Bioactive Molecular Networking for Mapping the Antimicrobial Constituents of the Baltic Brown Alga Fucus vesiculosus



Abstract
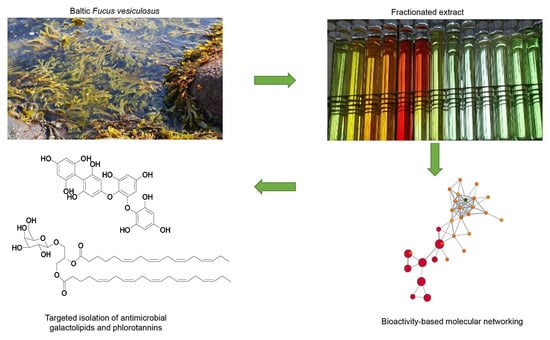
:
1. Introduction
2. Results
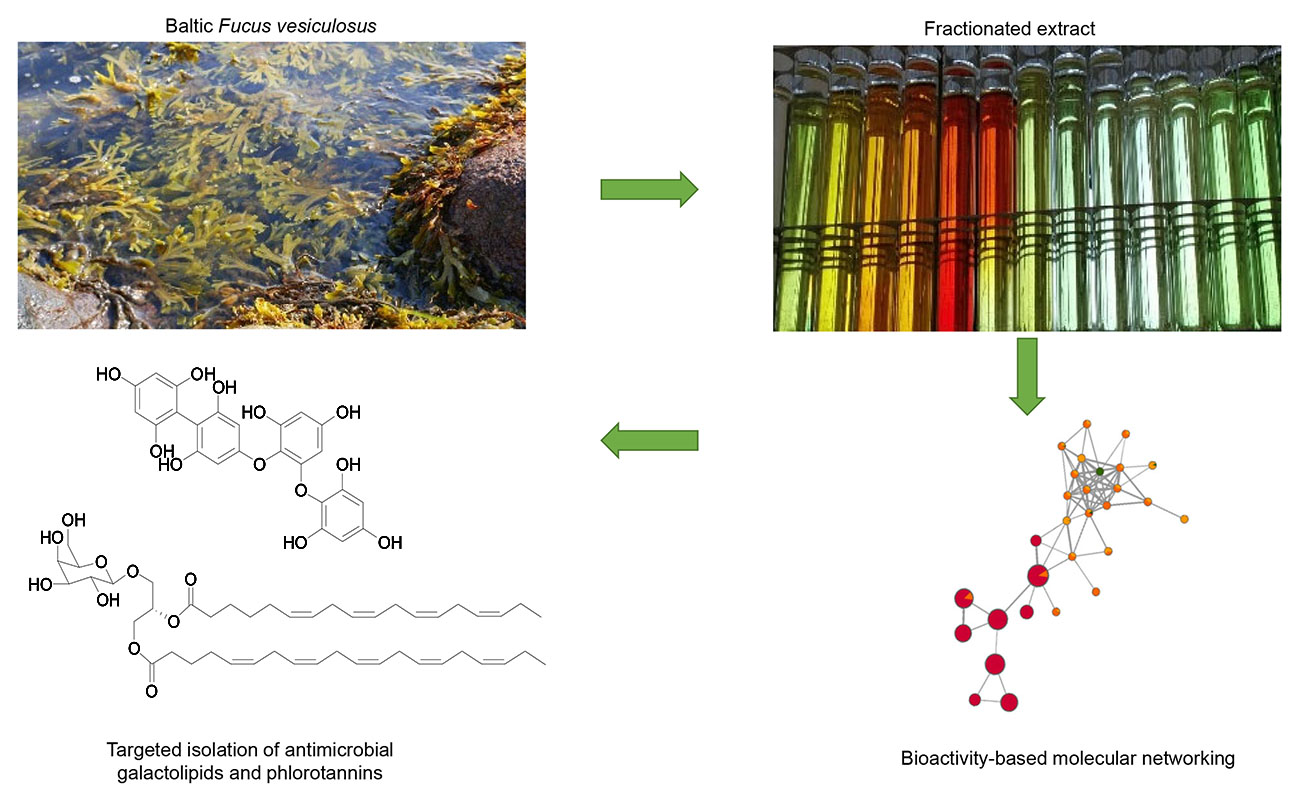
2.1. Bioactive Molecular Netwoking of the n-Hexane Subextract
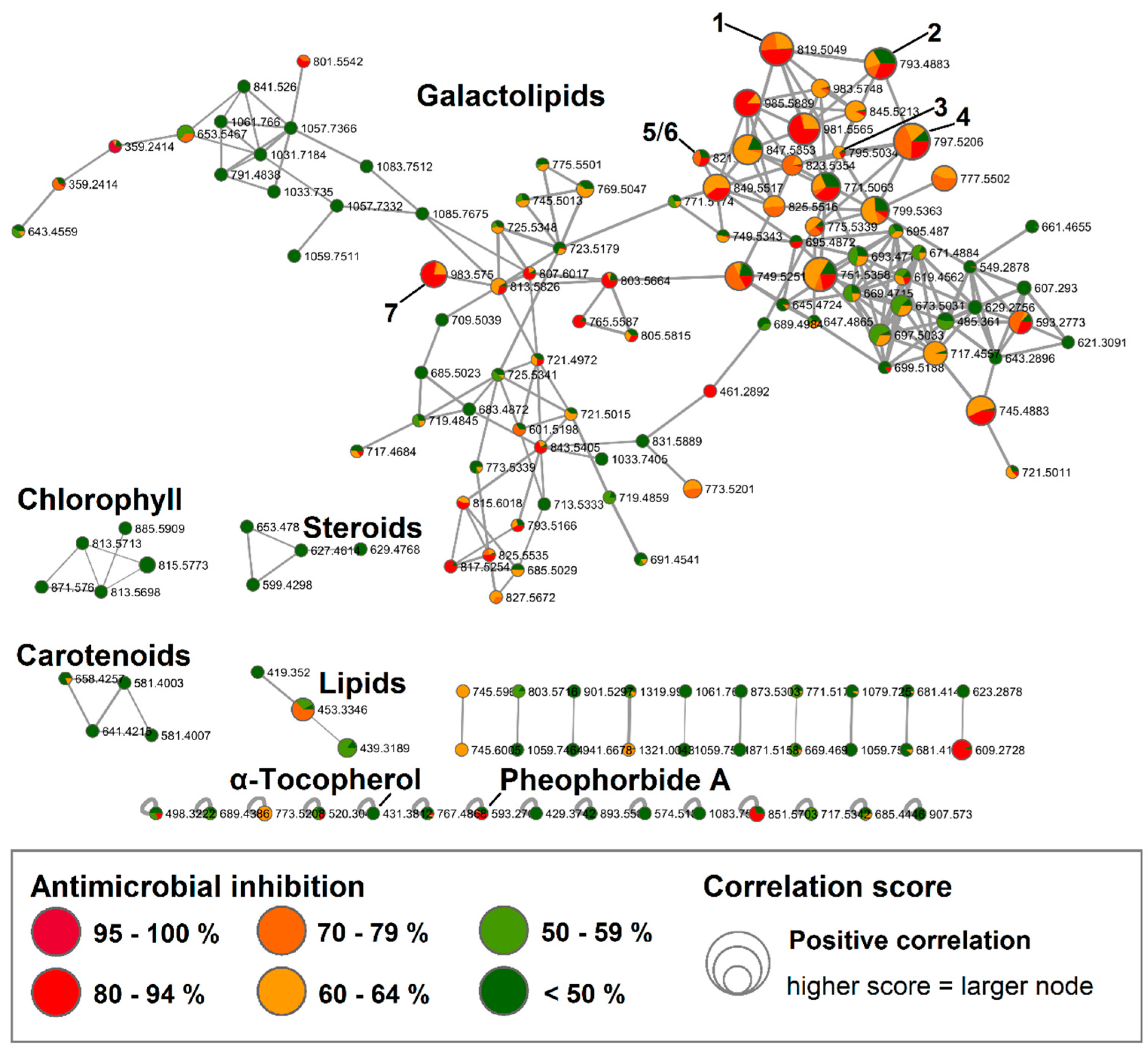
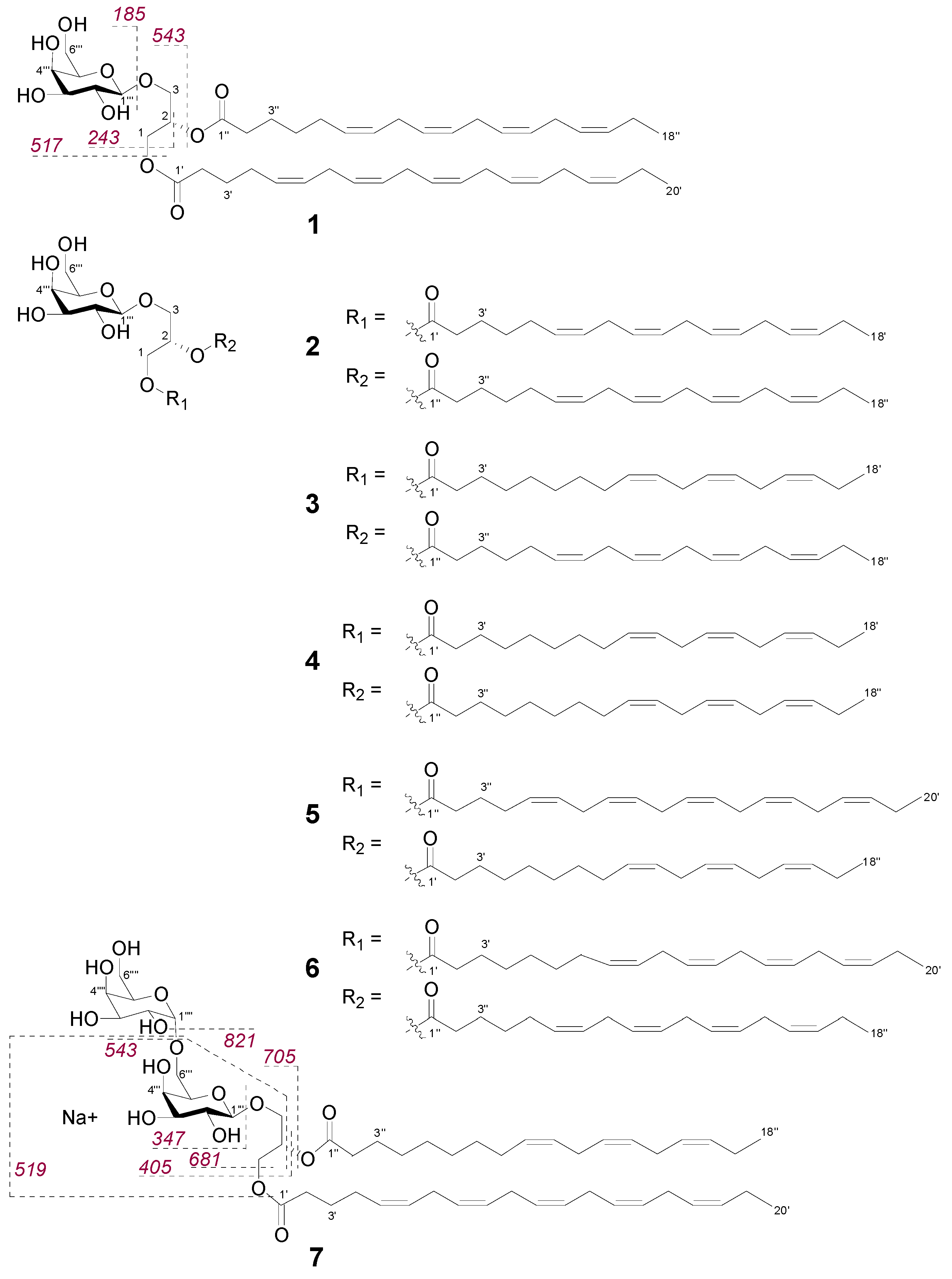
2.2. Bioactive Molecular Networking Guided Isolation of Galactolipids
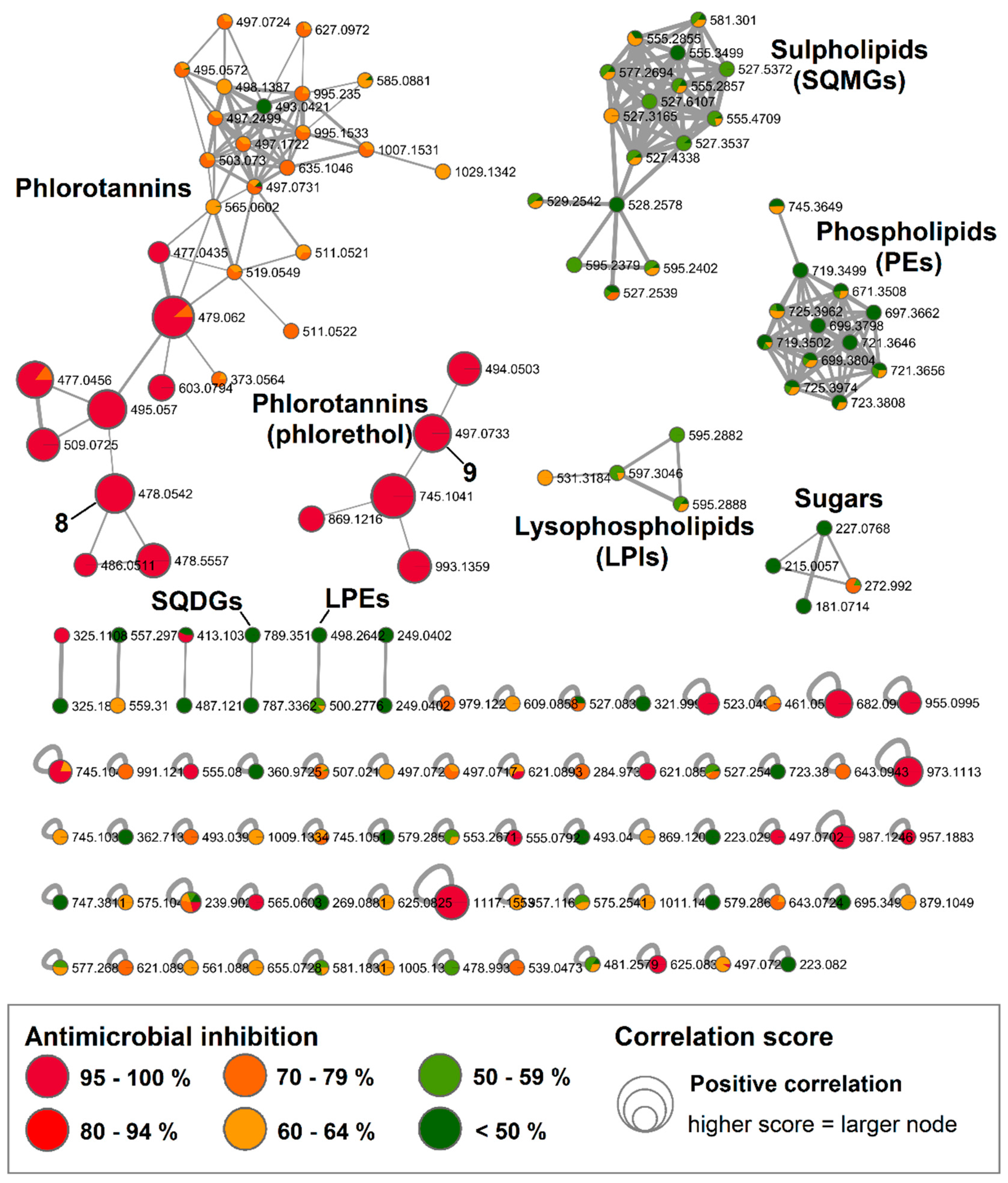
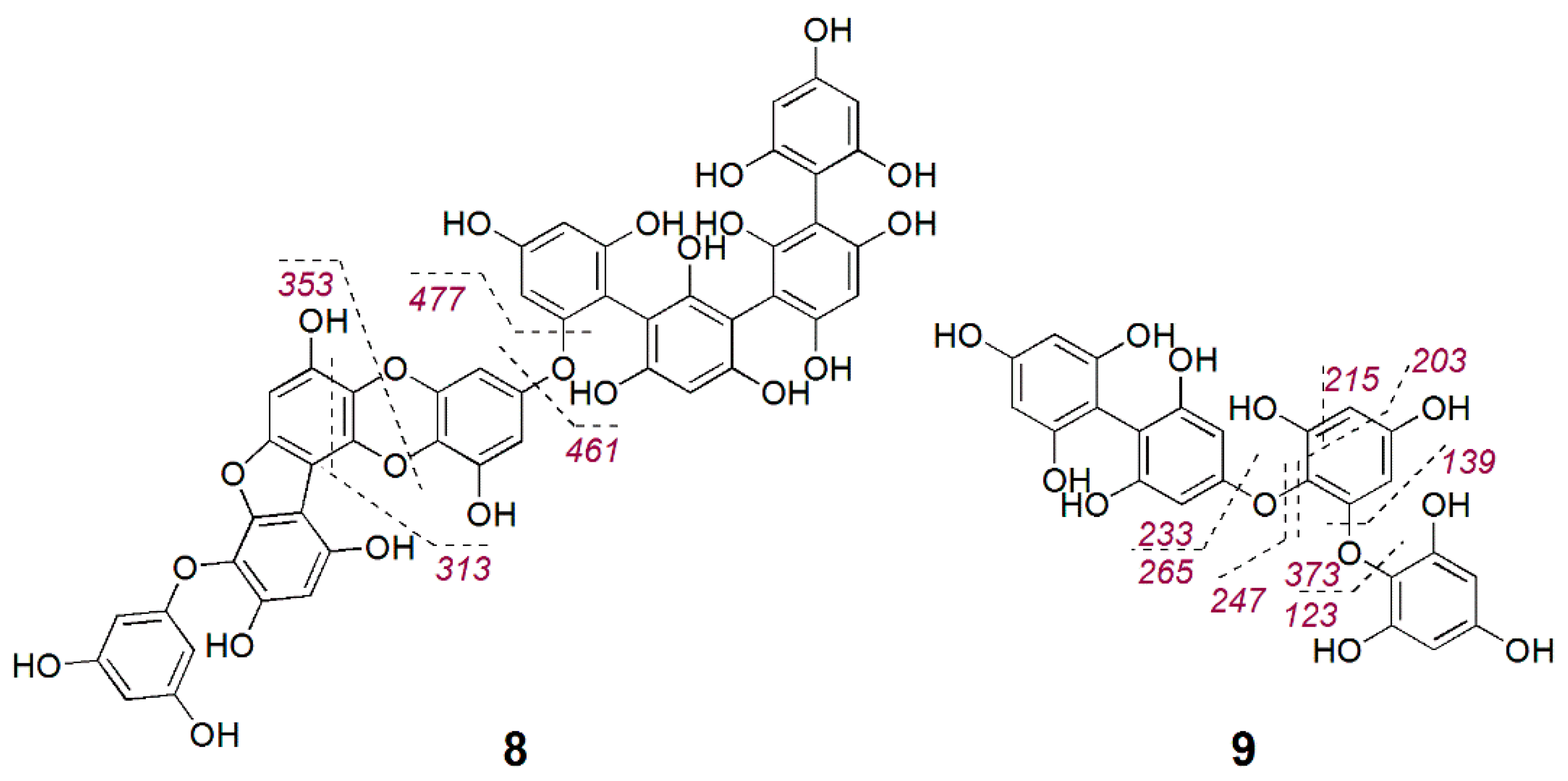
2.3. Bioactive Molecular Netwoking of the F. vesiculosus n-BuOH Subextract
3. Discussion
4. Materials and Methods
4.1. General Procedures
4.2. Biological Material
4.3. Extraction, Fractionation and Isolation
4.4. LC-MS/MS-Based Bioactivity Molecular Networking
4.5. UNPD in Silico MS/MS Database and Manual Dereplication
4.6. Antimicrobial Activity
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wahl, M.; Molis, M.; Hobday, A.J.; Dudgeon, S.; Neumann, R.; Steinberg, P.; Campbell, A.H.; Marzinelli, E.; Connell, S. The responses of brown macroalgae to environmental change from local to global scales: Direct versus ecologically mediated effects. Perspect. Psychol. Sci. 2015, 2, 11–29. [Google Scholar] [CrossRef] [Green Version]
- Wahl, M.; Shahnaz, L.; Dobretsov, S.; Saha, M.; Symanowski, F.; David, K.; Lachnit, T.; Vasel, M.; Weinberger, F. Ecology of antifouling resistance in the bladder wrack Fucus vesiculosus: Patterns of microfouling and antimicrobial protection. Mar. Ecol. Prog. Ser. 2010, 411, 33–48. [Google Scholar] [CrossRef]
- Rickert, E.; Lenz, M.; Barboza, F.; Gorb, S.; Wahl, M. Seasonally fluctuating chemical microfouling control in Fucus vesiculosus and Fucus serratus from the Baltic Sea. Mar. Biol. 2016, 163, 203. [Google Scholar] [CrossRef] [Green Version]
- Wahl, M.; Goecke, F.; Labes, A.; Dobretsov, S.; Weinberger, F. The second skin: Ecological role of epibiotic biofilms on marine organisms. Front. Microbiol. 2012, 3, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrot, D.; Blümel, M.; Utermann, C.; Chianese, G.; Krause, S.; Kovalev, A.; Gorb, S.N.; Tasdemir, D. Mapping the surface microbiome and metabolome of brown seaweed Fucus vesiculosus by amplicon sequencing, integrated metabolomics and imaging techniques. Sci. Rep. 2019, 9, 1061. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.J.; Falqué, E.; Domínguez, H. Antimicrobial action of compounds from marine seaweed. Mar. Drugs 2016, 14, 52. [Google Scholar] [CrossRef] [Green Version]
- Narayani, C.G.S.; Arulpriya, M.; Ruban, P.; Anantharaj, K.; Srinivasan, R. In vitro antimicrobial activities of seaweed extracts against human pathogens. J. Pharm. Res. 2011, 4, 2076–2077. [Google Scholar]
- Heavisides, E.; Rouger, C.; Reichel, A.F.; Ulrich, C.; Wenzel-Storjohann, A.; Sebens, S.; Tasdemir, D. Seasonal variations in the metabolome and bioactivity profile of Fucus vesiculosus extracted by an optimised, pressurised liquid extraction protocol. Mar. Drugs 2018, 16, 503. [Google Scholar] [CrossRef] [Green Version]
- Sandsdalen, E.; Haug, T.; Stensvåg, K.; Styrvold, O.B. The antibacterial effect of a polyhydroxylated fucophlorethol from the marine brown alga, Fucus vesiculosus. World J. Microbiol. Biotechnol. 2003, 19, 777–782. [Google Scholar] [CrossRef]
- Saha, M.; Rempt, M.; Grosser, K.; Pohnert, G.; Weinberger, F. Surface-associated fucoxanthin mediates settlement of bacterial epiphytes on the rockweed Fucus vesiculosus. Biofouling 2011, 27, 423–433. [Google Scholar] [CrossRef]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nothias, L.F.; Petras, D.; Schmid, R.; Dührkop, K.; Rainer, J.; Sarvepalli, A.; Protsyuk, I.; Ernst, M.; Tsugawa, H.; Fleischauer, M.; et al. Feature-based molecular networking in the GNPS analysis environment. bioRxiv 2019, 812404. [Google Scholar]
- Nothias, L.-F.L.; Nothias-Esposito, M.L.; da Silva, R.; Wang, M.; Protsyuk, I.; Zhang, Z.; Sarvepalli, A.; Leyssen, P.; Touboul, D.; Costa, J.; et al. Bioactivity-based molecular networking for the discovery of drug leads in natural product bioassay-guided fractionation. J. Nat. Prod. 2018, 81, 758–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kupchan, S.M.; Britton, R.W.; Ziegler, M.F.; Sigel, C.W. Bruceantin, a new potent antileukemic simaroubolide from Brucea antidysenterica. J. Org. Chem. 1973, 38, 178–179. [Google Scholar] [CrossRef]
- Allard, P.-M.; Péresse, T.; Bisson, J.; Gindro, K.; Marcourt, L.; Pham, V.C.; Roussi, F.; Litaudon, M.; Wolfender, J.-L. Integration of molecular networking and in-silico MS/MS fragmentation for natural products dereplication. Anal. Chem. 2016, 88, 3317–3323. [Google Scholar] [CrossRef]
- Taylor, R. Interpretation of the correlation coefficient: A basic review. J. Diagn. Med. Sonogr. 1990, 6, 35–39. [Google Scholar] [CrossRef]
- Da Costa, E.; Silva, J.; Mendonça, S.H.; Abreu, M.H.; Domingues, M.R. Lipidomic approaches towards deciphering glycolipids from microalgae as a reservoir of bioactive lipids. Mar. Drugs 2016, 14, 101. [Google Scholar] [CrossRef] [Green Version]
- IUPAC-IUB Commission on Biochemical Nomenclature. The nomenclature of lipids: Recommendations (1976). Lipids 1977, 12, 455–468. [Google Scholar] [CrossRef]
- Guella, G.; Frassanito, R.; Mancini, I. A new solution for an old problem: The regiochemical distribution of the acyl chains in galactolipids can be established by electrospray ionization tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2003, 17, 1982–1994. [Google Scholar] [CrossRef]
- Cholewski, M.; Tomczykowa, M.; Tomczyk, M. A comprehensive review of chemistry, sources and bioavailability of omega-3 fatty acids. Nutrients 2018, 10, 1662. [Google Scholar] [CrossRef] [Green Version]
- Gunstone, F.; Pollard, M.; Scrimgeour, C.; Vedanayagam, H. Fatty acids. Part 50. 13C nuclear magnetic resonance studies of olefinic fatty acids and esters. Chem. Phys. Lipids 1977, 18, 115–129. [Google Scholar] [CrossRef]
- Alexandri, E.; Ahmed, R.; Siddiqui, H.; Choudhary, M.I.; Tsiafoulis, C.G.; Gerothanassis, I.P. High resolution NMR spectroscopy as a structural and analytical tool for unsaturated lipids in solution. Molecules 2017, 22, 1663. [Google Scholar] [CrossRef] [PubMed]
- Imbs, T.I.; Ermakova, S.P.; Fedoreyev, S.A.; Anastyuk, S.D.; Zvyagintseva, T.N. Isolation of fucoxanthin and highly unsaturated monogalactosyldiacylglycerol from brown alga Fucus evanescens C Agardh and in vitro investigation of their antitumor activity. Mar. Biotechnol. 2013, 15, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Kim, E.H.; Lee, C.; Kim, M.H.; Rho, J.R. Two new monogalactosyl diacylglycerols from brown alga Sargassum thunbergii. Lipids 2007, 42, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Banskota, A.H.; Stefanova, R.; Gallant, P.; McGinn, P.J. Mono-and digalactosyldiacylglycerols: Potent nitric oxide inhibitors from the marine microalga Nannochloropsis granulata. J. Appl. Phycol. 2013, 25, 349–357. [Google Scholar] [CrossRef]
- Hauksson, J.B.; Bergqvist, M.H.; Rilfors, L. Structure of digalactosyldiacylglycerol from oats. Chem. Phys. Lipids 1995, 78, 97–102. [Google Scholar] [CrossRef]
- Kobayashi, M.; Hayashi, K.; Kawazoe, K.; Kitagawa, I. Marine natural products. XXIX. Heterosigma-glycolipids I, II, III, and IV, four diacylglyceroglycolipids possessing ω3-polyunsaturated fatty acid residues, from the raphidopycean dinoflagellate Heterosigma akashiwo. Chem. Pharm. Bull. 1992, 40, 1404–1410. [Google Scholar] [CrossRef] [Green Version]
- Larsen, E.; Kharazmi, A.; Christensen, L.P.; Christensen, S.B. An antiinflammatory galactolipid from rose hip (Rosa canina) that inhibits chemotaxis of human peripheral blood neutrophils in vitro. J. Nat. Prod. 2003, 66, 994–995. [Google Scholar] [CrossRef]
- Da Costa, E.; Domingues, P.; Melo, T.; Coelho, E.; Pereira, R.; Calado, R.; Abreu, M.H.; Domingues, M.R. Lipidomic signatures reveal seasonal shifts on the relative abundance of high-valued lipids from the brown alga Fucus vesiculosus. Mar. Drugs 2019, 17, 335. [Google Scholar] [CrossRef] [Green Version]
- Lopes, G.; Barbosa, M.; Vallejo, F.; Gil-Izquierdo, Á.; Andrade, P.B.; Valentão, P.; Pereira, D.M.; Ferreres, F. Profiling phlorotannins from Fucus spp. of the Northern Portuguese coastline: Chemical approach by HPLC-DAD-ESI/MSn and UPLC-ESI-QTOF/MS. Algal Res. 2018, 29, 113–120. [Google Scholar] [CrossRef]
- Catarino, M.D.; Silva, A.; Mateus, N.; Cardoso, S.M. Optimization of phlorotannins extraction from Fucus vesiculosus and evaluation of their potential to prevent metabolic disorders. Mar. Drugs 2019, 17, 162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, K.A.; Lee, K.H.; Chae, S.; Koh, Y.S.; Yoo, B.-S.; Kim, J.H.; Ham, Y.M.; Baik, J.S.; Lee, N.H.; Hyun, J.W. Triphlorethol-A from Ecklonia cava protects V79-4 lung fibroblast against hydrogen peroxide induced cell damage. Free Radic. Res. 2005, 39, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Glombitza, K.-W.; Zieprath, G. Phlorotannins from the brown alga Analipus japonicus. Planta Med. 1989, 55, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Yotsu-Yamashita, M.; Kondo, S.; Segawa, S.; Lin, Y.-C.; Toyohara, H.; Ito, H.; Konoki, K.; Cho, Y.; Uchida, T. Isolation and structural determination of two novel phlorotannins from the brown alga Ecklonia kurome Okamura, and their radical scavenging activities. Mar. Drugs 2013, 11, 165–183. [Google Scholar] [CrossRef] [Green Version]
- Ford, L.; Theodoridou, K.; Sheldrake, G.N.; Walsh, P.J. A critical review of analytical methods used for the chemical characterisation and quantification of phlorotannin compounds in brown seaweeds. Phytochem. Analysis 2019, 30, 587–599. [Google Scholar] [CrossRef]
- Kirke, D.; Smyth, T.; Rai, D.; Kenny, O.; Stengel, D. The chemical and antioxidant stability of isolated low molecular weight phlorotannins. Food Chem. 2017, 221, 1104–1112. [Google Scholar] [CrossRef]
- Wang, T.; Jónsdóttir, R.S.; Liu, H.; Gu, L.; Kristinsson, H.G.; Raghavan, S.; Ólafsdóttir, G.N. Antioxidant capacities of phlorotannins extracted from the brown algae Fucus vesiculosus. J. Agric. Food. Chem. 2012, 60, 5874–5883. [Google Scholar] [CrossRef]
- Kellogg, J.J.; Todd, D.A.; Egan, J.M.; Raja, H.A.; Oberlies, N.H.; Kvalheim, O.M.; Cech, N.B. Biochemometrics for natural products research: Comparison of data analysis approaches and application to identification of bioactive compounds. J. Nat. Prod. 2016, 79, 376–386. [Google Scholar] [CrossRef] [Green Version]
- Fan, B.; Dewapriya, P.; Li, F.; Blümel, M.; Tasdemir, D. Pyrenosetins A–C, new decalinoylspirotetramic acid derivatives isolated by bioactivity-based molecular networking from the seaweed-derived fungus Pyrenochaetopsis sp. FVE-001. Mar. Drugs 2020, 18, 47. [Google Scholar] [CrossRef] [Green Version]
- Gounaris, K.; Barber, J. Monogalactosyldiacylglycerol: The most abundant polar lipid in nature. Trends Biochem. Sci. 1983, 8, 378–381. [Google Scholar] [CrossRef]
- Jones, A.L.; Harwood, J.L. Lipid composition of the brown algae Fucus vesiculosus and Ascophyllum nodosum. Phytochemistry 1992, 31, 3397–3403. [Google Scholar] [CrossRef]
- Logvinov, S.; Gerasimenko, N.; Esipov, A.; Denisenko, V.A. Examination of the structures of several glycerolipids from marine macroalgae by NMR and GC-MS. J. Phycol. 2015, 51, 1066–1074. [Google Scholar] [CrossRef]
- Deal, M.S.; Hay, M.E.; Wilson, D.; Fenical, W. Galactolipids rather than phlorotannins as herbivore deterrents in the brown seaweed Fucus vesiculosus. Oecologia 2003, 136, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Bruno, A.; Rossi, C.; Marcolongo, G.; Di Lena, A.; Venzo, A.; Berrie, C.P.; Corda, D. Selective in vivo anti-inflammatory action of the galactolipid monogalactosyldiacylglycerol. Eur. J. Pharmacol. 2005, 524, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Ahamed, A.A.P.; Rasheed, M.U.; Noorani, K.P.M.; Reehana, N.; Santhoshkumar, S.; Imran, Y.M.M.; Alharbi, N.S.; Arunachalam, C.; Alharbi, S.A.; Akbarsha, M.A.; et al. In vitro antibacterial activity of MGDG-palmitoyl from Oscillatoria acuminata NTAPC05 against extended-spectrum β-lactamase producers. J. Antibiot. 2017, 70, 754–762. [Google Scholar] [CrossRef]
- Martins, A.; Vasas, A.; Viveiros, M.; Molnár, J.; Hohmann, J.; Amaral, L. Antibacterial properties of compounds isolated from Carpobrotus edulis. Int. J. Antimicrob. Agents 2011, 37, 438–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, K.; Lee, J.-B.; Atsumi, K.; Kanazashi, M.; Shibayama, T.; Okamoto, K.; Kawahara, T.; Hayashi, T. In vitro and in vivo anti-herpes simplex virus activity of monogalactosyl diacylglyceride from Coccomyxa sp. KJ (IPOD FERM BP-22254), a green microalga. PLoS ONE 2019, 14, e0219305. [Google Scholar] [CrossRef] [Green Version]
- Maeda, N.; Kokai, Y.; Hada, T.; Yoshida, H.; Mizushina, Y. Oral administration of monogalactosyl diacylglycerol from spinach inhibits colon tumor growth in mice. Exp. Ther. Med. 2013, 5, 17–22. [Google Scholar] [CrossRef]
- Morimoto, T.; Nagatsu, A.; Murakami, N.; Sakakibara, J.; Tokuda, H.; Nishino, H.; Iwashima, A. Anti-tumour-promoting glyceroglycolipids from the green alga, Chlorella vulgaris. Phytochemistry 1995, 40, 1433–1437. [Google Scholar] [CrossRef]
- Zhang, J.; Li, C.; Yu, G.; Guan, H. Total synthesis and structure-activity relationship of glycoglycerolipids from marine organisms. Mar. Drugs 2014, 12, 3634–3659. [Google Scholar] [CrossRef] [Green Version]
- Colombo, D.; Compostella, F.; Ronchetti, F.; Scala, A.; Toma, L.; Mukainaka, T.; Nagatsu, A.; Konoshima, T.; Tokuda, H.; Nishino, H. Inhibitory effects of monoacylated 2-O-β-galactosylglycerols on Epstein–Barr virus activation: The significant role of the hexanoyl chain. Cancer Lett. 1999, 143, 1–4. [Google Scholar] [CrossRef]
- Caesar, L.K.; Kellogg, J.J.; Kvalheim, O.M.; Cech, N.B. Opportunities and limitations for untargeted mass spectrometry metabolomics to identify biologically active constituents in complex natural product mixtures. J. Nat. Prod. 2019, 82, 469–484. [Google Scholar] [CrossRef]
- Peng, J.; Yuan, J.-P.; Wu, C.-F.; Wang, J.-H. Fucoxanthin, a marine carotenoid present in brown seaweeds and diatoms: Metabolism and bioactivities relevant to human health. Mar. Drugs 2011, 9, 1806–1828. [Google Scholar] [CrossRef]
- Karpiński, T.M.; Adamczak, A. Fucoxanthin-An antibacterial carotenoid. Antioxidants 2019, 8, 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoenwaelder, M.E.; Clayton, M.N. The role of the cytoskeleton in brown algal physode movement. Eur. J. Pharmacol. 1999, 34, 223–229. [Google Scholar] [CrossRef]
- Schoenwaelder, M.E.; Wiencke. Phenolic compounds in the embryo development of several northern hemisphere fucoids. Plant Biol. 2000, 2, 24–33. [Google Scholar] [CrossRef]
- Halm, H.; Lüder, U.H.; Wiencke, C. Induction of phlorotannins through mechanical wounding and radiation conditions in the brown macroalga Laminaria hyperborea. Eur. J. Phycol. 2011, 46, 16–26. [Google Scholar] [CrossRef] [Green Version]
- Koivikko, R.; Loponen, J.; Honkanen, T.; Jormalainen, V. Contents of soluble, cell-wall-bound and exuded phlorotannins in the brown alga Fucus vesiculosus, with implications on their ecological functions. J. Chem. Ecol. 2005, 31, 195–212. [Google Scholar] [CrossRef] [Green Version]
- Amsler, C.D.; Fairhead, V.A. Defensive and sensory chemical ecology of brown algae. Adv. Bot. Res. 2005, 43, 1–91. [Google Scholar]
- Nagayama, K.; Iwamura, Y.; Shibata, T.; Hirayama, I.; Nakamura, T. Bactericidal activity of phlorotannins from the brown alga Ecklonia kurome. J. Antimicrob. Chemother. 2002, 50, 889–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eom, S.-H.; Kim, Y.-M.; Kim, S.-K. Antimicrobial effect of phlorotannins from marine brown algae. Food Chem. Toxicol. 2012, 50, 3251–3255. [Google Scholar] [CrossRef] [PubMed]
- Shannon, E.; Abu-Ghannam, N. Antibacterial derivatives of marine algae: An overview of pharmacological mechanisms and applications. Mar. Drugs 2016, 14, 81. [Google Scholar] [CrossRef] [PubMed]
- Glombitza, K.-W.; Schmidt, A. Nonhalogenated and halogenated phlorotannins from the brown alga Carpophyllum angustifolium. J. Nat. Prod. 1999, 62, 1238–1240. [Google Scholar] [CrossRef] [PubMed]
- Glombitza, K.-W.; Schmidt, A. Trihydroxyphlorethols from the brown alga Carpophyllum angustifolium. Phytochemistry 1999, 51, 1095–1100. [Google Scholar] [CrossRef]
- Glombitza, K.-W.; Pauli, K. Fucols and phlorethols from the brown alga Scytothamnus australis Hook. et Harv. (Chnoosporaceae). Bot. Mar. 2003, 46, 315–320. [Google Scholar] [CrossRef]
- Ham, Y.M.; Baik, J.S.; Hyun, J.W.; Lee, N.H. Isolation of a new phlorotannin, fucodiphlorethol G, from a brown alga Ecklonia cava. Bull. Korean Chem. Soc. 2007, 28, 1595. [Google Scholar]
- Li, Y.; Lee, S.-H.; Le, Q.-T.; Kim, M.-M.; Kim, S.-K. Anti-allergic effects of phlorotannins on histamine release via binding inhibition between IgE and FcεRI. J. Agric. Food. Chem. 2008, 56, 12073–12080. [Google Scholar] [CrossRef]
- Parys, S.; Kehraus, S.; Krick, A.; Glombitza, K.-W.; Carmeli, S.; Klimo, K.; Gerhäuser, C.; König, G.M. In vitro chemopreventive potential of fucophlorethols from the brown alga Fucus vesiculosus L. by anti-oxidant activity and inhibition of selected cytochrome P450 enzymes. Phytochemistry 2010, 71, 221–229. [Google Scholar] [CrossRef]
- Kurihara, H.; Konno, R.; Takahashi, K. Fucophlorethol C, a phlorotannin as a lipoxygenase inhibitor. Biosci. Biotechnol. Biochem. 2015, 79, 1954–1956. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Yi, M.; Ding, L.; He, S.; Yan, X. Isolation and purification of a neuroprotective phlorotannin from the marine algae Ecklonia maxima by size exclusion and high-speed counter-current chromatography. Mar. Drugs 2019, 17, 212. [Google Scholar] [CrossRef] [Green Version]
- Zani, C.L.; Carroll, A.R. Database for rapid dereplication of known natural products using data from MS and fast NMR experiments. J. Nat. Prod. 2017, 80, 1758–1766. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δH Multiplicity (J in Hz) | δC | HMBC | NOE |
|---|---|---|---|---|
| 1 | 4.40 dd (12.1, 3.5) | 62.4 | C2, C1′ | H2 |
| 4.21 dd (12.1, 6.5) | ||||
| 2 | 5.31 m | 69.9 | - | H1, H3 |
| 3 | 3.98 dd (12.1, 5.8) 3.75 (11.3, 6.3) | 62.8 | C1, C2, C3‴ | H2, H1‴ |
| 1′ | - | 173.5 | - | |
| 2′ | 2.33 m | 33.2 | C1′, C3′ | |
| 3′ | 1.69 m | 24.4 | C1′, C5′, C2′ | |
| 4′ | 2.07 m | 26.5 | C2′, cluster1 | |
| 5′, 6′, 8′, 9′, 11′, 12′, 14′, 15′, 17′, 18′ (cluster1) | 5.31–5.39 m | 126.8–131.8 | cluster2 | |
| 7′, 10′, 13′, 16′ (cluster2) | 2.81 m | 25.3–26.6 | cluster1 | |
| 19′ | 2.08 m | 20.3 | C20′, cluster1 | |
| 20′ | 0.97 t (7.5) | 14.0 | C18′, C19′ | |
| 1″ | - | 173.3 | - | |
| 2″ | 2.35 m | 33.8 | C1″, C3″, C4″ | |
| 3″ | 1.65 m | 24.5 | C1″, C2″, C4″ | |
| 4″ | 1.38 m | 28.7 | C5″, cluster 1 | |
| 5″ | 2.07 m | 27.0 | cluster 1 | |
| 6″, 7″, 9″, 10″, 12″, 13″, 15″, 16″ (cluster1) | 5.31–5.39 m | 126.8–131.8 | cluster2 | |
| 8″, 11″, 14″ (cluster2) | 2.81 m | 25.3–26.6 | cluster1 | |
| 17″ | 2.07 m | 20.3 | C18′, cluster1 | |
| 18″ | 0.97 t (7.5) | 14.0 | C16″, C17″ | |
| 1‴ | 4.28 d (7.2) | 103.7 | C3 | H3‴, H5‴, H1, H3 |
| 2‴ | 3.63 dd (9.3, 7.2) | 71.4 | C1‴, C3‴ | H3 |
| 3‴ | 3.59 dd (9.3, 3.5) | 73.2 | C2‴ | H1‴, H4‴ |
| 4‴ | 4.01 dd (3.5, 0.6) | 69.3 | C2‴, C3‴ | H3‴, H5‴ |
| 5‴ | 3.55 m | 74.3 | - | H1‴, H4‴, H6‴ |
| 6‴ | 3.98 m 3.86 dd (11.4, 6.8) | 62.8 | - | H5‴ |
| Compound | MRSA | S. aureus (DSM346) | |
|---|---|---|---|
| Inhibition Rate (%, 100 µg/mL) | Inhibition Rate (%, 200 µg/mL) | IC50 (µg/mL) | |
| 1 | - | 40 | >200 |
| 2 | - | 49 | >200 |
| 3 | - | 32 | >200 |
| 4 | - | 49 | >200 |
| 5 | - | 60 | 96 (±4) |
| 6 | - | 62 | 123 (±8) |
| 7 | - | 66 | 87 (±4) |
| Positive control | 100 | 100 | 0.5 (±0.0) |
| δH Multiplicity (J in Hz) | δC | HMBC H→C |
|---|---|---|
| 5.65 d (2.1) | 94.1 | |
| 5.70 d (2.1) | 94.3 | 94.5; 157.7 |
| 5.71 d (2.1) | 94.3 | |
| 5.77 d (2.1) | 94.2 | 94.5; 157.5 |
| 5.78 d (2.3) | 95.6 | |
| 5.83 d (2.3) | 94.2 | 158.5 |
| 5.85 d (2.3) | 94.2 | |
| 8.4 s | - | 94.5; 157.7 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buedenbender, L.; Astone, F.A.; Tasdemir, D. Bioactive Molecular Networking for Mapping the Antimicrobial Constituents of the Baltic Brown Alga Fucus vesiculosus. Mar. Drugs 2020, 18, 311. https://doi.org/10.3390/md18060311
Buedenbender L, Astone FA, Tasdemir D. Bioactive Molecular Networking for Mapping the Antimicrobial Constituents of the Baltic Brown Alga Fucus vesiculosus. Marine Drugs. 2020; 18(6):311. https://doi.org/10.3390/md18060311
Chicago/Turabian StyleBuedenbender, Larissa, Francesca Anna Astone, and Deniz Tasdemir. 2020. "Bioactive Molecular Networking for Mapping the Antimicrobial Constituents of the Baltic Brown Alga Fucus vesiculosus" Marine Drugs 18, no. 6: 311. https://doi.org/10.3390/md18060311
APA StyleBuedenbender, L., Astone, F. A., & Tasdemir, D. (2020). Bioactive Molecular Networking for Mapping the Antimicrobial Constituents of the Baltic Brown Alga Fucus vesiculosus. Marine Drugs, 18(6), 311. https://doi.org/10.3390/md18060311