclic peptide. Asian J. Chem. 2007, 19, 1499–1505. [Google Scholar]
- Dahiya, R.; Pathak, D. Synthetic studies on a natural cyclic tetrapeptide-halolitoralin C. J. Pharm. Res. 2006, 5, 69–73. [Google Scholar]
- Dahiya, R.; Pathak, D.; Bhatt, S. Synthesis and biological evaluation of a novel series of 2-(2’-isopropyl-5’-methylphenoxy) acetyl amino acids and dipeptides. Bull. Chem. Soc. Ethiop. 2006, 20, 235–245. [Google Scholar] [CrossRef] [Green Version]
- Dahiya, R.; Pathak, D. Synthetic studies on novel benzimidazolopeptides with antimicrobial, cytotoxic and anthelmintic potential. Eur. J. Med. Chem. 2007, 42, 772–798. [Google Scholar] [CrossRef]
- Dahiya, R.; Kumar, A.; Yadav, R. Synthesis and biological activity of peptide derivatives of iodoquinazolinones/nitroimidazoles. Molecules 2008, 13, 958–976. [Google Scholar] [CrossRef] [Green Version]
- Dahiya, R. Synthesis, characterization and antimicrobial studies on some newer imidazole analogs. Sci. Pharm. 2008, 76, 217–240. [Google Scholar] [CrossRef] [Green Version]
- Rajiv, M.H.; Ramana, M.V. Synthesis of 6-nitrobenzimidazol-1-acetyl amino acids and peptides as potent anthelmintic agents. Indian J. Heterocycl. Chem. 2002, 12, 121–124. [Google Scholar]
- Dahiya, R.; Mourya, R.; Agrawal, S.C. Synthesis and antimicrobial screening of peptidyl derivatives of bromocoumarins/methylimidazoles. Afr. J. Pharma. Pharmacol. 2010, 4, 214–225. [Google Scholar]
- Dahiya, R.; Kumar, A. Synthesis, spectral and anthelmintic activity studies on some novel imidazole derivatives. E-J. Chem. 2008, 5, 1133–1143. [Google Scholar] [CrossRef]
- Himaja, M.; Rajiv; Ramana, M.V.; Poojary, B.; Satyanarayana, D.; Subrahmanyam, E.V.; Bhat, K.I. Synthesis and biological activity of a novel series of 4-[2’-(6’-nitro) benzimidazolyl] benzoyl amino acids and peptides. Boll. Chim. Farmac. 2003, 142, 450–453. [Google Scholar]
- Dahiya, R.; Kaur, R. Synthesis and anthelmintic potential of a novel series of 2-mercaptobenzimidazolopeptides. Biosci. Biotech. Res. Asia 2007, 4, 561–566. [Google Scholar]
- Singh, A.P.; Ramadan, W.M.; Dahiya, R.; Sarpal, A.S.; Pathak, K. Product development studies of amino acid conjugate of aceclofenac. Curr. Drug Deliv. 2009, 6, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, R.; Mourya, R. Synthesis of peptide analogs of 4-[2-(3-bromophenyl)-7-nitro-4-oxo-3, 4-dihydro-3-quinazolinyl] benzoic acids as potent antifungal agents. Indian J. Heterocycl. Chem. 2013, 22, 407–412. [Google Scholar]
- Dahiya, R.; Pathak, D. Synthesis of heterocyclic analogs of 5-(4-methylcarboxamidophenyl)-2- furoic acid as potent antimicrobial agents. Indian J. Heterocycl. Chem. 2006, 16, 53–56. [Google Scholar]
- Dahiya, R.; Mourya, R. Synthetic studies on novel nitroquinazolinone analogs with antimicrobial potential. Bull. Pharm. Res. 2013, 3, 51–57. [Google Scholar]
- Dahiya, R.; Kaur, R. Synthesis of some 1, 2, 5-trisubstituted benzimidazole analogs as possible anthelmintic and antimicrobial agents. Int. J. Biol. Chem. Sci. 2008, 2, 1–13. [Google Scholar] [CrossRef]
- Dahiya, R.; Bansal, Y. Synthesis and antimicrobial potential of novel quinoxalinopeptide analogs. Res. J. Chem. Environ. 2008, 12, 52–58. [Google Scholar]
- Dahiya, R. Synthesis of 4-(2-methyl-1H-5-imidazolyl) benzoyl amino acids and peptides as possible anthelmintic agents. Ethiop. Pharm. J. 2008, 26, 17–26. [Google Scholar] [CrossRef]
- Dahiya, R.; Kumar, S.; Khokra, S.L.; Gupta, S.V.; Sutariya, V.B.; Bhatia, D.; Sharma, A.; Singh, S.; Maharaj, S. Toward the synthesis and improved biopotential of an N-methylated analog of a proline-rich cyclic tetrapeptide from marine bacteria. Mar. Drugs 2018, 16, 305. [Google Scholar] [CrossRef] [Green Version]
- Dahiya, R.; Singh, S. Synthesis, characterization, and biological activity studies on fanlizhicyclopeptide A. Iran. J. Pharm. Res. 2017, 16, 1176–1184. [Google Scholar] [PubMed]
- Dahiya, R.; Singh, S.; Sharma, A.; Chennupati, S.V.; Maharaj, S. First total synthesis and biological screening of a proline-rich cyclopeptide from a Caribbean marine sponge. Mar. Drugs 2016, 14, 228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahiya, R.; Gautam, H. Total synthesis and antimicrobial activity of a natural cycloheptapeptide of marine origin. Mar. Drugs 2010, 8, 2384–2394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahiya, R.; Kumar, A.; Gupta, R. Synthesis, cytotoxic and antimicrobial screening of a proline-rich cyclopolypeptide. Chem. Pharm. Bull. (Tokyo) 2009, 57, 214–217. [Google Scholar] [CrossRef] [Green Version]
- Dahiya, R. Total synthesis and biological potential of psammosilenin A. Arch. Pharm. (Weinheim) 2008, 341, 502–509. [Google Scholar] [CrossRef]
- Dahiya, R. Synthesis of a phenylalanine-rich peptide as potential anthelmintic and cytotoxic agent. Acta Pol. Pharm. 2007, 64, 509–516. [Google Scholar]
- Dahiya, R. Synthetic and pharmacological studies on longicalycinin A. Pak. J. Pharm. Sci. 2007, 20, 317–323. [Google Scholar]
- Dahiya, R.; Pathak, D.; Himaja, M.; Bhatt, S. First total synthesis and biological screening of hymenamide E. Acta Pharm. 2006, 56, 399–415. [Google Scholar]
- Dahiya, R.; Gautam, H. Solution phase synthesis and bioevaluation of cordyheptapeptide B. Bull. Pharm. Res. 2011, 1, 1–10. [Google Scholar]
- Dahiya, R. Synthesis, characterization and biological evaluation of a glycine-rich peptide- cherimolacyclopeptide E. J. Chil. Chem. Soc. 2007, 52, 1224–1229. [Google Scholar] [CrossRef]
- Dahiya, R. Synthesis and in vitro cytotoxic activity of a natural peptide of plant origin. J. Iran. Chem. Soc. 2008, 5, 445–452. [Google Scholar] [CrossRef]
- Dahiya, R. Synthesis, spectroscopic and biological investigation of cyclic octapeptide: Cherimolacyclopeptide G. Turk. J. Chem. 2008, 32, 205–215. [Google Scholar]
- Dahiya, R.; Maheshwari, M.; Kumar, A. Toward the synthesis and biological evaluation of hirsutide. Monatsh. Chem. 2009, 140, 121–127. [Google Scholar] [CrossRef]
- Dahiya, R. Synthesis and biological activity of a cyclic hexapeptide from Dianthus superbus. Chem. Pap. 2008, 62, 527–535. [Google Scholar] [CrossRef]
- Dahiya, R.; Gautam, H. Synthesis and pharmacological studies on a cyclooligopeptide from marine bacteria. Chin. J. Chem. 2011, 29, 1911–1916. [Google Scholar]
- Dahiya, R.; Singh, S.; Kaur, K.; Kaur, R. Total synthesis of a natural cyclooligopeptide from fruits of sugar-apples. Bull. Pharm. Res. 2017, 7, 151. [Google Scholar]
- Dahiya, R.; Singh, S. First total synthesis and biological potential of a heptacyclopeptide of plant origin. Chin. J. Chem. 2016, 34, 1158–1164. [Google Scholar] [CrossRef]
- Bruno, P.; Peña, S.; Just-Baringo, X.; Albericio, F.; Álvarez, M. Total synthesis of aeruginazole A. Org. Lett. 2011, 13, 4648–4651. [Google Scholar] [CrossRef] [Green Version]
- You, S.-L.; Kelly, J.W. Total synthesis of didmolamides A and B. Tetrahedron Lett. 2005, 46, 2567–2570. [Google Scholar] [CrossRef]
- Zhou, W.; Nie, X.-D.; Zhang, Y.; Si, C.-M.; Zhou, Z.; Sun, X.; Wei, B.-G. A practical approach to asymmetric synthesis of dolastatin 10. Org. Biomol. Chem. 2017, 15, 6119–6131. [Google Scholar] [CrossRef] [Green Version]
- Sellanes, D.; Manta, E.; Serra, G. Toward the total synthesis of scleritodermin A: Preparation of the C1–N15 fragment. Tetrahedron Lett. 2007, 48, 1827–1830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Ma, Z.-H.; Mei, D.; Li, C.-X.; Zhang, X.-L.; Li, Y.-X. Total synthesis and reassignment of stereochemistry of obyanamide. Tetrahedron 2006, 62, 9966–9972. [Google Scholar] [CrossRef]
- Zhang, W.; Ding, N.; Li, Y. Synthesis and biological evaluation of analogues of the marine cyclic depsipeptide obyanamide. J. Pept. Sci. 2011, 17, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Islam, M.A.; McAlpine, S.R. Synthesis of the natural product marthiapeptide A. Org. Lett. 2015, 17, 5149–5151. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, R.; Singh, S.; Varghese Gupta, S.; Sutariya, V.B.; Bhatia, D.; Mourya, R.; Chennupati, S.V.; Sharma, A. First total synthesis and pharmacological potential of a plant based hexacyclopeptide. Iran. J. Pharm. Res. 2019, 18, 938–947. [Google Scholar]
- Dahiya, R.; Singh, S. Synthesis, characterization and biological screening of diandrine A. Acta Pol. Pharm. 2017, 74, 873–880. [Google Scholar]
- Dahiya, R.; Kumar, A. Synthetic and biological studies on a cyclopolypeptide of plant origin. J. Zhejiang Univ. Sci. B 2008, 9, 391–400. [Google Scholar] [CrossRef] [Green Version]
- Dahiya, R.; Kaur, K. Synthesis and pharmacological investigation of segetalin C as a novel antifungal and cytotoxic agent. Arzneimittelforschung 2008, 58, 29–34. [Google Scholar] [CrossRef]
- Dahiya, R.; Kaur, K. Synthetic and biological studies on natural cyclic heptapeptide: Segetalin E. Arch. Pharm. Res. 2007, 30, 1380–1386. [Google Scholar] [CrossRef]
- Dahiya, R.; Maheshwari, M.; Yadav, R. Synthetic and cytotoxic and antimicrobial activity studies on annomuricatin B. Z. Naturforsch. B 2009, 64, 237–244. [Google Scholar] [CrossRef]
- Dahiya, R.; Gautam, H. Toward the first total synthesis of gypsin D: A natural cyclopolypeptide from Gypsophila arabica. Am. J. Sci. Res. 2010, 11, 150–158. [Google Scholar]
- Wipf, P.; Fritch, P.C.; Geib, S.J.; Sefler, A.M. Conformational studies and structure−activity analysis of lissoclinamide 7 and related cyclopeptide alkaloids. J. Am. Chem. Soc. 1998, 120, 4105–4112. [Google Scholar] [CrossRef]
- Fennell, B.J.; Carolan, S.; Pettit, G.R.; Bell, A. Effects of the antimitotic natural product dolastatin 10, and related peptides, on the human malarial parasite Plasmodium falciparum. J. Antimicrb. Chemother. 2003, 51, 833–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mooberry, S.L.; Leal, R.M.; Tinley, T.L.; Luesch, H.; Moore, R.E.; Corbett, T.H. The molecular pharmacology of symplostatin 1: A new antimitotic dolastatin 10 analog. Int. J. Cancer 2003, 104, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Salvador, L.A.; Byeon, S.; Ying, Y.; Kwan, J.C.; Law, B.K.; Hong, J.; Luesch, H. Anticolon cancer activity of largazole, a marine-derived tunable histone deacetylase inhibitor. J. Pharmacol. Exp. Ther. 2010, 335, 351–361. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.K.; Choi, M.C.; Seo, C.H.; Park, Y. Therapeutic properties and biological benefits of marine-derived anticancer peptides. Int. J. Mol. Sci. 2018, 19, 919. [Google Scholar] [CrossRef] [Green Version]
- Espiritu, R.A.; Cornelio, K.; Kinoshita, M.; Matsumori, N.; Murata, M.; Nishimura, S.; Kakeya, H.; Yoshida, M.; Matsunaga, S. Marine sponge cyclic peptide theonellamide A disrupts lipid bilayer integrity without forming distinct membrane pores. Biochim. Biophys. Acta 2016, 1858, 1373–1379. [Google Scholar] [CrossRef]
- Mahaffy, R.E.; Pollard, T.D. Influence of phalloidin on the formation of actin filament branches by Arp2/3 Complex. Biochemistry 2008, 47, 6460–6467. [Google Scholar] [CrossRef] [Green Version]
- Odaka, C.; Sanders, M.L.; Crews, P. Jasplakinolide induces apoptosis in various transformed cell lines by a caspase-3-like protease-dependent pathway. Clin. Diagn. Lab. Immun. 2000, 7, 947–952. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.-X.; Crews, M.S.; Draskovic, M.; Sohn, J.; Johnson, T.A.; Tenney, K.; Valeriote, F.A.; Yao, X.-J.; Bjeldanes, L.F.; Crews, P. Azonazine, a novel dipeptide from a Hawaiian marine sediment-derived fungus, Aspergillus insulicola. Org. Lett. 2010, 12, 4458–4461. [Google Scholar] [CrossRef] [Green Version]
- Dahiya, S.; Pathak, K. Physicochemical characterization and dissolution enhancement of aceclofenac-hydroxypropyl beta-cyclodextrin binary systems. PDA J. Pharm. Sci. Technol. 2006, 60, 378–388. [Google Scholar] [PubMed]
- Dahiya, S.; Pathak, K. Influence of amorphous cyclodextrin derivatives on aceclofenac release from directly compressible tablets. Pharmazie 2007, 62, 278–283. [Google Scholar] [PubMed]
- Dahiya, S.; Kaushik, A.; Pathak, K. improved pharmacokinetics of aceclofenac immediate release tablets incorporating its inclusion complex with hydroxypropyl-β-cyclodextrin. Sci. Pharm. 2015, 83, 501–510. [Google Scholar] [CrossRef] [Green Version]
- Dahiya, S. Studies on formulation development of a poorly water-soluble drug through solid dispersion technique. Thai J. Pharm. Sci. 2010, 34, 77–87. [Google Scholar]
- Dahiya, S.; Kaushik, A. Effect of water soluble carriers on dissolution enhancement of aceclofenac. Asian J. Pharm. 2010, 4, 34–40. [Google Scholar] [CrossRef]
- Dahiya, S.; Tayde, P. Binary and ternary solid systems of carvedilol. Bull. Pharm. Res. 2013, 3, 128–134. [Google Scholar]
- Dahiya, R.; Dahiya, S. Ocular delivery of peptides and proteins. In Drug Delivery for the Retina and Posterior Segment Disease; Patel, J.K., Sutariya, V., Kanwar, J.R., Pathak, Y.V., Eds.; Springer: Cham, Switzerland, 2018; pp. 411–437, Chapter 24. [Google Scholar]
- Dahiya, S.; Dahiya, R. Recent nanotechnological advancements in delivery of peptide and protein macromolecules. In Nanotechnology in Biology and Medicine: Research Advancements and Future Perspectives, 1st ed.; Rauta, P.R., Mohanta, Y.K., Nayak, D., Eds.; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2019; pp. 143–157, Chapter 11. [Google Scholar]
- Otvos, L., Jr.; Wade, J.D. Current challenges in peptide-based drug discovery. Front Chem. 2014, 2, 62. [Google Scholar] [CrossRef]
- Ayoub, M.; Scheidegger, D. Peptide drugs, overcoming the challenges, a growing business. Chim. Oggi. 2006, 24, 46–48. [Google Scholar]
- Usmani, S.S.; Bedi, G.; Samuel, J.S.; Singh, S.; Kalra, S.; Kumar, P.; Ahuja, A.A.; Sharma, M.; Gautam, A.; Raghava, G.P.S. THPdb: Database of FDA-approved peptide and protein therapeutics. PLoS ONE 2017, 12, e0181748. [Google Scholar] [CrossRef] [Green Version]
- Perez, E.A.; Hillman, D.W.; Fishkin, P.A.; Krook, J.E.; Tan, W.W.; Kuriakose, P.A.; Alberts, S.R.; Dakhil, S.R. Phase II trial of dolastatin-10 in patients with advanced breast cancer. Investig. New Drugs 2005, 23, 257–261. [Google Scholar] [CrossRef]
- Mita, A.C.; Hammond, L.A.; Bonate, P.L.; Weiss, G.; McCreery, H.; Syed, S.; Garrison, M.; Chu, Q.S.; DeBono, J.S.; Jones, C.B.; et al. Phase I and pharmacokinetic study of tasidotin hydrochloride (ILX651), a third-generation dolastatin-15 analogue, administered weekly for 3 weeks every 28 days in patients with advanced solid tumors. Clin. Cancer Res. 2006, 12, 5207–5215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smyth, J.; Boneterre, M.E.; Schellens, J.; Calvert, H.; Greim, G.; Wanders, J.; Hanauske, A. Activity of the dolastatin analogue, LU103793, in malignant melanoma. Ann. Oncol. 2001, 12, 509–511. [Google Scholar] [CrossRef] [PubMed]
- Riely, G.J.; Gadgeel, S.; Rothman, I.; Saidman, B.; Sabbath, K.; Feit, K.; Kris, M.G.; Rizvi, N.A. A phase 2 study of TZT-1027, administered weekly to patients with advanced non-small cell lung cancer following treatment with platinum-based chemotherapy. Lung Cancer 2007, 55, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Ebbinghaus, S.; Hersh, E.; Cunningham, C.C.; O’Day, S.; McDermott, D.; Stephenson, J.; Richards, D.A.; Eckardt, J.; Haider, O.L.; Hammond, L.A. Phase II study of synthadotin (SYN-D.; ILX651) administered daily for 5 consecutive days once every 3 weeks (qdx5q3w) in patients (Pts) with inoperable locally advanced or metastatic melanoma. J. Clin. Oncol. 2004, 22, 7530. [Google Scholar] [CrossRef]
- Supko, J.G.; Lynch, T.J.; Clark, J.W.; Fram, R.; Allen, L.F.; Velagapudi, R.; Kufe, D.W.; Eder, J.P., Jr. A phase I clinical and pharmacokinetic study of the dolastatin analogue cemadotin administered as a 5-day continuous intravenous infusion. Cancer Chemother. Pharmacol. 2000, 46, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Giddings, L.A.; Newman, D.J. Microbial natural products: Molecular blueprints for antitumor drugs. J. Ind. Microbiol. Biotechnol. 2013, 40, 1181–1210. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Current status of marine-derived compounds as warheads in anti-tumor drug candidates. Mar. Drugs 2017, 15, 99. [Google Scholar] [CrossRef] [Green Version]






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Cyclic Peptide | Molecular Formula | Composition | Heterocyclic Ring (s) * |
|---|---|---|---|---|
| 1980 | Ulicyclamide [53] | C33H39N7O5S2 | cyclooligopeptide | Tzl, mOzn |
| 1980 | Ulithiacyclamide [53] | C32H42N8O6S4 | bicyclic peptide | Tzl, mOzn |
| 1982 | Patellamide A [39] | C35H50N8O6S2 | cyclooctapeptide | Tzl, Ozn, mOzn |
| 1982 | Patellamide B [39] | C38H48N8O6S2 | cyclooctapeptide | Tzl, mOzn |
| 1982 | Patellamide C [39] | C37H46N8O6S2 | cyclooctapeptide | Tzl, mOzn |
| 1983 | Ascidiacyclamide [106] | C36H52N8O6S2 | cyclopolypeptide | Tzl, mOzn |
| 1989 | Lissoclinamide 4 [56] | C38H43N7O5S2 | cycloheptapeptide | Tzl, Tzn, mOzn |
| 1989 | Lissoclinamide 5 [56] | C38H41N7O5S2 | cycloheptapeptide | Tzl, mOzn |
| 1989 | Ulithiacyclamide B [57] | C35H40N8O6S4 | bicycle peptide | Tzl, mOzn |
| 1989 | Patellamide D [80] | C38H48N8O6S2 | cyclooctapeptide | Tzl, mOzn |
| 1990 | Lissoclinamide 8 [55] | C38H43N7O5S2 | cycloheptapeptide | Tzl, Tzn, mOzn |
| 1990 | Lissoclinamide 7 [55] | C38H45N7O5S2 | cycloheptapeptide | Tzn, mOzn |
| 1992 | Tawicyclamide A [41] | C39H51N8O5S3 | cyclooctapeptide | Tzl, Tzn |
| 1992 | Tawicyclamide B [41] | C36H53N8O5S3 | cyclooctapeptide | Tzl, Tzn |
| 1992 | Patellamide E [58] | C39H50N8O6S2 | cyclooctapeptide | Tzl, mOzn |
| 1992 | Bistratamide C [59] | C22H26N6O4S2 | cyclohexapeptide | Tzl, Ozl |
| 1992 | Bistratamide D [59] | C25H34N6O5S | cyclohexapeptide | Tzl, Ozl, mOzn |
| 1995 | Keramamide J [67] | C33H58N10O11S | cyclopolypeptide | Tzl, Trp |
| 1995 | Keramamide G [67] | C43H56N10O11S | cyclopolypeptide | Tzl, Htrp |
| 1995 | Keramamide H [67] | C43H57N10O12BrS | cyclopolypeptide | Tzl, Bhtrp |
| 1995 | Cyclodidemnamide [62] | C34H43N7O5S2 | cycloheptapeptide | Tzl, Tzn, Ozn |
| 1995 | Dolastatin E [76] | C21H26N6O4S2 | cyclohexapeptide | Tzl, Tzn, Ozl |
| 1995 | Lissoclinamide 3 [54] | C33H41N7O5S2 | cycloheptapeptide | Tzl, mOzn |
| 1995 | Patellamide F [54] | C37H46N8O6S2 | cyclooctapeptide | Tzl, Ozn, mOzn |
| 1995 | Nostocyclamide [107] | C27H32N6O6S | cyclohexapeptide | Tzl, mOzl |
| 1996 | Waiakeamide [66,108] | C37H49N7O8S3 | cyclohexapeptide | Tzl |
| 1996 | Raocyclamide B [32] | C27H32N6O6S | cyclohexapeptide | Tzl, Ozl |
| 1996 | Raocyclamide A [32] | C27H30N6O5S | cyclohexapeptide | Tzl, Ozl, Ozn |
| 1996 | Dendramide A [40] | C21H24N6O4S2 | cyclohexapeptide | Tzl, mOzl |
| 1996 | Dendramide B [40] | C21H24N6O4S3 | cyclohexapeptide | Tzl, mOzl |
| 1996 | Dendramide C [40] | C21H24N6O5S3 | cyclohexapeptide | Tzl, mOzl |
| 1997 | Oriamide [65] | C44H54N15O9S2Na | cyclopolypeptide | Tzl |
| 1997 | Dolastatin I [75] | C24H32N6O5S | cyclohexapeptide | Tzl, mOzl, Ozn |
| 1998 | Ulithiacyclamide E [51] | C35H44N8O8S4 | bicyclic peptide | Tzl |
| 1998 | Comoramide B [45] | C34H50N6O7S | cyclohexapeptide | Tzn |
| 1998 | Mayotamide A [45] | C30H43N7O4S4 | cycloheptapeptide | Tzl, Tzn |
| 1998 | Mayotamide B [45] | C29H41N7O4S4 | cycloheptapeptide | Tzl, Tzn |
| 1998 | Keramamide K [109] | C44H60N10O11S | cyclopolypeptide | Tzl, Metrp |
| 1998 | Ulithiacyclamide F [51] | C35H42N8O7S4 | bicycle peptide | Tzl, mOzn |
| 1998 | Ulithiacyclamide G [51] | C35H42N8O7S4 | bicycle peptide | Tzl, mOzn |
| 1998 | Comoramide A [45] | C34H48N6O6S | cyclohexapeptide | Tzn, mOzn |
| 1998 | Patellamide G [51] | C38H50N8O7S2 | cyclooctapeptide | Tzl, mOzn |
| 1998 | Tenuecyclamide A [105] | C19H20N6O4S2 | cyclohexapeptide | Tzl, mOzl |
| 1998 | Tenuecyclamide C [105] | C20H22N6O4S3 | cyclohexapeptide | Tzl, mOzl |
| 1998 | Tenuecyclamide D [105] | C20H22N6O5S3 | cyclohexapeptide | Tzl, mOzl |
| 2000 | Haligramide A [63] | C37H49N7O6S | cyclohexapeptide | Tzl |
| 2000 | Haligramide B [63] | C37H49N7O7S | cyclohexapeptide | Tzl |
| 2000 | Dolastatin 3 [9] | C25H36N6O5S2 | cyclopentapeptide | Tzl |
| 2000 | Homodolastatin 3 [9] | C30H42N8O6S2 | cyclopentapeptide | Tzl |
| 2000 | Lyngbyabellin A [27] | C29H40N4O7S2Cl2 | cyclodepsipeptide | Tzl |
| 2000 | Lyngbyabellin B [86] | C28H40N4O7S2Cl2 | cyclodepsipeptide | Tzl, Tzn |
| 2000 | Kororamide [9] | C45H64N10O10S2 | cyclononapeptide | Tzl, Tzn |
| 2000 | Lissoclinamide 9 [52] | C35H45N7O5S2 | cycloheptapeptide | Tzl, Tzn, mOzn |
| 2000 | Ceratospongamide [77] | C41H49N7O6S | cycloheptapeptide | Tzl, mOzn |
| 2000 | Microcyclamide [35] | C26H30N8O4S2 | cyclohexapeptide | Tzl, mOzl, mImz |
| 2001 | Nostocyclamide M [36] | C20H22N6O4S3 | cyclohexapeptide | Tzl, mOzl |
| 2002 | Cyclodidemnamide B [42] | C32H47N7O6S2 | cycloheptapeptide | Tzl |
| 2002 | Obyanamide [12] | C30H41N5O6S | cyclodepsipeptide | Tzl |
| 2002 | Ulongamide A [13] | C32H45N5O6S | cyclodepsipeptide | Tzl |
| 2002 | Ulongamide D [13] | C34H49N5O7S | cyclodepsipeptide | Tzl |
| 2002 | Ulongamide E [13] | C35H51N5O7S | cyclodepsipeptide | Tzl |
| 2002 | Ulongamide B [13] | C32H45N5O7S | cyclodepsipeptide | Tzl |
| 2002 | Ulongamide C [13] | C36H45N5O7S | cyclodepsipeptide | Tzl |
| 2002 | Ulongamide F [13] | C30H49N5O6S | cyclodepsipeptide | Tzl |
| 2002 | Banyascyclamide B [11] | C22H30N6O5S2 | cyclohexapeptide | Tzl |
| 2002 | Banyascyclamide C [11] | C25H28N6O5S2 | cyclohexapeptide | Tzl |
| 2002 | Banyascyclamide A [11] | C25H26N6O4S2 | cyclohexapeptide | Tzl, mOzn |
| 2002 | Leucamide A [70] | C29H37N7O6S | cycloheptapeptide | Tzl, Ozl, mOzl |
| 2003 | Guineamide A [14] | C31H44N5O6S | cyclodepsipeptide | Tzl |
| 2003 | Guineamide B [14] | C32H45N5O6S | cyclodepsipeptide | Tzl |
| 2003 | Didmolamide A [48] | C25H26N6O4S2 | cyclohexapeptide | Tzl |
| 2003 | Didmolamide B [48] | C25H28N6O5S2 | cyclohexapeptide | Tzl |
| 2003 | Bistratamide J [50] | C25H36N6O5S2 | cyclohexapeptide | Tzl |
| 2003 | Bistratamide I [50] | C25H36N6O5S2 | cyclohexapeptide | Tzl, Ozl |
| 2003 | Bistratamide H [50] | C25H32N6O4S2 | cyclohexapeptide | Tzl, mOzl |
| 2003 | Bistratamide E [50] | C25H34N6O4S2 | cyclohexapeptide | Tzl, mOzn |
| 2003 | Bistratamide G [50] | C25H32N6O5S | cyclohexapeptide | Tzl, Ozl, mOzl |
| 2003 | Bistratamide F [50] | C26H36N6O5S | cyclohexapeptide | Tzl, Ozn, mOzn |
| 2003 | Myriastramide C [69] | C42H53N9O7S | cyclooctapeptide | Tzl, Ozl, Trp |
| 2003 | Bistratamide B [60] | C27H32N6O4S2 | cyclohexapeptide | Tzl, Tzn, mOzn |
| 2004 | Scleritodermin A [64] | C42H54N7O10SNa | cyclopolypeptide | Tzl |
| 2005 | Lyngbyabellin E [28] | C37H51N3O12S2Cl2 | cyclodepsipeptide | Tzl |
| 2005 | Lyngbyabellin H [28] | C37H51N3O11S2Cl2 | cyclodepsipeptide | Tzl |
| 2005 | Mechercharmycin A [79] | C35H32N8O7S | cyclooligopeptide | Tzl, Ozl |
| 2006 | Trichamide [17] | C44H66N16O12S2 | cyclopolypeptide | Tzl, His |
| 2007 | Urukthapelstatin A [78] | C34H30N8O6S2 | cyclooligopeptide | Tzl, Ozl |
| 2007 | Venturamide A [34] | C21H24N6O4S2 | cyclohexapeptide | Tzl, mOzl |
| 2007 | Venturamide B [34] | C22H26N6O5S2 | cyclohexapeptide | Tzl, mOzl |
| 2008 | Mollamide C [46] | C30H46N6O6S | cyclohexapeptide | Tzl |
| 2008 | Aerucyclamide B [37] | C24H33N6O4S2 | cyclohexapeptide | Tzl, mOzn |
| 2008 | Aerucyclamide A [37] | C24H34N6O4S2 | cyclohexapeptide | Tzl, Tzn, mOzn |
| 2008 | Aerucyclamide D [38] | C26H31N6O4S3 | cyclohexapeptide | Tzl, Tzn, mOzn |
| 2008 | Aerucyclamide C [38] | C24H32N6O5S | cyclohexapeptide | Tzl, Ozl, mOzn |
| 2009 | Sanguinamide A [73] | C37H52N7O6S | cycloheptapeptide | Tzl |
| 2009 | Sanguinamide B [73] | C33H43N8O6S2 | cyclooctapeptide | Tzl, Ozl |
| 2010 | Microcyclamide MZ602 [18] | C28H38N6O7S | cyclohexapeptide | Tzl |
| 2010 | Microcyclamide MZ568 [18] | C25H40N6O7S | cyclohexapeptide | Tzl |
| 2010 | Aeruginazole A [91] | C53H66N13O11S3 | cyclododecapeptide | Tzl |
| 2010 | Lyngbyabellin J [30] | C37H51N3O12S2Cl2 | cyclodepsipeptide | Tzl |
| 2010 | 27-deoxylyngbyabellin A [30] | C29H40N4O6S2Cl2 | cyclodepsipeptide | Tzl |
| 2012 | Aeruginazole DA1497 [8] | C68H91N17NaO14S4 | cyclopolypeptide | Tzl |
| 2012 | Aeruginazole DA1304 [8] | C61H72N14NaO13S3 | cyclopolypeptide | Tzl |
| 2012 | Aeruginazole DA1274 [8] | C60H70N14NaO12S3 | cyclopolypeptide | Tzl |
| 2012 | Lyngbyabellin N [29] | C40H58N4O11S2Cl2 | cyclodepsipeptide | Tzl |
| 2012 | Largazole [16] | C29H38N4O5S3 | cyclodepsipeptide | Tzl, Tzn |
| 2012 | Marthiapeptide A [74] | C30H31N7O3S4 | cyclooligopeptide | Tzl, Tzn |
| 2012 | Calyxamide A [110] | C45H61N11O12S | cyclooligopeptide | Tzl, Htrp |
| 2012 | Calyxamide B [110] | C45H61N11O12S | cyclooligopeptide | Tzl, Htrp |
| 2013 | Aestuaramide A [10] | C40H51N7O6S3 | cyclopolypeptide | Tzl |
| 2013 | Aestuaramide B [10] | C35H43N7O6S3 | cyclopolypeptide | Tzl |
| 2013 | Aestuaramide C [10] | C40H51N7O6S3 | cyclopolypeptide | Tzl |
| 2014 | Balgacyclamide A [33] | C25H37N6O5S | cyclooligopeptide | Tzl, mOzn |
| 2014 | Balgacyclamide B [33] | C25H39N6O6S | cyclooligopeptide | Tzl, mOzn |
| 2014 | Balgacyclamide C [33] | C28H37N6O6S | cyclooligopeptide | Tzl, mOzn |
| 2016 | Jamaicensamide A [89] | C45H61N9O10S | cyclooligopeptide | Tzl, Htrp |
| 2017 | Cyclotheonellazole A [68] | C44H54N9O14S2Na2 | cyclopolypeptide | Tzl |
| 2017 | Cyclotheonellazole B [68] | C45H57N9O14S2Na | cyclopolypeptide | Tzl |
| 2017 | Cyclotheonellazole C [68] | C43H52N9O14S2Na2 | cyclopolypeptide | Tzl |
| 2017 | Bistratamide M, N [61] | C21H24N6O4S2 | cyclohexapeptide | Tzl, Ozl |
| TBPs | Resource | Bioactivity | |
|---|---|---|---|
| Susceptibilty | MICa Value | ||
| Haligramide A [63] | marine sponge Haliclona nigra | Cytotoxicity against A-549 (lung), HCT-15 (colon), SF-539 (CNSb), and SNB-19 (CNS) human tumor cell lines | 5.17–15.62 μg/mL |
| Haligramide B [63] | marine sponge Haliclona nigra | Cytotoxicity against A-549 (lung), HCT-15 (colon), SF-539 (CNS), and SNB-19 (CNS) human tumor cells | 3.89–8.82 μg/mL |
| Scleritodermin A [64] | marine sponge Scleritoderma nodosum | Cytotoxicity against colon HCT116, ovarian A2780, and breast SKBR3 cell lines | 0.67–1.9 μM |
| Obyanamide [12] | marine cyanobacterium Lyngbya confervoides | Cytotoxicity against KBc and LoVo cells | 0.58 and 3.14 µg/mL |
| Waiakeamide [66] | marine sponge Ircinia dendroides | Anti-TB activity against Mycobacterium tuberculosis | 7.8 μg/mL |
| Ulongamide A [13] | marine cyanobacterium Lyngbya sp. | Cytotoxicity against KB and LoVo cells | 1 and 5 µM |
| Guineamide B [14] | marine cyanobacterium Lyngbya majuscula | Cytotoxicity against mouse neuroblastoma cell line | 15 µM |
| Calyxamide A [110] | marine sponge Discodermia calyx | Cytotoxicity against P388 murine leukemia cells | 3.9 and 0.9 μM |
| Bistratamide J [50] | marine ascidian Lissoclinum bistratum | Cytotoxic activity against the human colon tumor (HCT-116) cell line | 1.0 µg/mL |
| Didmolamide A and B [48] | marine tunicate Didemnum molle | Cytotoxicity against several cultured tumor cell lines (A549, HT29, and MEL28) | 10–20 µg/mL |
| Aeruginazole A [91] | freshwater cyanobacterium Microcystis sp. | Antibacterial activity againt B. subtilis and S. albus Cytotoxicity against MOLT-4 human leukemia cell line and peripheral blood lymphocytes | 2.2 and 8.7 μM 41 and 22.5 μM |
| Cyclotheonellazole A, B and C [68] | marine sponge Theonella aff. swinhoei | Inhibitory activity against serine protease enzyme chymotrypsin Inhibitory activity against serine protease enzyme elastase | 0.62, 2.8, and 2.3 nM 0.034, 0.10, and 0.099 nM |
| Microcyclamide MZ602 [18] | cyanobacterium Microcystis sp. | Inhibition activity of chymotrypsin | 75 μM |
| Dolastatin 3 [9] | marine cyanobacterium Lyngbya majuscula | Inhibition of HIV-1 integrase (for the terminal-cleavage and strand- transfer reactions) | 5 mM and 4.1 mM |
| Lyngbyabellin A [27] | marine cyanobacterium Lyngbya majuscula | Cytotoxicity against KB cells (human nasopharyngeal carcinoma cell line) and LoVo cells (human colon adenocarcinoma cell line) Cytotoxicity against HT29 colorectal adenocarcinoma and HeLa cervical carcinoma cells Cytoskeletal-disrupting effects in A-10 cells | 0.03 and 0.50 μg/mL 1.1 and 0.71 μM 0.01–5.0 μg/mL |
| Lyngbyabellin B [86] | marine cyanobacterium Lyngbya majuscula | Toxicity to brine shrimp (Artemia salina) Antifungal activity against Candida albicans (ATCC 14053) in a disk diffusion assay Cytotoxicity against HT29 colorectal adenocarcinoma and HeLa cervical carcinoma cells | 3.0 ppm 100 μg/disk 1.1 and 0.71 μM |
| Lyngbyabellin E [28] | marine cyanobacterium Lyngbya majuscula | Cytotoxicity against NCI-H460 human lung tumor and neuro-2a mouse neuroblastoma cells Cytoskeletal-disrupting effects in A-10 cells | 0.4 and 1.2 μM 0.01–6.0 μM |
| Lyngbyabellin H [28] | marine cyanobacterium Lyngbya majuscula | Cytotoxicity against NCI-H460 human lung tumor and neuro-2a mouse neuroblastoma cells | 0.2 and 1.4 μM |
| Lyngbyabellin N [29] | marine cyanobacterium Moorea bouilloni | Cytotoxic activity against HCT116 colon cancer cell line | 40.9 nM |
| 27-Deoxy- lyngbyabellin A [30] | marine cyanobacterium Lyngbya bouillonii | Cytotoxicity against HT29 colorectal adenocarcinoma and HeLa cervical carcinoma cells | 0.012 and 0.0073 μM |
| Lyngbyabellin J [30] | marine cyanobacterium Lyngbya bouillonii | Cytotoxicity against HT29 colorectal adenocarcinoma and HeLa cervical carcinoma cells | 0.054 and 0.041 μM |
| Raocyclamide A [32] | filamentous cyanobacterium Oscillatoria raoi | Cytotoxicity against embryos of sea urchin Paracentrotus lividus | 30 μg/mL (ED100)d |
| Tenuecyclamide A, C and D [105] | cultured cyanobacterium Nostoc spongiaeforme var. tenue | Cytotoxicity against embryos of sea urchin Paracentrotus lividus | 10.8, 9.0, and 19.1 μM (ED100) |
| Dolastatin I [75] | sea hare Dolabella auricularia | Cytotoxicity against HeLa S3 cells | 12 μg/mL |
| Marthiapeptide A [74] | marine actinomycete Marinactinospora thermotolerans SCSIO 00652 | Antibacterial activities against Micrococcus luteus, Staphylococcus aureus, Bacillus subtilis, and Bacillus thuringiensis Cytotoxicity against SF-268 (human glioblastoma) cell line, MCF-7 (human breast adenocarcinoma) cell line, NCI-H460 (human lung carcinoma) cell line, and HepG2 (human hepatocarcinoma) cancer cell line | 2.0, 8.0, 4.0, and 2.0 μg/mL 0.38, 0.43, 0.47, and 0.52 μM |
| Keramamide G, H and J [67] | marine sponge Theonella sp. | Cytotoxicity against L1210 murine leukemia cells and KB human epidermoid carcinoma cells | 10 µg/mL |
| Keramamide K [109] | marine sponge Theonella sp. | Cytotoxicity against L1210 murine leukemia cells and KB human epidermoid carcinoma cells | 0.72 and 0.42 µg/mL |
| Lissoclinamide 8 [55] | sea squirt Lissoclinum patella | Cytotoxicity against T24 (bladder carcinoma cells), MRC5CV1 (fibroblasts), and lymphocytes | 6, 1, and 8 μg/mL |
| Mechercharmycin A [79] | marine bacterium Thermoactinomyces sp. YM3-251 | Cytotoxic activity against A549 (human lung cancer) cells and Jurkat cells (human leukemia) | 4.0 × 10−8 M and 4.6 × 10−8 M |
| Leucamide A [70] | marine sponge Leucetta microraphis | Cytotoxicity against HM02, HepG2, and Huh7 tumor cell lines | 5.2, 5.9, and 5.1 μg/mL |
| Bistratamide H [50] | marine ascidian Lissoclinum bistratum | Cytotoxic activity against the human colon tumor (HCT-116) cell line | 1.7 µg/mL |
| Patellamide E [58] | marine ascidian Lissoclinum patella | Cytotoxicity against human colon tumor cells in vitro | 125 µg/mL |
| Microcyclamide [35] | cultured cyanobacterium Microcystis aeruginosa | Cytotoxicity against P388 murine leukemia cells | 1.2 µg/mL |
| Dolastatin E [76] | sea hare Dolabella auricularia | Cytotoxicity against HeLa-S3 cells | 22–40 μg/mL |
| Aerucyclamide A [38] | freshwater cyanobacterium Microcystis aeruginosa PCC 7806 | Antiparasite activity against Plasmodium falciparum K1 and Trypanosoma brucei rhodesiense STIB 900 | 5.0 and 56.3 μM |
| Aerucyclamide B [38] | freshwater cyanobacterium Microcystis aeruginosa PCC 7806 | Antiparasite activity against Plasmodium falciparum K1 and Trypanosoma brucei rhodesiense STIB 900 | 0.7 and 15.9 μM |
| Aerucyclamide C [38] | freshwater cyanobacterium Microcystis aeruginosa PCC 7806 | Antiparasite activity against Plasmodium falciparum K1 and Trypanosoma brucei rhodesiense STIB 900 | 2.3 and 9.2 μM |
| Aerucyclamide D [38] | freshwater cyanobacterium Microcystis aeruginosa PCC 7806 | Antiparasite activity against Plasmodium falciparum K1 and Trypanosoma brucei rhodesiense STIB 900 | 6.3 and 50.1 μM |
| Aerucyclamide A, B and C [37,38] | freshwater cyanobacterium Microcystis aeruginosa PCC 7806 | Grazer toxicity against the freshwater crustacean Thamnocephalus platyurus | 30.5, 33.8, and 70.5 μM |
| Aerucyclamide B and C [38] | freshwater cyanobacterium Microcystis aeruginosa PCC 7806 | Cytotoxic activity against Rat Myoblast L6 cells | 120 and 106 μM |
| Urukthapelstatin A [78] | marine-derived bacterium Mechercharimyces asporophorigenens YM11-542 | Cytotoxicity against A549 human lung cancer cells | 12 nM |
| Mechercharmycin A [79] | marine-derived bacterium Thermoactinomyces sp. | Cytotoxicity against A549 human lung cancer cells and Jurkat cells | 4.0 × 10-8 M and 4.6 × 10-8 M |
| Ulithiacyclamide [56,117] | marine tunicate Lissoclinum patella | Cytotoxic activity against L1210, MRC5CV1, T24, and CEM cell lines (continuous exposure) | 0.35, 0.04, 0.10, and 0.01 μg/mL |
| Ulicyclamide [117] | marine tunicate Lissoclinum patella | Cytotoxic activity against L1210 murine leukemia cells | 7.2 μg/mL |
| Patellamide A [117] | marine tunicate Lissoclinum patella | Cytotoxic activity against L1210 murine leukemia and human ALL cell line (CEM) | 3.9 and 0.028 μg/mL |
| Patellamide B, C [117] | marine tunicate Lissoclinum patella | Cytotoxic activity against L1210 murine leukemia cells | 2.0 and 3.2 μg/mL |
| Venturamide A [34] | marine cyanobacterium Oscillatoria sp. | Antiparasitic activity against Plasmodium falciparum, Trypanasoma cruzi Cytotoxicity against mammalian Vero cells and MCF-7 cancer cells | 8.2 and 14.6 μM 86 and 13.1 μM |
| Venturamide B [34] | marine cyanobacterium Oscillatoria sp. | Antiparasitic activity against Plasmodium falciparum, Trypanasoma cruzi Cytotoxicity against mammalian Vero cells | 5.2 and 15.8 μM 56 μM |
| Bistratamides A and B [60] | aplousobranch ascidian Lissoclinum bistratum | Cytotoxicity against MRC5CV1 fibroblasts and T24 bladder carcinoma cells | 50 and 100 µg/mL |
| Bistratamide M [61] | marine ascidian Lissoclinum bistratum | Cytotoxicity against breast, colon, lung, and pancreas cell lines | 18, 16, 9.1, and 9.8 μM |
| Balgacyclamide A [33] | freshwater cyanobacterium Microcystis aeruguinosa EAWAG 251 | Antimalarial activity against Plasmodium falciparum K1 | 9 and 59 μM |
| Balgacyclamide B [33] | freshwater cyanobacterium Microcystis aeruguinosa EAWAG 251 | Antiparasitic activity against Trypanosoma brucei rhodesiense STIB 900 | 8.2 and 51 μM |
| Sr. No. | Associated Issue |
|---|---|
| 1. | Low bioavailability and short half-life due to instability of peptides in the body |
| 2. | Formulation challenges and synthesis challenges including aggregation and solubility problems |
| 3. | Difficulty optimizing peptide length to pharmacologically useful levels for receptor activation |
| 4. | Expensive synthesis and manufacturing cost |
| 5. | Difficulty in delivering expected purities and yields |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dahiya, R.; Dahiya, S.; Fuloria, N.K.; Kumar, S.; Mourya, R.; Chennupati, S.V.; Jankie, S.; Gautam, H.; Singh, S.; Karan, S.K.; et al. Natural Bioactive Thiazole-Based Peptides from Marine Resources: Structural and Pharmacological Aspects. Mar. Drugs 2020, 18, 329. https://doi.org/10.3390/md18060329
Dahiya R, Dahiya S, Fuloria NK, Kumar S, Mourya R, Chennupati SV, Jankie S, Gautam H, Singh S, Karan SK, et al. Natural Bioactive Thiazole-Based Peptides from Marine Resources: Structural and Pharmacological Aspects. Marine Drugs. 2020; 18(6):329. https://doi.org/10.3390/md18060329
Chicago/Turabian StyleDahiya, Rajiv, Sunita Dahiya, Neeraj Kumar Fuloria, Suresh Kumar, Rita Mourya, Suresh V. Chennupati, Satish Jankie, Hemendra Gautam, Sunil Singh, Sanjay Kumar Karan, and et al. 2020. "Natural Bioactive Thiazole-Based Peptides from Marine Resources: Structural and Pharmacological Aspects" Marine Drugs 18, no. 6: 329. https://doi.org/10.3390/md18060329
APA StyleDahiya, R., Dahiya, S., Fuloria, N. K., Kumar, S., Mourya, R., Chennupati, S. V., Jankie, S., Gautam, H., Singh, S., Karan, S. K., Maharaj, S., Fuloria, S., Shrivastava, J., Agarwal, A., Singh, S., Kishor, A., Jadon, G., & Sharma, A. (2020). Natural Bioactive Thiazole-Based Peptides from Marine Resources: Structural and Pharmacological Aspects. Marine Drugs, 18(6), 329. https://doi.org/10.3390/md18060329