Bioactive Bianthraquinones and Meroterpenoids from a Marine-Derived Stemphylium sp. Fungus

 , ,
, ,
Abstract
:
1. Introduction
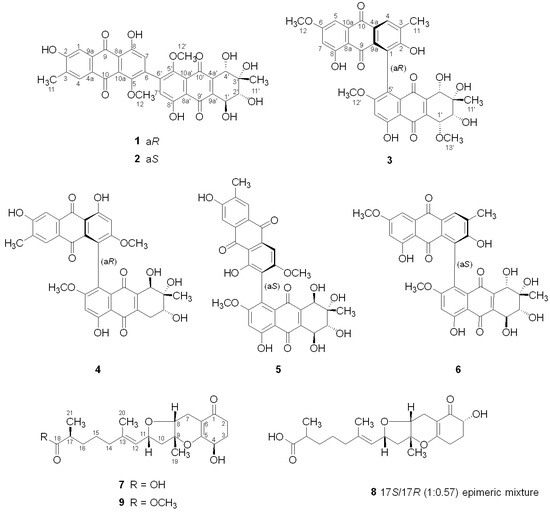
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Fermentation
3.4. Extraction and Isolation
3.5. Preparations of the (S)- and (R)-MTPA Esters of Compounds 7 and 8
3.5.1. (S)-MTPA Ester of 7 (7-4S)
3.5.2. (R)-MTPA Ester of 7 (7-4R)
3.5.3. (S)-MTPA Ester of 8 (8-2S)
3.5.4. (R)-MTPA Ester of 8 (8-2R)
3.6. Preparations of the (S)- and (R)-PGME Amides of Compounds 7 and 8
3.6.1. (S)-PGME Amide of 7 (7-18S)
3.6.2. (R)-PGME Amide of 7 (7-18R)
3.6.3. (S)-PGME Amide of 8 (8-18S)
3.6.4. (R)-PGME Amide of 8 (8-18R)
3.7. ECD Calculations
3.8. DP4 Analysis
3.9. Cytotoxic, Antibacterial, and Enzyme-Inhibitory Activities Assays
3.10. RAW 264.7 Cell Culture
3.11. Nitrite Production Measurement
3.12. Western Blotting Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cox, R.J.; Simpson, T.J. Fungal type I polyketide synthases. Methods Enzymol. 2009, 459, 49–78. [Google Scholar] [CrossRef]
- Cox, R.J. Polyketides, proteins and genes in fungi: Programmed nano-machines begin to reveal their secrets. Org. Biomol. Chem. 2007, 5, 2010–2026. [Google Scholar] [CrossRef]
- Fouillaud, M.; Venkatachalam, M.; Girard-Valenciennes, E.; Caro, Y.; Dufosse, L. Anthraquinones and derivatives from marine-derived fungi: Structural diversity and selected biological activities. Mar. Drugs 2016, 14, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, H.; Al-Sadi, A.M.; Schulz, B.; Steinert, M.; Khan, A.; Green, I.R.; Ahmed, I. A fruitful decade for fungal polyketides from 2007 to 2016: Antimicrobial activity, chemotaxonomy and chemodiversity. Future Med. Chem. 2017, 9, 1631–1648. [Google Scholar] [CrossRef] [PubMed]
- Stoessl, A.; Unwin, C.H.; Sthothers, J.B. On the biosynthesis of some polyketide metabolites in Alternaria solani: 13C and 2Hmr studies. Can. J. Chem. 1983, 61, 372–377. [Google Scholar] [CrossRef]
- Bringmann, G.; Irmer, A.; Feineis, D.; Gulder, T.A.M.; Fiedler, H.-P. Convergence in the biosynthesis of acetogenic natural products from plants, fungi, and bacteria. Phytochemistry 2009, 70, 1776–1786. [Google Scholar] [CrossRef]
- Caro, Y.; Anamale, L.; Fouillaud, M.; Laurent, P.; Petit, T.; Dufosse, L. Natural hydroxyanthraquinoid pigments as potent food grade colorants: An overview. Nat. Prod. Bioprospect. 2012, 2, 174–193. [Google Scholar] [CrossRef]
- Ge, X.; Sun, C.; Feng, Y.; Wang, L.; Peng, J.; Che, Q.; Gu, Q.; Zhu, T.; Li, D.; Zhang, G. Anthraquinone derivatives from a marine-derived fungus Sporendonema casei HDN16-802. Mar. Drugs 2019, 17, 334. [Google Scholar] [CrossRef] [Green Version]
- Gessler, N.N.; Egorova, A.S.; Belozerskaia, T.A. Fungal anthraquinones. Appl. Biochem. Microbiol. 2013, 49, 85–99. [Google Scholar] [CrossRef]
- Fouillaud, M.; Caro, Y.; Venkatachalam, M.; Grondin, I.; Dufossé, L. Anthraquinones. In Phenolic Compounds in Food Characterization and Analysis; Nollet, L.M.L., Gutierrez-Uribe, J.A., Eds.; CRC Press: Boca Raton, FL, USA, 2018; pp. 130–170. [Google Scholar]
- Griffiths, S.; Mesarich, C.H.; Saccomanno, B.; Vaisberg, A.; Wit, P.J.G.M.; Cox, R.; Collemare, J. Elucidation of cladofulvin biosynthesis reveals a cytochrome P450 monooxygenase required for anthraquinone dimerization. Proc. Natl. Acad. Sci. USA 2016, 113, 6851–6856. [Google Scholar] [CrossRef] [Green Version]
- Bräse, S.; Encinas, A.; Keck, J.; Nising, C.F. Chemistry and biology of mycotoxins and related fungal metabolites. Chem. Rev. 2009, 109, 3903–3990. [Google Scholar] [CrossRef] [PubMed]
- Malik, E.M.; Muller, C.E. Anthraquinones as pharmacological tools and drugs. Med. Res. Rev. 2016, 36, 705–748. [Google Scholar] [CrossRef] [PubMed]
- Geris, R.; Simpson, T.J. Meroterpenoids produced by fungi. Nat. Prod. Rep. 2009, 26, 1063–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Z.Z.; Miao, F.P.; Fang, S.T.; Liu, X.H.; Yin, X.L.; Ji, N.Y. Sesteralterin and tricycloalterfurenes A–D: terpenes with rarely occurring frameworks from the marine-alga-epiphytic fungus Alternaria alternata k21-1. J. Nat. Prod. 2017, 80, 2524–2529. [Google Scholar] [CrossRef]
- Zhang, G.J.; Wu, G.W.; Zhu, T.J.; Kurtán, T.; Mándi, A.; Jiao, J.J.; Li, J.; Qi, X.; Gu, Q.Q.; Li, D.H. Meroterpenoids with diverse ring systems from the sponge-associated fungus Alternaria sp. JJY-32. J. Nat. Prod. 2013, 76, 1946–1957. [Google Scholar] [CrossRef] [PubMed]
- Bai, Z.Q.; Lin, X.P.; Wang, J.F.; Zhou, X.F.; Liu, J.; Yang, B.; Yang, X.W.; Liao, S.R.; Wang, L.S.; Liu, Y.H. New meroterpenoids from the endophytic fungus Aspergillus flavipes AIL8 derived from the mangrove plant Acanthus ilicifolius. Mar. Drugs 2015, 13, 237–248. [Google Scholar] [CrossRef]
- Lou, J.; Fu, L.; Peng, Y.; Zhou, L. Metabolites from Alternaria fungi and their bioactivities. Molecules 2013, 18, 5891–5935. [Google Scholar] [CrossRef]
- Shen, L.; Tian, S.-J.; Song, H.-L.; Chen, X.; Guo, H.; Wan, D.; Wang, Y.-R.; Wang, F.-W.; Liu, L.-J. Cytotoxic tricycloalternarene compounds from endophyte Alternaria sp. W-1 associated with Laminaria japonica. Mar. Drugs 2018, 16, 402. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Zhao, Z.; Chen, J.; Bai, X.; Wang, H. Tricycloalternarene analogs from a symbiotic fungus Aspergillus sp. D and their antimicrobial and cytotoxic effects. Molecules 2018, 23, 855. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.-H.; Pan, J.-H.; Chen, B.; Yu, M.; Huang, H.-B.; Zhu, X.; Lu, Y.-J.; She, Z.-G.; Lin, Y.-C. Three bianthraquinone derivatives from the mangrove endophytic fungus Alternaria sp. ZJ9-6B from the South China Sea. Mar. Drugs 2011, 9, 832–843. [Google Scholar] [CrossRef]
- Zhou, X.-M.; Zheng, C.-J.; Chen, G.-Y.; Song, X.-P.; Han, C.-R.; Li, G.-N.; Fu, Y.-H.; Chen, W.-H.; Niu, Z.-G. Bioactive anthraquinone derivatives from the mangrove-derived fungus Stemphylium sp. 33231. J. Nat. Prod. 2014, 77, 2021–2028. [Google Scholar] [CrossRef] [PubMed]
- Debbab, A.; Aly, A.H.; Edrada-Ebel, R.; Wray, V.; Pretsch, A.; Pescitelli, G.; Kurtan, T.; Proksch, P. New anthracene derivatives—Structure elucidation and antimicrobial activity. Eur. J. Org. Chem. 2012, 2012, 1351–1359. [Google Scholar] [CrossRef]
- Phuwapraisirisan, P.; Rangsan, J.; Siripong, P.; Tip-pyang, S. New antitumour fungal metabolites from Alternaria porri. Nat. Prod. Res. 2009, 23, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Debbab, A.; Aly, A.H.; Edrada-Ebel, R.; Wray, V.; Müller, W.E.G.; Totzke, F.; Zirrgiebel, U.; Schächtele, C.; Kubbutat, M.H.G.; Lin, W.H.; et al. Bioactive metabolites from the endophytic fungus Stemphylium globuliferum isolated from Mentha pulegium. J. Nat. Prod. 2009, 72, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Kwon, O.-S.; Kim, C.-K.; Byun, W.S.; Oh, J.; Lee, Y.-J.; Lee, H.-S.; Sim, C.J.; Oh, D.-C.; Lee, S.K.; Oh, K.-B.; et al. Cyclopeptides from the Sponge Stylissa flabelliformis. J. Nat. Prod. 2018, 81, 1426–1434. [Google Scholar] [CrossRef]
- Lee, H.-S.; Lee, T.-H.; Yang, S.H.; Shin, H.J.; Shin, J.; Oh, K.-B. Sesterterpene sulfates as isocitrate lyase inhibitors from tropical sponge Hippospongia sp. Bioorg. Med. Chem. Lett. 2007, 17, 2483–2486. [Google Scholar] [CrossRef]
- Oh, K.-B.; Kim, S.-H.; Lee, J.; Cho, W.-J.; Lee, T.; Kim, S. Discovery of diarylacrylonitriles as a novel series of small molecule sortase A inhibitors. J. Med. Chem. 2004, 47, 2418–2421. [Google Scholar] [CrossRef]
- Oh, K.-B.; Lee, J.H.; Chung, S.-C.; Shin, J.; Shin, H.J.; Kim, H.-K.; Lee, H.-S. Antimicrobial activities of the bromophenols from the red alga Odonthalia corymbifera and some synthetic derivatives. Bioorg. Med. Chem. Lett. 2008, 18, 104–108. [Google Scholar] [CrossRef]
- Hong, S.-H.; Ban, Y.H.; Byun, W.S.; Kim, D.; Jang, Y.-J.; An, J.S.; Shin, B.; Lee, S.K.; Shin, J.; Yoon, Y.J.; et al. Camporidines A and B: antimetastatic and anti-inflammatory polyketide alkaloids from a gut bacterium of Camponotus kiusiuensis. J. Nat. Prod. 2019, 82, 903–910. [Google Scholar] [CrossRef]
- Byun, W.S.; Kim, W.K.; Han, H.J.; Chung, H.-J.; Jang, K.; Kim, H.S.; Kim, S.; Kim, D.; Bae, E.S.; Park, S.; et al. Targeting histone methyltransferase DOT1L by a novel psammaplin A analog inhibits growth and metastasis of triple-negative breast cancer. Mol. Ther. Oncolytics 2019, 15, 140–152. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 a | 2 b | 3 c | |||
|---|---|---|---|---|---|---|
| δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | |
| 1 | 111.7, CH | 7.51, d (0.5) | 111.8, CH | 7.51, d (0.5) | 129.0, C | |
| 2 | 164.0, C | 163.7, C | 165.5, C d | |||
| 3 | 133.8, C | 133.8, C | 134.5, C | |||
| 4 | 131.2, CH | 7.65, d (0.5) | 131.3, CH | 7.65, d (0.5) | 130.5, CH | 8.00, s |
| 4a | 126.8, C | 127.1, C | 131.2, C | |||
| 5 | 165.8, C | 166.2, C | 106.9, CH | 7.20, d (2.5) | ||
| 6 | 125.3, C | 125.7, C | 167.1, C | |||
| 7 | 104.6, CH | 6.78, s | 104.3, CH | 6.80, s | 105.9, CH | 6.52, d (2.5) |
| 8 | 166.9, C | 167.1, C | 166.0, C | |||
| 8a | 111.9, C | 111.7, C | 112.1, C | |||
| 9 | 188.7, C | 188.6, C | 191.0, C | |||
| 9a | 134.7, C | 134.6, C | 132.8, C | |||
| 10 | 183.5, C | 183.5, C | 182.3, C | |||
| 10a | 133.4, C | 132.8, C | 137.8, C | |||
| 11 | 16.6, CH3 | 2.23, s | 16.6, CH3 | 2.23, s | 17.9, CH3 | 2.21, s |
| 12 | 56.9, CH3 | 3.69, s | 56.9, CH3 | 3.70, s | 56.3, CH3 | 3.89, s |
| 1′ | 70.6, CH | 4.73, d (7.5) | 70.6, CH | 4.73, d (7.4) | 75.2, CH | 4.58, d (4.7) |
| 2′ | 75.2, CH | 3.79, d (7.5) | 75.2, CH | 3.76, d (7.4) | 70.7, CH | 3.93, d (4.7) |
| 3′ | 74.6, C | 74.7, C | 75.4, C | |||
| 4′ | 70.1, CH | 4.26, s | 70.2, CH | 4.26, s | 70.6, CH | 4.39, s |
| 4a’ | 143.8, C | 143.9, C | 144.7, C | |||
| 5′ | 166.3, C | 166.5, C | 130.7, C | |||
| 6′ | 123.4, C | 123.9, C | 166.8, C | |||
| 7′ | 104.6, CH | 6.81, s | 104.6, CH | 6.82, s | 104.5, CH | 6.79, s |
| 8′ | 166.1, C | 166.4, C | 165.9, C | |||
| 8a’ | 111.0, C | 111.0, C | 110.9, C | |||
| 9′ | 190.5, C | 190.6, C | 189.6, C | |||
| 9a’ | 143.9, C | 143.7, C | 141.0, C | |||
| 10′ | 185.7, C | 185.5, C | 185.7, C | |||
| 10a’ | 130.8, C | 130.7, C | n.d. e | |||
| 11′ | 22.3, CH3 | 1.33, s | 22.3, CH3 | 1.32, s | 21.6, CH3 | 1.33, s |
| 12′ | 57.0, CH3 | 3.70, s | 57.0, CH3 | 3.71, s | 56.8, CH3 | 3.71, s |
| 13′ | 62.8, CH3 | 3.77, s | ||||
| No. | 7 a | 8 a | 9 b | |||
|---|---|---|---|---|---|---|
| δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | |
| 1 | 200.4, C | 199.9, C | 200.4, C | |||
| 2 | 33.6, CH2 | 2.62, m 2.32, ddd (16.9, 6.9, 4.9) | 72.4, CH | 4.07, dd (12.6, 4.9) | 33.6, CH2 | 2.62, m 2.32, ddd (17.0, 6.8, 5.1) |
| 3 | 30.5, CH2 | 2.18, m 1.97, m | 30.7, CH2 | 2.24, m1.83, m | 30.5, CH2 | 2.19, m 1.97, m |
| 4 | 66.8, CH | 4.30, t (5.1) | 28.8, CH2 | 2.58, m2.44, m | 66.8, CH | 4.29, t (5.3) |
| 5 | 170.4, C | 170.8, C | 170.4, C | |||
| 6 | 107.5, C | 105.6, C | 107.5, C | |||
| 7 | 19.8, CH2 | 2.59, d (17.8) 2.22, dd (17.7, 4.2) | 19.8, CH2 | 2.68, d (17.8) 2.20, dd (17.7, 4.2) | 19.8, CH2 | 2.59, d (17.8) 2.22, dd (17.7, 4.2) |
| 8 | 78.1, CH | 3.95, dd (4.3, 1.2) | 77.9, CH | 3.95, dd (4.3, 1.2) | 78.1, CH | 3.95, dd (4.5, 1.3) |
| 9 | 85.8, C | 85.7, C | 85.8, C | |||
| 10 | 46.8, CH2 | 2.42, dd (13.6, 9.4) 2.07, dd (13.6, 3.9) | 46.9, CH2 | 2.39, m 2.00, dd (13.7, 3.7) | 46.8, CH2 | 2.42, dd (13.7, 9.5) 2.07, dd (13.6, 3.9) |
| 11 | 74.5, CH | 4.80, td (9.0, 3.9) | 74.5, CH | 4.80, td (9.0, 3.9) | 74.4, CH | 4.79, m |
| 12 | 127.5, CH | 5.17, br d (8.8) | 127.5, CH | 5.17, br d (8.8) | 127.7, CH | 5.17, br d (8.5) |
| 13 | 139.7, C | 139.7, C | 139.6, C | |||
| 14 | 40.2, CH2 | 1.99, m | 40.3, CH2 | 1.98, m | 40.1, CH2 | 1.98, m |
| 15 | 26.3, CH2 | 1.42, m | 26.5, CH2 | 1.42, m | 26.1, CH2 | 1.39, m |
| 16 | 34.5, CH2 | 1.59, m 1.38, m | 34.9, CH2 | 1.58, m 1.35, m | 34.4, CH2 | 1.58, m 1.38, m |
| 17 | 40.8, CH | 2.37, dq (13.8, 7.0) | 41.6, CH | 2.37, m | 40.5, CH | 2.46, m |
| 18 | 180.9, C | 182.1, C | 178.9, C | |||
| 19 | 20.1, CH3 | 1.35, s | 19.9, CH3 | 1.30, s | 20.1, CH3 | 1.35, s |
| 20 | 16.4, CH3 | 1.65, s | 16.4, CH3 | 1.64, s | 16.3, CH3 | 1.65, s |
| 21 | 17.7, CH3 | 1.11, d (7.0) | 18.0, CH3 | 1.11, d (7.0) | 17.5, CH3 | 1.11, d (7.0) |
| 22 | 52.1, OCH3 | 3.65, s | ||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hwang, J.-Y.; Park, S.C.; Byun, W.S.; Oh, D.-C.; Lee, S.K.; Oh, K.-B.; Shin, J. Bioactive Bianthraquinones and Meroterpenoids from a Marine-Derived Stemphylium sp. Fungus. Mar. Drugs 2020, 18, 436. https://doi.org/10.3390/md18090436
Hwang J-Y, Park SC, Byun WS, Oh D-C, Lee SK, Oh K-B, Shin J. Bioactive Bianthraquinones and Meroterpenoids from a Marine-Derived Stemphylium sp. Fungus. Marine Drugs. 2020; 18(9):436. https://doi.org/10.3390/md18090436
Chicago/Turabian StyleHwang, Ji-Yeon, Sung Chul Park, Woong Sub Byun, Dong-Chan Oh, Sang Kook Lee, Ki-Bong Oh, and Jongheon Shin. 2020. "Bioactive Bianthraquinones and Meroterpenoids from a Marine-Derived Stemphylium sp. Fungus" Marine Drugs 18, no. 9: 436. https://doi.org/10.3390/md18090436
APA StyleHwang, J. -Y., Park, S. C., Byun, W. S., Oh, D. -C., Lee, S. K., Oh, K. -B., & Shin, J. (2020). Bioactive Bianthraquinones and Meroterpenoids from a Marine-Derived Stemphylium sp. Fungus. Marine Drugs, 18(9), 436. https://doi.org/10.3390/md18090436