Inspired by Sea Urchins: Warburg Effect Mediated Selectivity of Novel Synthetic Non-Glycoside 1,4-Naphthoquinone-6S-Glucose Conjugates in Prostate Cancer

 ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. Results
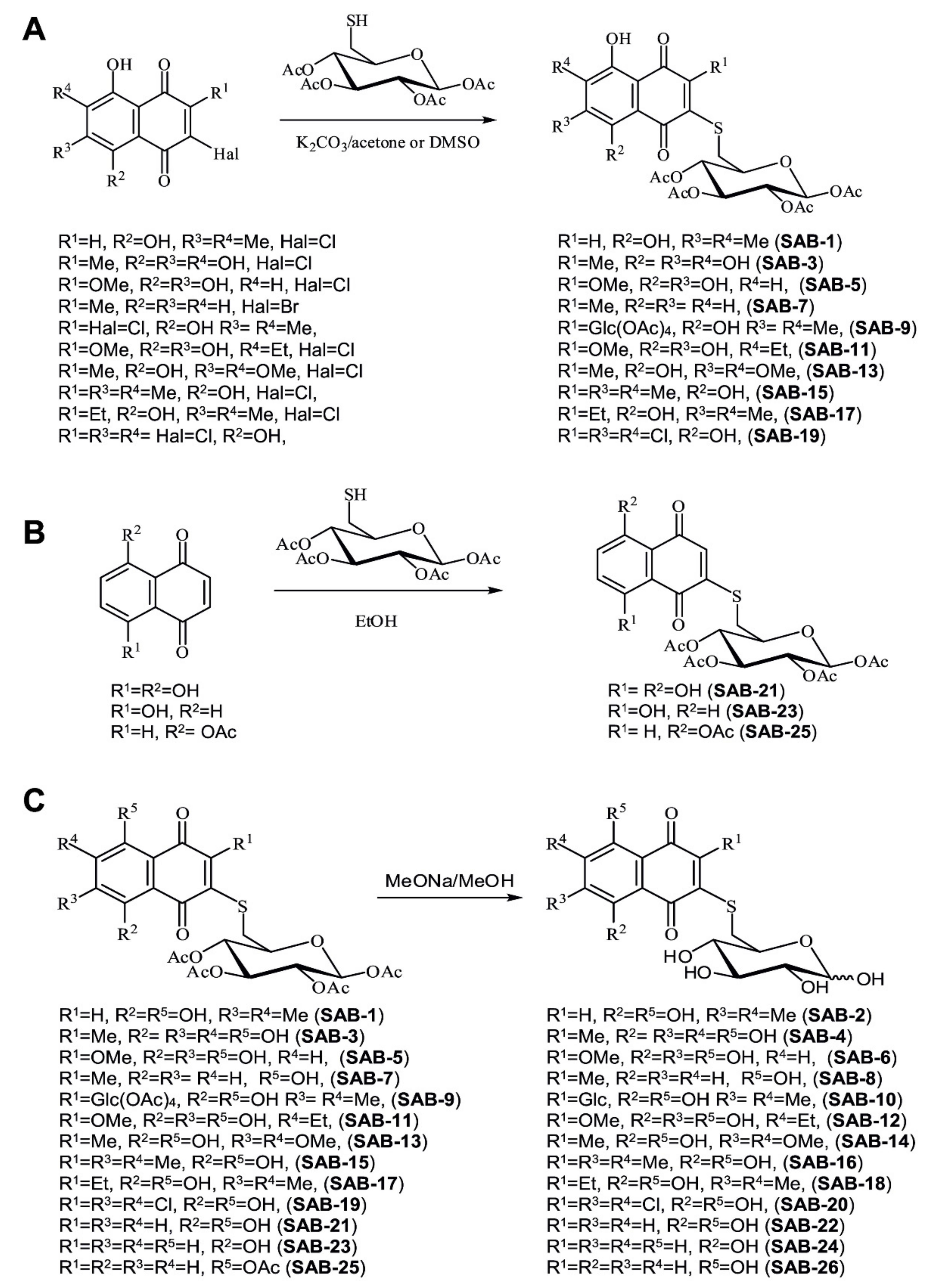
2.1. Design and Synthesis of the 6-S-(1,4-Naphthoquinon-2-yl)-d-Glucose Chimera Molecules
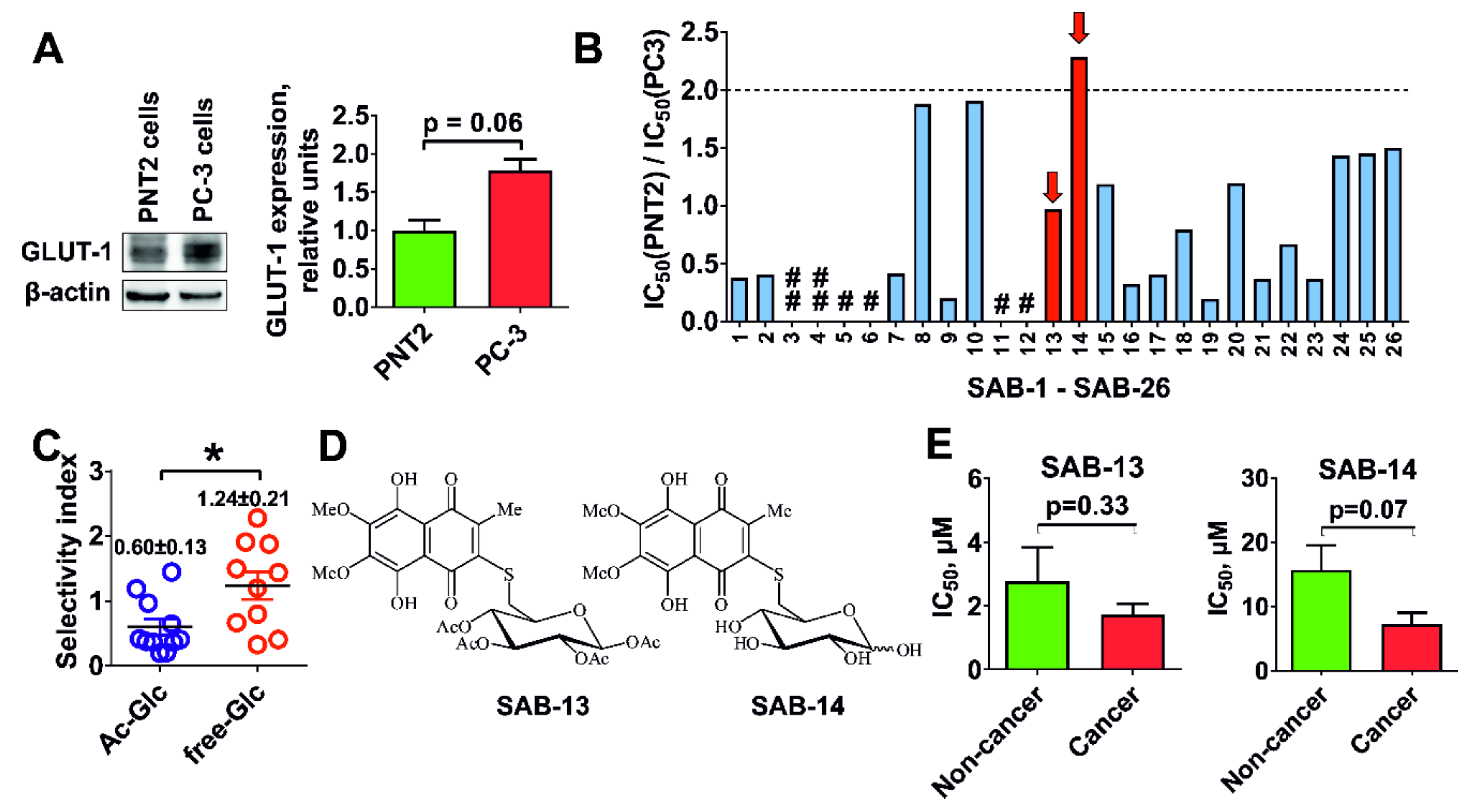
2.2. Evaluation of Cytotoxicity and Selectivity
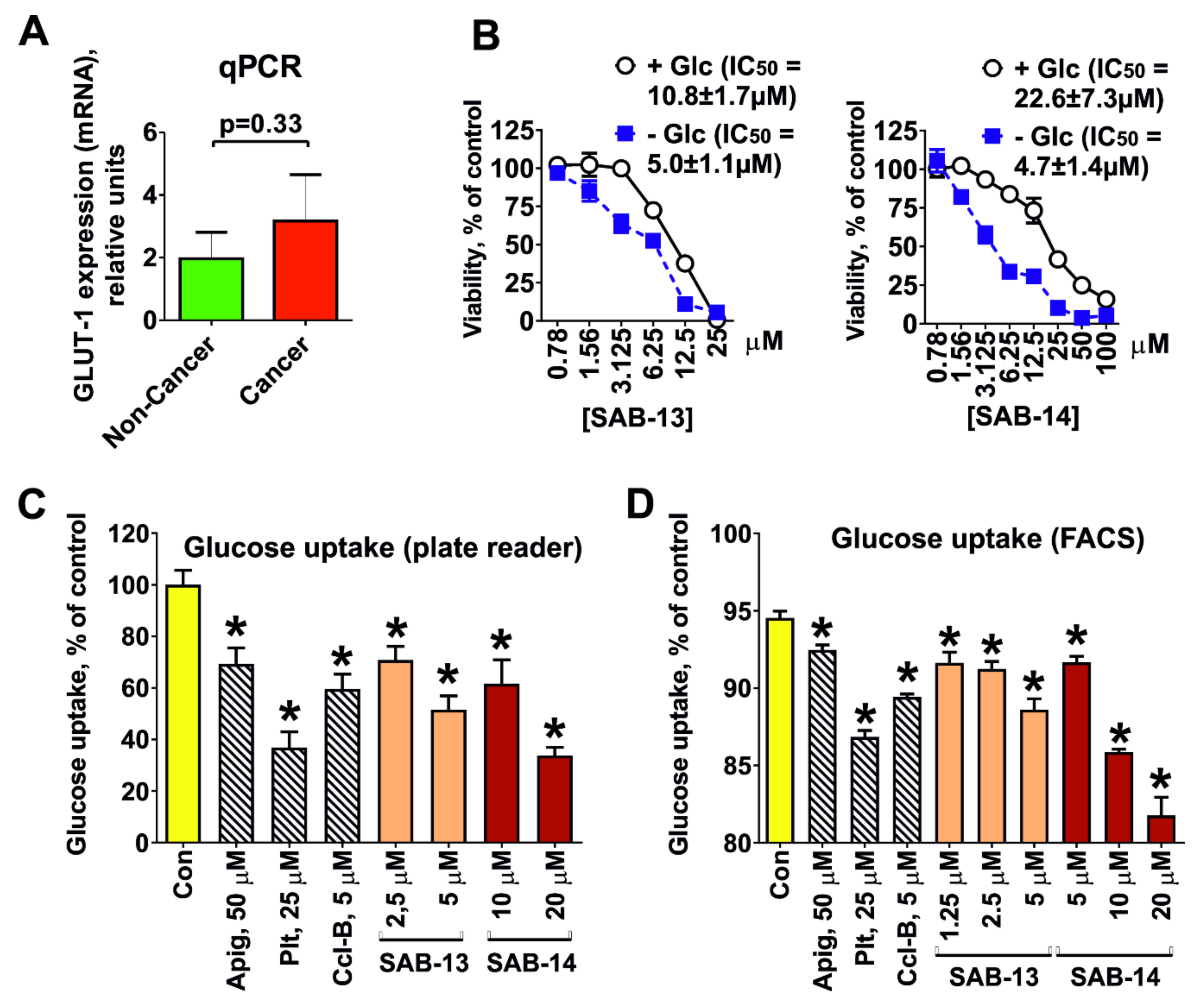
2.3. Cytotoxicity Correlates with Glucose Uptake Rate in Prostate Cancer Cells
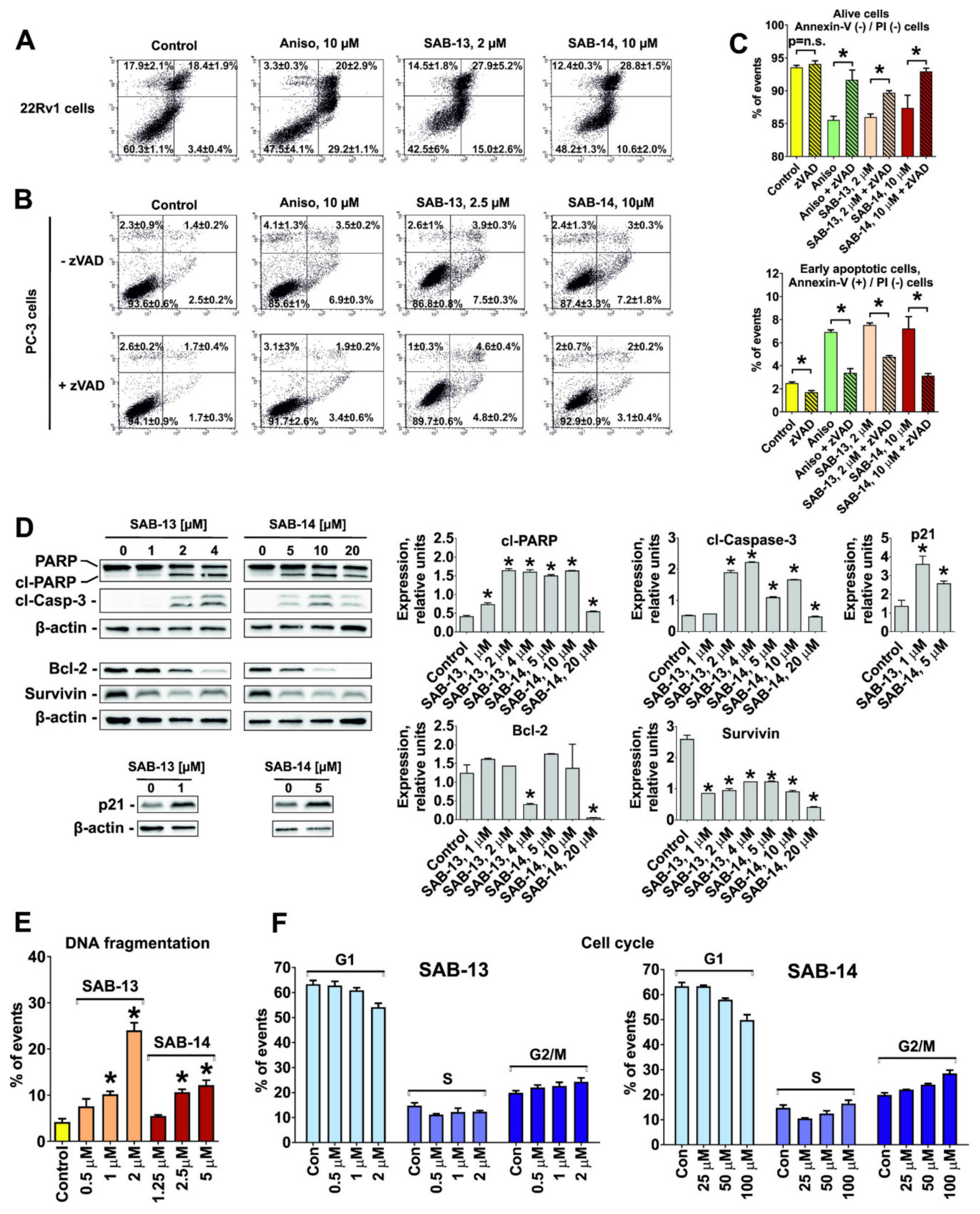
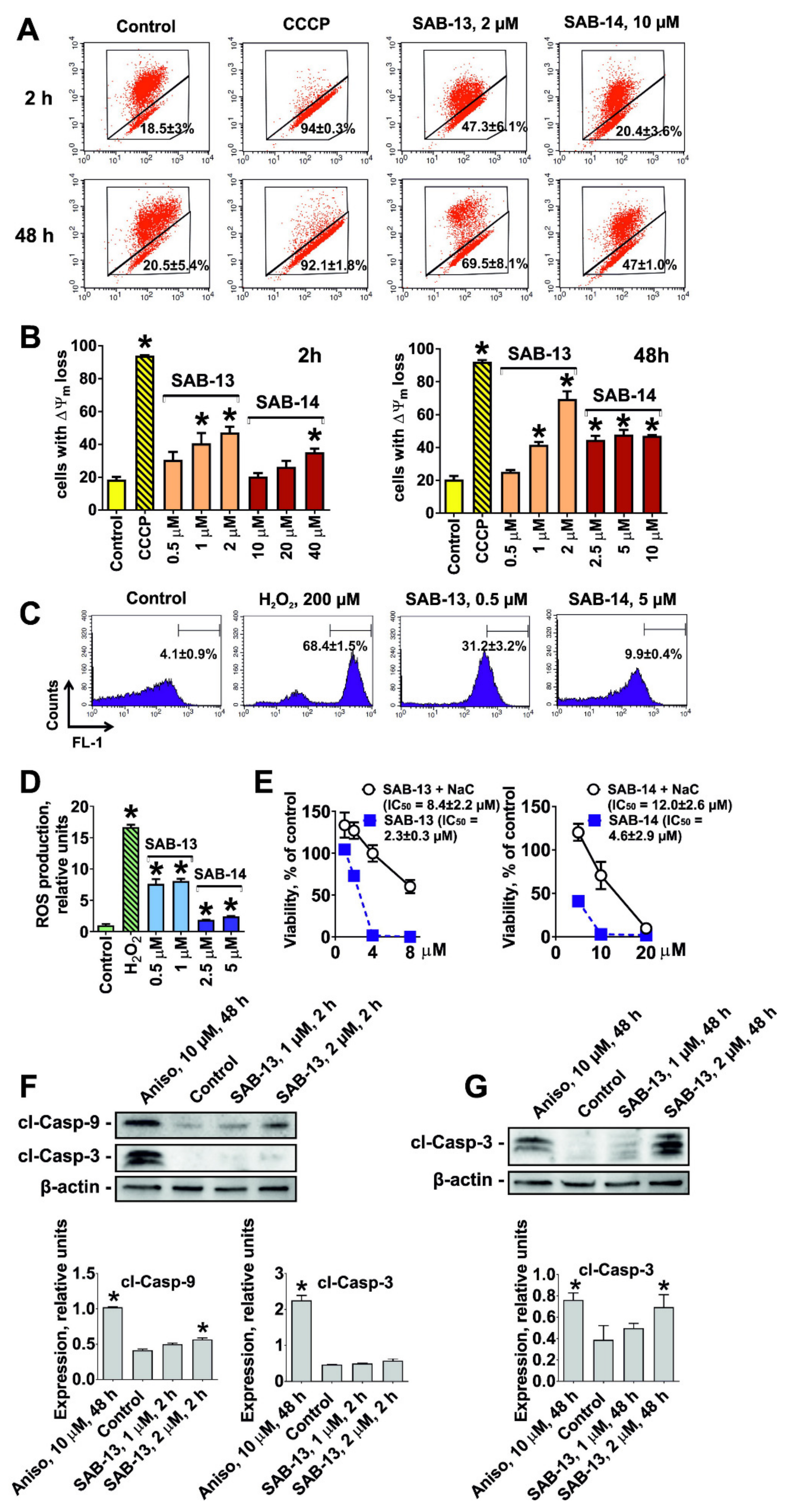
2.4. SAB-13 and -14 Induce Caspase-Dependent Apoptosis
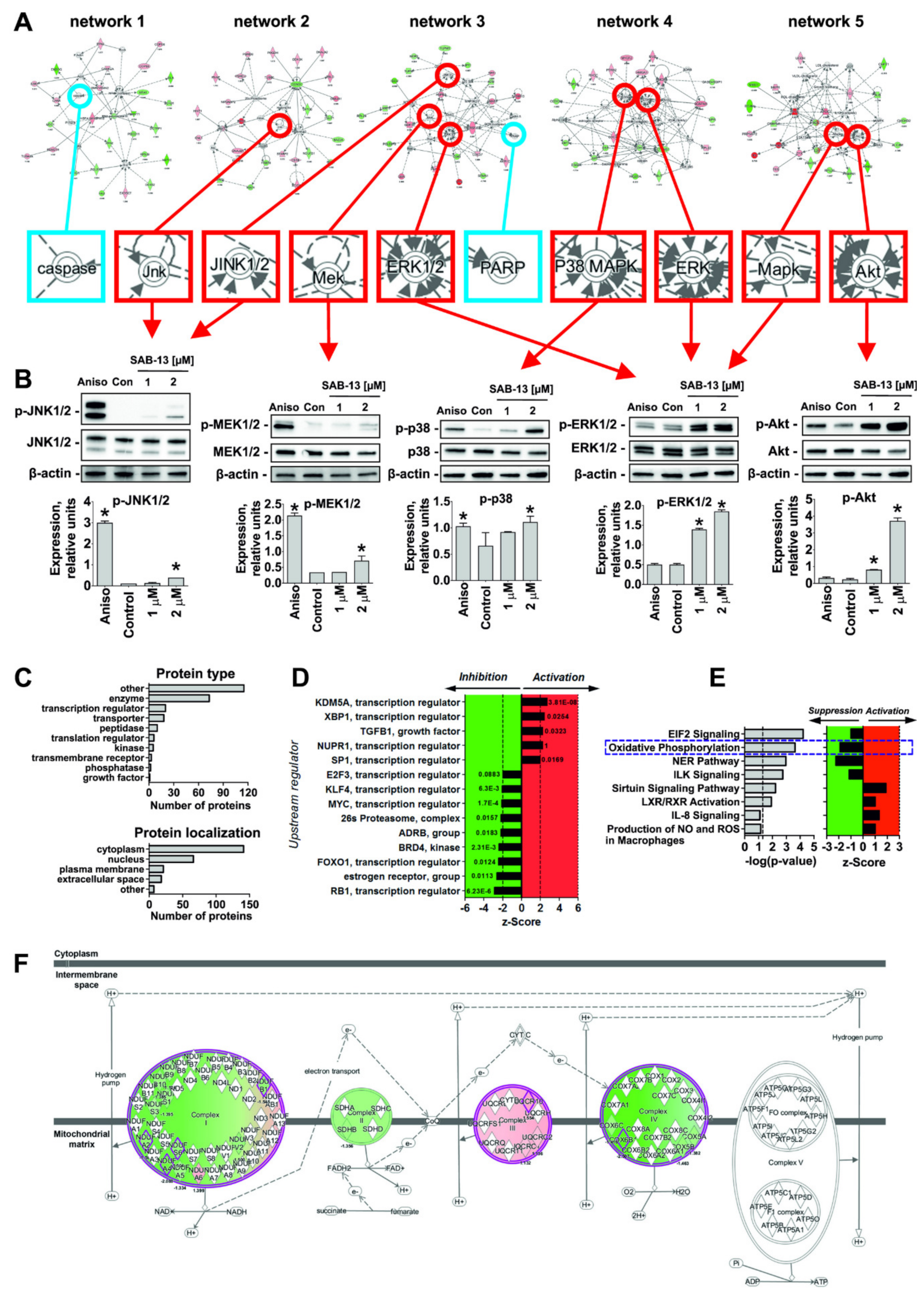
2.5. Effect of SAB-13 on the Proteome of Prostate Cancer Cells
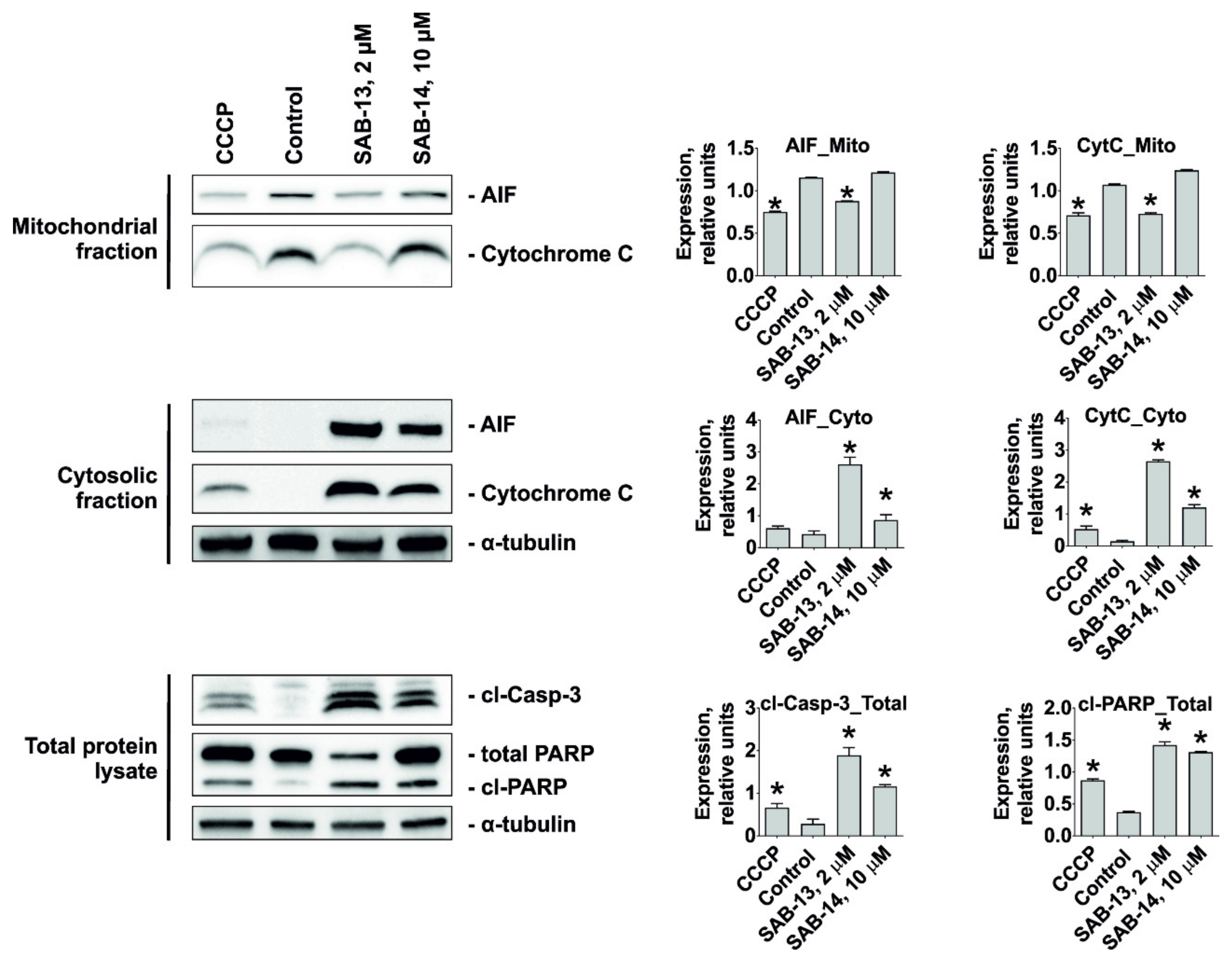
2.6. SAB-13 and SAB-14 Induce Apoptosis via Mitochondria Targeting
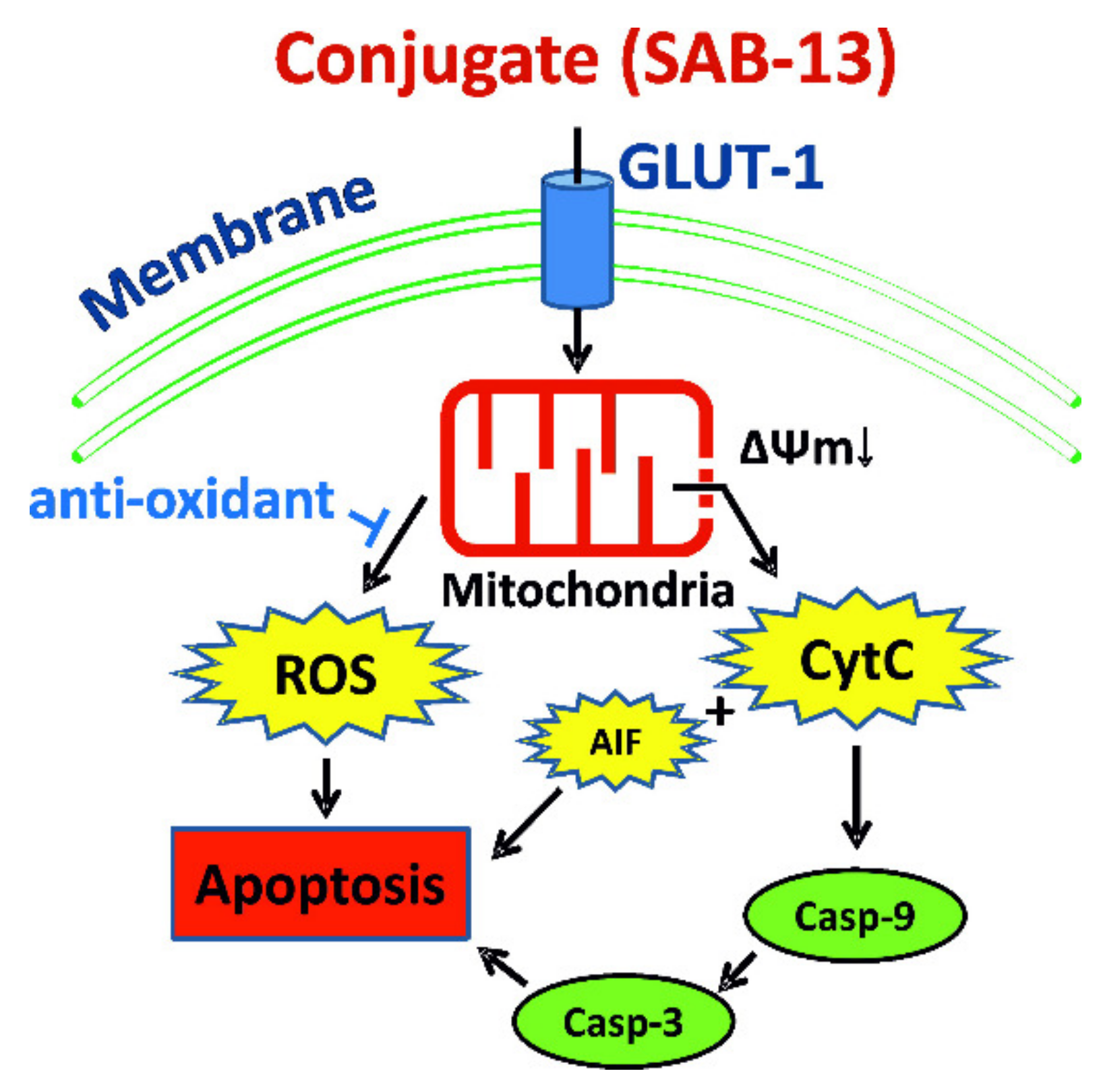
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. General Chemistry (Reagents, Solvents and Equipment)
4.1.2. General Procedure for Synthesis of the Acetylated Conjugates SAB-1, -7, -9, -13, -15, -17, and -19 in Acetone Solution (Figure 1A)
4.1.3. General Procedure for Synthesis of the Acetylated Conjugates SAB-3, -5, and -11 in DMSO Solution (Figure 1A)
4.1.4. Procedure for Synthesis of Naphthazarin Derivative SAB-21 (Figure 1B)
4.1.5. General Procedure for Synthesis of Juglone Derivatives 23 and 25 (Figure 1B)
4.1.6. General Procedure for Synthesis of Deacetylated Conjugates SAB-2, -4, -6, -8, -10, -12, -14, -16, -18, -20, -22, -24, and -26 (Figure 1C, Saponification of the Acetylated Conjugates)
4.2. Biology
4.2.1. General Biology (Reagents and Antibodies)
4.2.2. Cell Lines and Culture Conditions
4.2.3. MTT Assay
4.2.4. Quantitative Real-Time PCR (qPCR)
4.2.5. Glucose Uptake Assay
4.2.6. Analysis of Cell Cycle Progression and DNA Fragmentation
4.2.7. Analysis of Apoptosis (Annexin-V-FITC/PI Double Staining)
4.2.8. Western Blotting
4.2.9. Proteomics
4.2.10. Bioinformatical Analysis
4.2.11. Analysis of Intracellular ROS Level
4.2.12. Analysis of Mitochondrial Membrane Potential (ΔΨm)
4.2.13. Cell Fractionation
4.2.14. Data and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Peng, X.; Pentassuglia, L.; Sawyer, D.B. Emerging anticancer therapeutic targets and the cardiovascular system: Is there cause for concern? Circ. Res. 2010, 106, 1022–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coats, S.; Williams, M.; Kebble, B.; Dixit, R.; Tseng, L.; Yao, N.-S.; Tice, D.A.; Soria, J.-C. Antibody–Drug Conjugates: Future Directions in Clinical and Translational Strategies to Improve the Therapeutic Index. Clin. Cancer Res. 2019, 25, 5441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGregor, B.A.; Sonpavde, G. Enfortumab Vedotin, a fully human monoclonal antibody against Nectin 4 conjugated to monomethyl auristatin E for metastatic urothelial Carcinoma. Expert Opin. Investig. Drugs 2019, 28, 821–826. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, R.; Siamof, C.M.; Dash, A.; Cai, W. Targeted α-therapy of prostate cancer using radiolabeled PSMA inhibitors: A game changer in nuclear medicine. Am. J. Nucl. Med. Mol. Imag. 2018, 8, 247–267. [Google Scholar]
- Adashek, J.J.; Jain, R.K.; Zhang, J. Clinical Development of PARP Inhibitors in Treating Metastatic Castration-Resistant Prostate Cancer. Cells 2019, 8, 860. [Google Scholar] [CrossRef] [Green Version]
- Calvaresi, E.C.; Hergenrother, P.J. Glucose conjugation for the specific targeting and treatment of cancer. Chem. Sci. 2013, 4, 2319–2333. [Google Scholar] [CrossRef] [Green Version]
- Hossain, F.; Andreana, P.R. Developments in carbohydrate-based cancer therapeutics. Pharmaceuticals 2019, 12, 84. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- NIH, Studies Found for: Glufosfamide. 2019. Available online: https://clinicaltrials.gov/ct2/results?cond=glufosfamide&term=&cntry=&state=&city=&dist= (accessed on 31 December 2019).
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [Green Version]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar] [CrossRef]
- Stonik, V.A. Marine natural products: A way to new drugs. Acta Nat. 2009, 2, 15–25. [Google Scholar] [CrossRef]
- Dyshlovoy, S.A.; Honecker, F. Marine Compounds and Cancer: The First Two Decades of XXI Century. Mar. Drugs 2019, 18, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, A. Marine Pharmaceutical: The Clinical Pipeline. 2019. Available online: https://www.midwestern.edu/departments/marinepharmacology/clinical-pipeline.xml (accessed on 16 April 2020).
- Shikov, A.N.; Pozharitskaya, O.N.; Krishtopina, A.S.; Makarov, V.G. Naphthoquinone pigments from sea urchins: Chemistry and pharmacology. Phytochem. Rev. 2018, 17, 509–534. [Google Scholar] [CrossRef]
- Aminin, D.; Polonik, S. 1,4-Naphthoquinones: Some Biological Properties and Application. Chem. Pharm. Bull. 2020, 68, 46–57. [Google Scholar] [CrossRef] [Green Version]
- Klotz, L.-O.; Hou, X.; Jacob, C. 1,4-naphthoquinones: From oxidative damage to cellular and inter-cellular signaling. Molecules 2014, 19, 14902–14918. [Google Scholar] [CrossRef] [Green Version]
- Kumagai, Y.; Shinkai, Y.; Miura, T.; Cho, A.K. The chemical biology of naphthoquinones and its environmental implications. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 221–247. [Google Scholar] [CrossRef]
- Acharya, B.R.; Bhattacharyya, S.; Choudhury, D.; Chakrabarti, G. The microtubule depolymerizing agent naphthazarin induces both apoptosis and autophagy in A549 lung cancer cells. Apoptosis 2011, 16, 924–939. [Google Scholar] [CrossRef]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef]
- Masuda, K.; Funayama, S.; Komiyama, K.; Umezawa, I. Constituents of Tritonia crocosmaeflora, II. Tricrozarin B, an antitumor naphthazarin derivative. J. Nat. Prod. 1987, 50, 958–960. [Google Scholar] [CrossRef]
- Pelageev, D.N.; Dyshlovoy, S.A.; Pokhilo, N.D.; Denisenko, V.A.; Borisova, K.L.; Keller-von Amsberg, G.; Bokemeyer, C.; Fedorov, S.N.; Honecker, F.; Anufriev, V.P. Quinone-carbohydrate nonglucoside conjugates as a new type of cytotoxic agents: Synthesis and determination of in vitro activity. Eur. J. Med. Chem. 2014, 77, 139–144. [Google Scholar] [CrossRef]
- Balaneva, N.N.; Shestak, O.P.; Anufriev, V.F.; Novikov, V.L. Synthesis of Spinochrome D, A Metabolite of Various Sea-Urchin Species. Chem. Nat. Compd. 2016, 52, 213–217. [Google Scholar] [CrossRef]
- Pelageev, D.N.; Anufriev, V.P. Synthesis of Mirabiquinone A: A Biquinone from the Sea Urchin Scaphechinus mirabilis and Related Compounds. Synthesis 2016, 48, 761–764. [Google Scholar]
- Sabutskii, Y.E.; Denisenko, V.A.; Polonik, S.G. The Acid-Catalyzed 2-O-Alkylation of Substituted 2-Hydroxy-1,4-naphthoquinones by Alcohols: Versatile Preparative Synthesis of Spinochrome D and Its 6-Alkoxy Derivatives. Synthesis 2018, 50, 3738–3748. [Google Scholar]
- Dyshlovoy, S.A.; Pelageev, D.N.; Hauschild, J.; Borisova, K.L.; Kaune, M.; Krisp, C.; Venz, S.; Sabutskii, Y.E.; Khmelevskaya, E.A.; Busenbender, T.; et al. Successful Targeting of the Warburg Effect in Prostate Cancer by Glucose-Conjugated 1,4-Naphthoquinones. Cancers 2019, 11, 1690. [Google Scholar] [CrossRef] [Green Version]
- Driguez, H. Thiooligosaccharides as Tools for Structural Biology. ChemBioChem 2001, 2, 311–318. [Google Scholar] [CrossRef]
- Thomson, R.H. Studies in the Juglone Series. III. Addition Reactions. J. Org. Chem. 1951, 16, 1082–1090. [Google Scholar] [CrossRef]
- Tamaki, H.; Harashima, N.; Hiraki, M.; Arichi, N.; Nishimura, N.; Shiina, H.; Naora, K.; Harada, M. Bcl-2 family inhibition sensitizes human prostate cancer cells to docetaxel and promotes unexpected apoptosis under caspase-9 inhibition. Oncotarget 2014, 5, 11399–11412. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Zhu, Y.; Lou, W.; Nadiminty, N.; Chen, X.; Zhou, Q.; Shi, X.B.; de Vere White, R.W.; Gao, A.C. Functional p53 determines docetaxel sensitivity in prostate cancer cells. Prostate 2013, 73, 418–427. [Google Scholar] [CrossRef] [Green Version]
- Karimian, A.; Ahmadi, Y.; Yousefi, B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair 2016, 42, 63–71. [Google Scholar] [CrossRef]
- Dyshlovoy, S.A.; Otte, K.; Tabakmakher, K.M.; Hauschild, J.; Makarieva, T.N.; Shubina, L.K.; Fedorov, S.N.; Bokemeyer, C.; Stonik, V.A.; von Amsberg, G. Synthesis and anticancer activity of the derivatives of marine compound rhizochalin in castration resistant prostate cancer. Oncotarget 2018, 9, 16962–16973. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Fedor, H.L.; Lotan, T.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, P.S. Targeting the androgen receptor in prostate cancer—A resilient foe. N. Engl. J. Med. 2014, 371, 1067–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, C.S.; Falconer, A.; Dodson, A.R.; Norman, A.R.; Dennis, N.; Fletcher, A.; Southgate, C.; Dowe, A.; Dearnaley, D.; Jhavar, S.; et al. factor E2F3 overexpressed in prostate cancer independently predicts clinical outcome. Oncogene 2004, 23, 5871–5879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braadland, P.R.; Ramberg, H.; Grytli, H.H.; Taskén, K.A. β-Adrenergic Receptor Signaling in Prostate Cancer. Front. Oncol. 2015, 4, 375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafran, J.S.; Andrieu, G.P.; Gyorffy, B.; Denis, G.V. BRD4 regulates metastatic potential of castration-resistant prostate cancer through AHNAK. Mol. Cancer Res. 2019, 17, 1627–1638. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.; Wang, L.; Du, Y.; Liu, X.; Chen, Z.; Weng, X.; Guo, J.; Chen, H.; Wang, M.; Wang, X. Inhibition of BRD4 suppresses tumor growth in prostate cancer via the enhancement of FOXO1 expression. Int. J. Oncol. 2018, 53, 2503–2517. [Google Scholar] [CrossRef] [Green Version]
- Frankland-Searby, S.; Bhaumik, S.R. The 26S proteasome complex: An attractive target for cancer therapy. Biochim. Biophys. Acta 2012, 1825, 64–76. [Google Scholar] [CrossRef] [Green Version]
- Koh, C.M.; Bieberich, C.J.; Dang, C.V.; Nelson, W.G.; Yegnasubramanian, S.; De Marzo, A.M. MYC and Prostate Cancer. Genes Cancer 2010, 1, 617–628. [Google Scholar] [CrossRef] [Green Version]
- Sica, V.; Bravo-San Pedro, J.M.; Stoll, G.; Kroemer, G. Oxidative phosphorylation as a potential therapeutic target for cancer therapy. Int. J. Cancer 2020, 146, 10–17. [Google Scholar] [CrossRef]
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X.F. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6, 19. [Google Scholar] [CrossRef] [Green Version]
- Horwich, A.; Hugosson, J.; de Reijke, T.; Wiegel, T.; Fizazi, K.; Kataja, V. Prostate cancer: ESMO consensus conference guidelines 2012. Ann. Oncol. 2013, 24, 1141–1162. [Google Scholar] [CrossRef] [PubMed]
- Caffo, O.; De Giorgi, U.; Fratino, L.; Alesini, D.; Zagonel, V.; Facchini, G.; Gasparro, D.; Ortega, C.; Tucci, M.; Verderame, F. Clinical outcomes of castration-resistant prostate cancer treatments administered as third or fourth line following failure of docetaxel and other second-line treatment: Results of an Italian multicentre study. Eur. Urol. 2015, 68, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, C.M.; Gao, A.C. Drug resistance in castration resistant prostate cancer: Resistance mechanisms and emerging treatment strategies. Am. J. Clin. Exp. Urol. 2015, 3, 64–76. [Google Scholar] [PubMed]
- Sabutskii, Y.E.; Denisenko, V.A.; Popov, R.S.; Polonik, S.G. Acid-Catalyzed Heterocyclization of Trialkylnaphthazarin Thioglucosides in Angular Quinone-Carbohydrate Tetracycles. Russ. J. Org. Chem. 2019, 55, 147–151. [Google Scholar] [CrossRef]
- Wellington, K.W. Understanding cancer and the anticancer activities of naphthoquinones—A review. RSC Adv. 2015, 5, 20309–20338. [Google Scholar] [CrossRef]
- Lown, J.W.; Sim, S.K.; Majumdar, K.C.; Chang, R.Y. Strand scission of DNA by bound adriamycin and daunorubicin in the presence of reducing agents. Biochem. Biophys. Res. Commun. 1977, 76, 705–710. [Google Scholar] [CrossRef]
- Leopold, W.R.; Shillis, J.L.; Mertus, A.E.; Nelson, J.M.; Roberts, B.J.; Jackson, R.C. Anticancer activity of the structurally novel antibiotic CI-920 and its analogues. Cancer Res. 1984, 44, 1928–1932. [Google Scholar]
- Tewey, K.M.; Chen, G.L.; Nelson, E.M.; Liu, L.F. Intercalative antitumor drugs interfere with the breakage-reunion reaction of mammalian DNA topoisomerase II. J. Biol. Chem. 1984, 259, 9182–9187. [Google Scholar]
- Chamberlain, G.R.; Tulumello, D.V.; Kelley, S.O. Targeted delivery of doxorubicin to mitochondria. ACS Chem. Biol. 2013, 8, 1389–1395. [Google Scholar] [CrossRef]
- Battogtokh, G.; Choi, Y.S.; Kang, D.S.; Park, S.J.; Shim, M.S.; Huh, K.M.; Cho, Y.Y.; Lee, J.Y.; Lee, H.S.; Kang, H.C. Mitochondria-targeting drug conjugates for cytotoxic, anti-oxidizing and sensing purposes: Current strategies and future perspectives. Acta Pharm. Sin. B 2018, 8, 862–880. [Google Scholar] [CrossRef]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative phosphorylation as an emerging target in cancer therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyshlovoy, S.A.; Fedorov, S.N.; Shubina, L.K.; Kuzmich, A.S.; Bokemeyer, C.; Keller-von Amsberg, G.; Honecker, F. Aaptamines from the marine sponge Aaptos sp. display anticancer activities in human cancer cell lines and modulate AP-1-, NF-kappa B-, and p53-dependent transcriptional activity in mouse JB6 Cl41 cells. Biomed Res. Int. 2014, 2014, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyshlovoy, S.A.; Tabakmakher, K.M.; Hauschild, J.; Shchekaleva, R.K.; Otte, K.; Guzii, A.G.; Makarieva, T.N.; Kudryashova, E.K.; Fedorov, S.N.; Shubina, L.K.; et al. Guanidine alkaloids from the marine sponge Monanchora pulchra show cytotoxic properties and prevent EGF-induced neoplastic transformation In Vitro. Mar. Drugs 2016, 14, 133. [Google Scholar] [CrossRef] [PubMed]
- Dyshlovoy, S.A.; Naeth, I.; Venz, S.; Preukschas, M.; Sievert, H.; Jacobsen, C.; Shubina, L.K.; Gesell Salazar, M.; Scharf, C.; Walther, R.M.; et al. Honecker, Proteomic profiling of germ cell cancer cells treated with aaptamine, a marine alkaloid with anti proliferative activity. J. Proteome Res. 2012, 11, 2316–2330. [Google Scholar] [CrossRef] [PubMed]
- Dyshlovoy, S.A.; Venz, S.; Shubina, L.K.; Fedorov, S.N.; Walther, R.; Jacobsen, C.; Stonik, V.A.; Bokemeyer, C.; Balabanov, S.; Honecker, F. Activity of aaptamine and two derivatives, demethyloxyaaptamine and isoaaptamine, in cisplatin-resistant germ cell cancer. J. Proteom. 2014, 96, 223–239. [Google Scholar] [CrossRef] [PubMed]
- Dyshlovoy, S.A.; Venz, S.; Hauschild, J.; Tabakmakher, K.M.; Otte, K.; Madanchi, R.; Walther, R.; Guzii, A.G.; Makarieva, T.N.; Shubina, L.K.; et al. Amsberg, Anti-migratory activity of marine alkaloid monanchocidin A—Proteomics-based discovery and confirmation. Proteomics 2016, 16, 1590–1603. [Google Scholar] [CrossRef]
- Dyshlovoy, S.; Rast, S.; Hauschild, J.; Otte, K.; Alsdorf, W.; Madanchi, R.; Kalinin, V.; Silchenko, A.; Avilov, S.; Dierlamm, J.; et al. Frondoside A induces AIF-associated caspase-independent apoptosis in Burkitt’s lymphoma cells. Leuk. Lymphoma 2017, 58, 2905–2915. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibodies | Clonality | Source | Cat.-No. | Dilution | Manufacturer |
|---|---|---|---|---|---|
| anti-AIF | mAb | rabbit | #5318 | 1:1000 | Cell Signaling |
| anti-Akt | pAb | rabbit | #9272 | 1:1000 | Cell Signaling |
| anti-Bcl-2 | pAb | rabbit | #2876 | 1:1000 | Cell Signaling |
| anti-cleaved Caspase-3 | mAb | rabbit | #9664 | 1:1000 | Cell Signaling |
| anti-cleaved Caspase-9 | mAb | rabbit | #20750 | 1:1000 | Cell Signaling |
| anti-cytochrome C | mAb | rabbit | #11940 | 1:1000 | Cell Signaling |
| anti-ERK1/2 | mAb | mouse | #9107 | 1:2000 | Cell Signaling |
| anti-GLUT1 | mAb | rabbit | #12939 | 1:1000 | Cell Signaling |
| anti-JNK1/2 | mAb | rabbit | #9258 | 1:1000 | Cell Signaling |
| anti-MEK1/2 | pAb | rabbit | #9122 | 1:1000 | Cell Signaling |
| anti-mouse IgG-HRP | sheep | NXA931 | 1:10,000 | GE Healthcare | |
| anti-p21Waf1/Cip1 | mAb | rabbit | #2947 | 1:1000 | Cell Signaling |
| anti-p38 | mAb | rabbit | #9212 | 1:1000 | Cell Signaling |
| anti-PARP | pAb | rabbit | #9542 | 1:1000 | Cell Signaling |
| anti-phospho-Akt | mAb | rabbit | #4058 | 1:1000 | Cell Signaling |
| anti-phospho-ERK1/2 | mAb | rabbit | #4377 | 1:1000 | Cell Signaling |
| anti-phospho-JNK1/2 | mAb | rabbit | #4668 | 1:1000 | Cell Signaling |
| anti-phospho-MEK1/2 | mAb | rabbit | #2338 | 1:1000 | Cell Signaling |
| anti-phospho-p38 | mAb | rabbit | #4511 | 1:1000 | Cell Signaling |
| anti-rabbit IgG-HRP | goat | #7074 | 1:5000 | Cell Signaling | |
| anti-Survivin | pAb | rabbit | NB500-201 | 1:1000 | Novus |
| anti-α-Tubulin | mAb | mouse | T5168 | 1:5000 | Sigma-Aldrich |
| anti-β-Actin-HRP | pAb | goat | sc-1616 | 1:10,000 | Santa Cruz |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dyshlovoy, S.A.; Pelageev, D.N.; Hauschild, J.; Sabutskii, Y.E.; Khmelevskaya, E.A.; Krisp, C.; Kaune, M.; Venz, S.; Borisova, K.L.; Busenbender, T.; et al. Inspired by Sea Urchins: Warburg Effect Mediated Selectivity of Novel Synthetic Non-Glycoside 1,4-Naphthoquinone-6S-Glucose Conjugates in Prostate Cancer. Mar. Drugs 2020, 18, 251. https://doi.org/10.3390/md18050251
Dyshlovoy SA, Pelageev DN, Hauschild J, Sabutskii YE, Khmelevskaya EA, Krisp C, Kaune M, Venz S, Borisova KL, Busenbender T, et al. Inspired by Sea Urchins: Warburg Effect Mediated Selectivity of Novel Synthetic Non-Glycoside 1,4-Naphthoquinone-6S-Glucose Conjugates in Prostate Cancer. Marine Drugs. 2020; 18(5):251. https://doi.org/10.3390/md18050251
Chicago/Turabian StyleDyshlovoy, Sergey A., Dmitry N. Pelageev, Jessica Hauschild, Yurii E. Sabutskii, Ekaterina A. Khmelevskaya, Christoph Krisp, Moritz Kaune, Simone Venz, Ksenia L. Borisova, Tobias Busenbender, and et al. 2020. "Inspired by Sea Urchins: Warburg Effect Mediated Selectivity of Novel Synthetic Non-Glycoside 1,4-Naphthoquinone-6S-Glucose Conjugates in Prostate Cancer" Marine Drugs 18, no. 5: 251. https://doi.org/10.3390/md18050251
APA StyleDyshlovoy, S. A., Pelageev, D. N., Hauschild, J., Sabutskii, Y. E., Khmelevskaya, E. A., Krisp, C., Kaune, M., Venz, S., Borisova, K. L., Busenbender, T., Denisenko, V. A., Schlüter, H., Bokemeyer, C., Graefen, M., Polonik, S. G., Anufriev, V. P., & von Amsberg, G. (2020). Inspired by Sea Urchins: Warburg Effect Mediated Selectivity of Novel Synthetic Non-Glycoside 1,4-Naphthoquinone-6S-Glucose Conjugates in Prostate Cancer. Marine Drugs, 18(5), 251. https://doi.org/10.3390/md18050251