
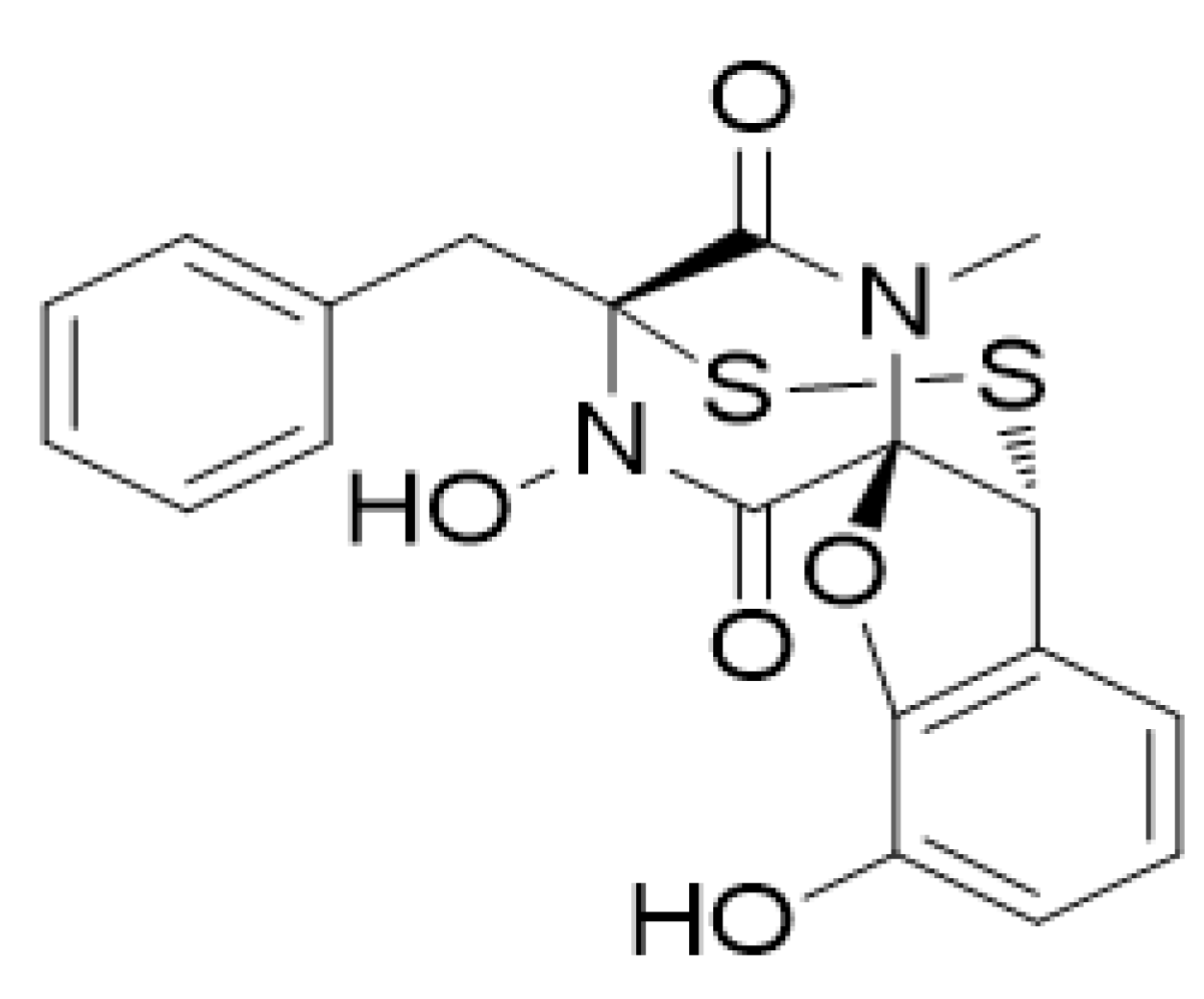
PNSA, a Novel C-Terminal Inhibitor of HSP90, Reverses Epithelial–Mesenchymal Transition and Suppresses Metastasis of Breast Cancer Cells In Vitro



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. PNSA Impairs the Migration and Invasion of Breast Cancer Cells In Vitro
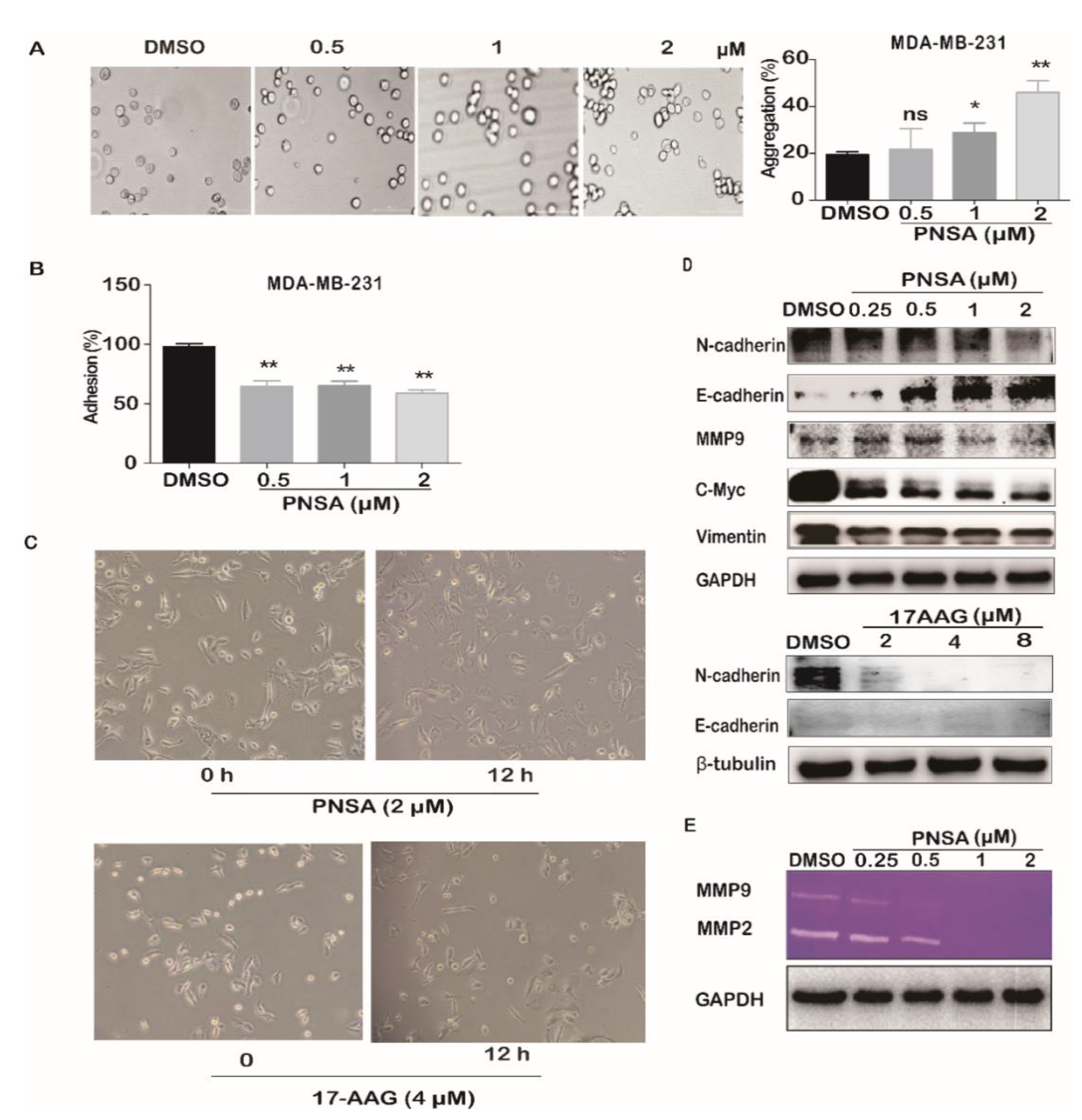
2.2. PNSA Reverses EMT of MDA-MB-231 Cells
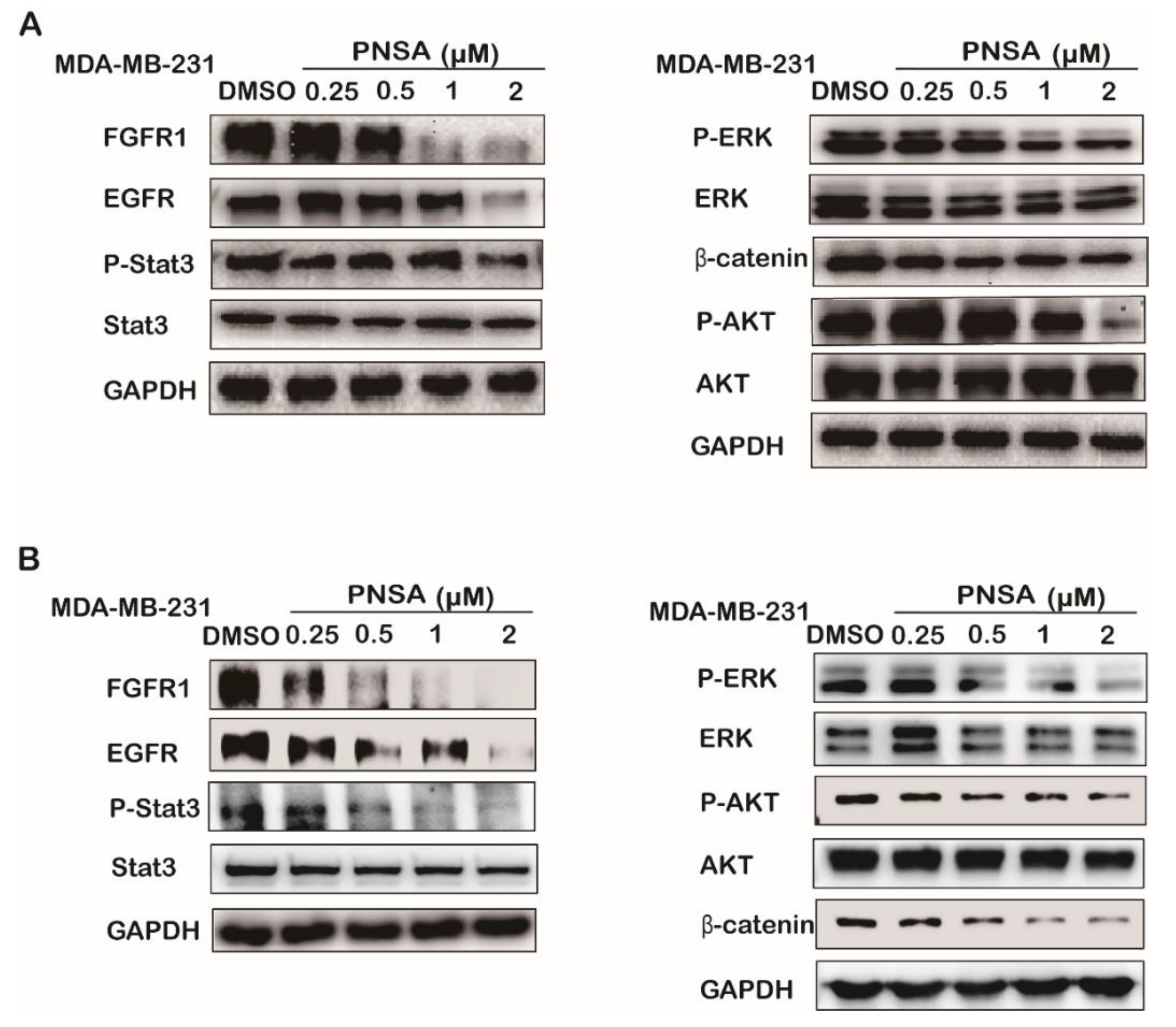
2.3. PNSA Inhibits Expressions and Activations of Signaling Molecules Related to EMT
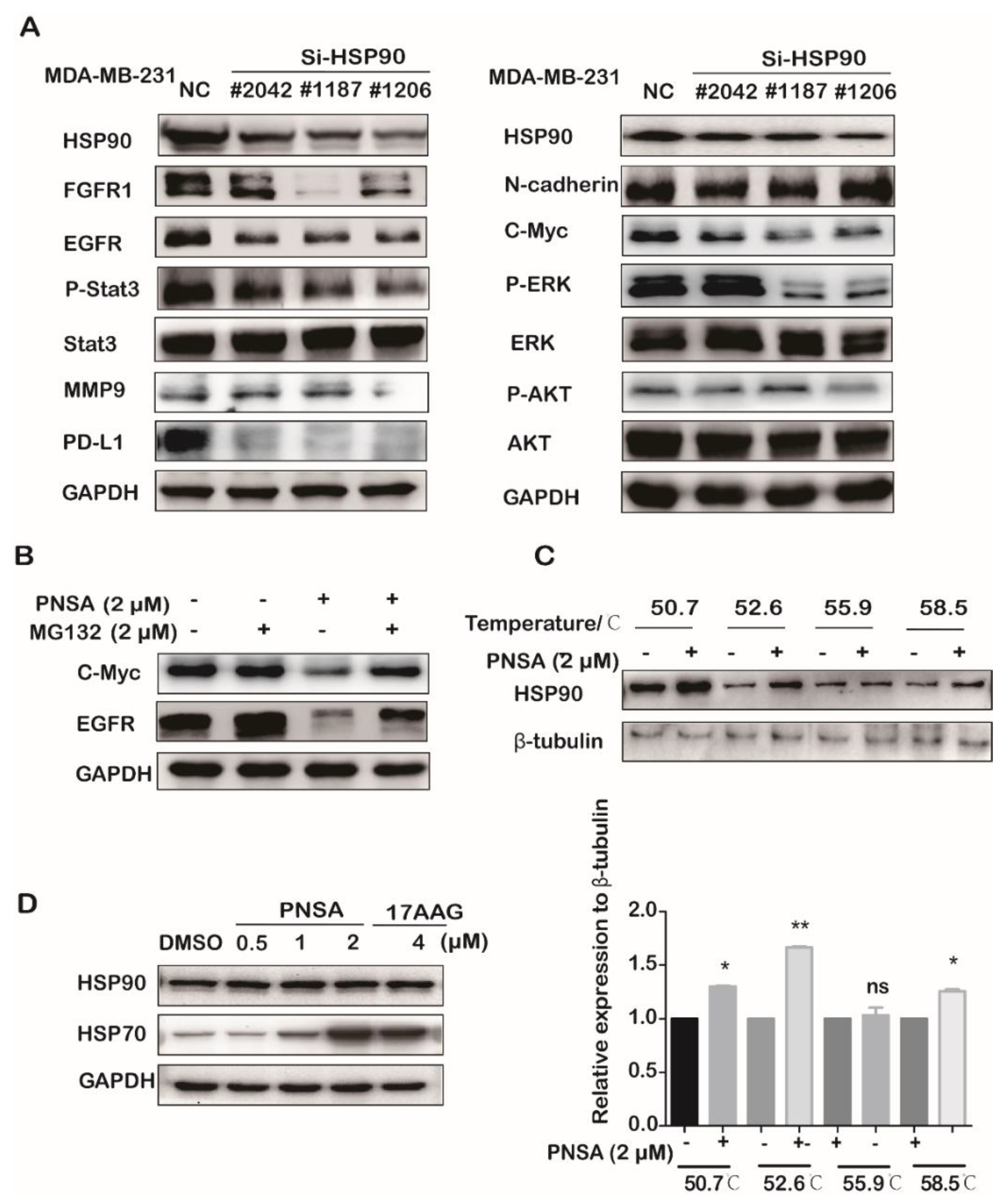
2.4. PNSA Inhibits the Protein Expressions and Activations in the Signaling Pathway by Targeting HSP90
2.5. PNSA Inhibits the Migration, Invasion, and EMT-related Signaling Pathway Proteins in JIMT-1 Cells
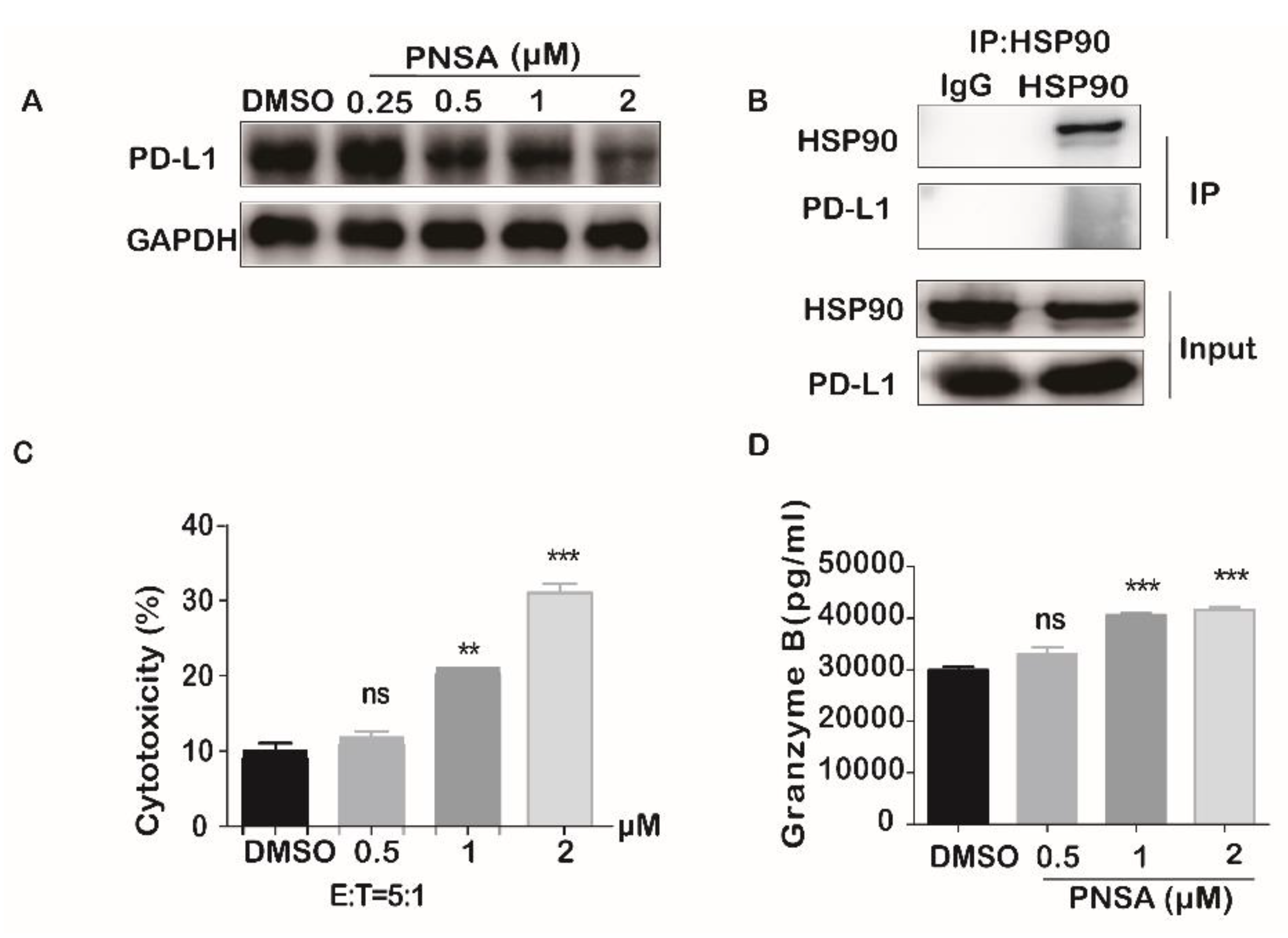
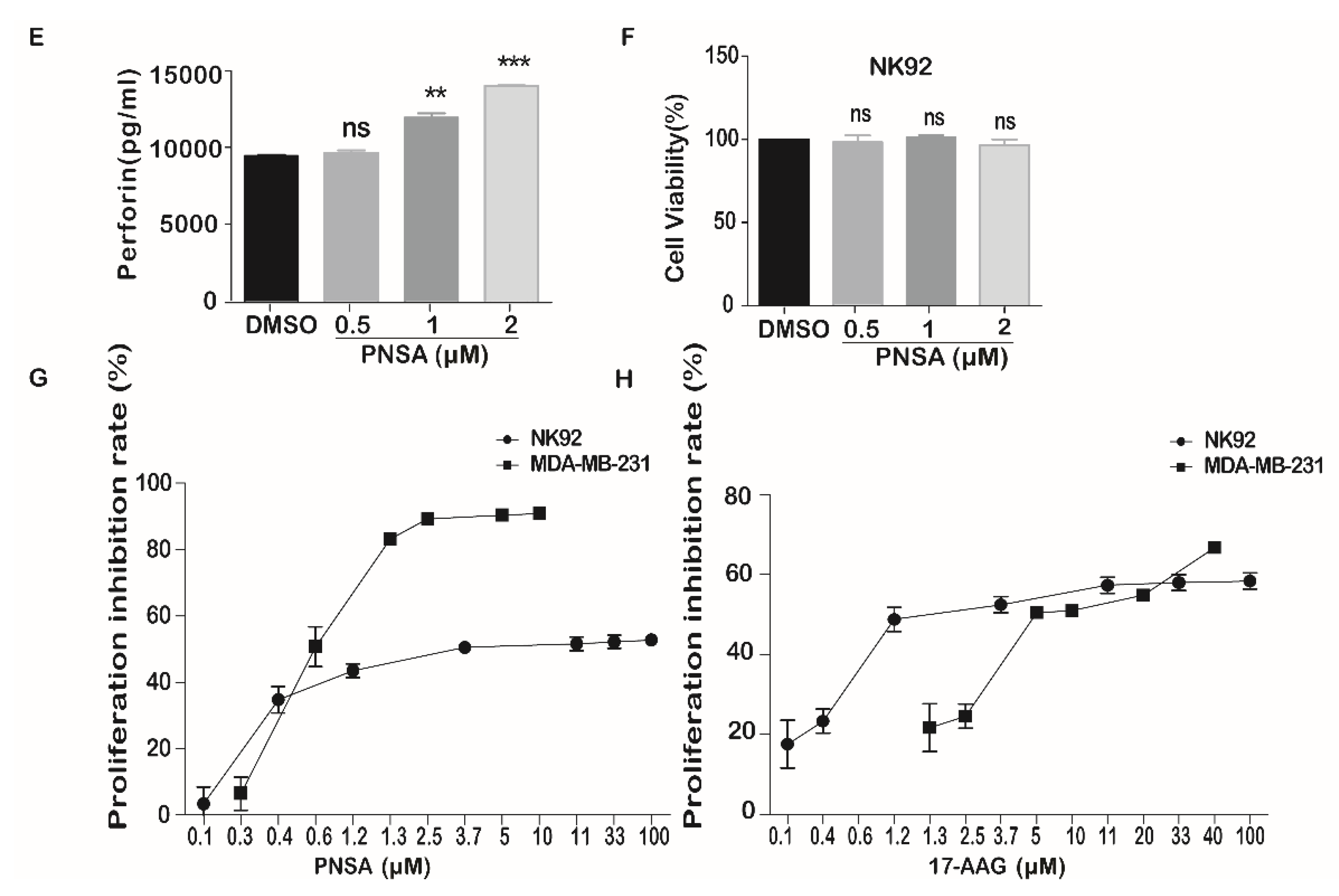
2.6. PNSA Inhibits PD-L1 Protein Expression in MDA-MB-231 and Enhances NK Cells Cytotoxic Activity
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Cell Proliferation Assay
4.4. Migration and Invasion Assay
4.5. Cell Aggregation Assay
4.6. Cell Adhesion Assay
4.7. Gelatin Zymography Assay
4.8. Western Blotting
4.9. Cell Transfection
4.10. Cellular Thermal Shift Assay (CETSA)
4.11. Immunoprecipitation
4.12. Cytotoxicity Assays of NK92 Cell
4.13. Analysis of Perforin and Granzyme by ELISA
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Han, B.; Siegel, E.; Cui, Y.; Giuliano, A.; Cui, X. Breast cancer lung metastasis: Molecular biology and therapeutic implications. Cancer Biol. Ther. 2018, 19, 858–868. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Soni, A.; Ren, Z.; Hameed, O.; Chanda, D.; Morgan, C.J.; Siegal, G.P.; Wei, S. Breast cancer subtypes predispose the site of distant metastases. Am. J. Clin. Pathol. 2015, 143, 471–478. [Google Scholar] [CrossRef]
- Marotti, J.D.; de Abreu, F.B.; Wells, W.A.; Tsongalis, G.J. Triple-Negative Breast Cancer: Next-Generation Sequencing for Target Identification. Am. J. Pathol. 2017, 187, 2133–2138. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.K.; Collier, A.L.; Lee, D.; Hoefer, R.A.; Zheleva, V.; van Reesema, L.L.S.; Tang-Tan, A.M.; Guye, M.L.; Chang, D.Z.; Winston, J.S.; et al. Perspectives on Triple-Negative Breast Cancer: Current Treatment Strategies, Unmet Needs, and Potential Targets for Future Therapies. Cancers (Basel) 2020, 12, 2392. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Zhang, H.; Song, X.; Yang, Q. Metastatic heterogeneity of breast cancer: Molecular mechanism and potential therapeutic targets. Semin. Cancer Biol. 2020, 60, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Liu, Q.; Han, X.; Qin, S.; Zhao, W.; Li, A.; Wu, K. Development and clinical application of anti-HER2 monoclonal and bispecific antibodies for cancer treatment. Exp. Hematol. Oncol. 2017, 6, 31. [Google Scholar] [CrossRef] [Green Version]
- Valabrega, G.; Montemurro, F.; Sarotto, I.; Petrelli, A.; Rubini, P.; Tacchetti, C.; Aglietta, M.; Comoglio, P.M.; Giordano, S. TGFalpha expression impairs Trastuzumab-induced HER2 downregulation. Oncogene 2005, 24, 3002–3010. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Pan, C.; Guo, L.; Wu, M.; Guo, J.; Peng, S.; Wu, Q.; Zuo, Q. A new mechanism of trastuzumab resistance in gastric cancer: MACC1 promotes the Warburg effect via activation of the PI3K/AKT signaling pathway. J. Hematol. Oncol. 2016, 9, 76. [Google Scholar] [CrossRef] [Green Version]
- Jolly, M.K.; Tripathi, S.C.; Somarelli, J.A.; Hanash, S.M.; Levine, H. Epithelial/mesenchymal plasticity: How have quantitative mathematical models helped improve our understanding? Mol. Oncol. 2017, 11, 739–754. [Google Scholar] [CrossRef] [Green Version]
- Jolly, M.K.; Boareto, M.; Huang, B.; Jia, D.; Lu, M.; Ben-Jacob, E.; Onuchic, J.N.; Levine, H. Implications of the Hybrid Epithelial/Mesenchymal Phenotype in Metastasis. Front. Oncol. 2015, 5, 155. [Google Scholar] [CrossRef] [Green Version]
- Loh, C.Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef] [Green Version]
- Onder, T.T.; Gupta, P.B.; Mani, S.A.; Yang, J.; Lander, E.S.; Weinberg, R.A. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008, 68, 3645–3654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santamaria, P.G.; Moreno-Bueno, G.; Portillo, F.; Cano, A. EMT: Present and future in clinical oncology. Mol. Oncol. 2017, 11, 718–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sankhala, K.K.; Mita, M.M.; Mita, A.C.; Takimoto, C.H. Heat shock proteins: A potential anticancer target. Curr. Drug Targets 2011, 12, 2001–2008. [Google Scholar] [CrossRef] [PubMed]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef]
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef]
- Zagouri, F.; Sergentanis, T.N.; Nonni, A.; Papadimitriou, C.A.; Michalopoulos, N.V.; Domeyer, P.; Theodoropoulos, G.; Lazaris, A.; Patsouris, E.; Zogafos, E.; et al. Hsp90 in the continuum of breast ductal carcinogenesis: Evaluation in precursors, preinvasive and ductal carcinoma lesions. BMC Cancer 2010, 10, 353. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.W.; Kim, K.M. Clinical significance of heat shock protein 90alpha expression as a biomarker of prognosis in patients with gastric cancer. Niger. J. Clin. Pract. 2019, 22, 1698–1705. [Google Scholar]
- Dithio-Bridged-Diketone Piperazine Compounds, Preparation Method and Application. Available online: https://worldwide.espacenet.com/patent/search/family/054444416/publication/CN105037396A?q=CN105037396A (accessed on 11 February 2021).
- Dai, J.; Zhu, M.; Qi, X.; Wang, Y.; Li, H.; Tang, S.; Wang, Q.; Chen, A.; Liu, M.; Gu, Q.; et al. Fungal mycotoxin penisuloxazin A, a novel C-terminal Hsp90 inhibitor and characteristics of its analogues on Hsp90 function related to binding sites. Biochem. Pharmacol. 2020, 182, 114218. [Google Scholar] [CrossRef]
- Glinsky, G.V.; Glinsky, V.V. Apoptosis amd metastasis: A superior resistance of metastatic cancer cells to programmed cell death. Cancer Lett. 1996, 101, 43–51. [Google Scholar] [CrossRef]
- Huang, H. Matrix Metalloproteinase-9 (MMP-9) as a Cancer Biomarker and MMP-9 Biosensors: Recent Advances. Sensors (Basel) 2018, 18, 3249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meskyte, E.M.; Keskas, S.; Ciribilli, Y. MYC as a Multifaceted Regulator of Tumor Microenvironment Leading to Metastasis. Int. J. Mol. Sci. 2020, 21, 7710. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, Z.; Lu, N. A new role for the PI3K/Akt signaling pathway in the epithelial-mesenchymal transition. Cell Adhes. Migr. 2015, 9, 317–324. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Chen, J.; Feng, J.; Xu, Y.; Zheng, W.; Wang, J. SOSTDC1 inhibits follicular thyroid cancer cell proliferation, migration, and EMT via suppressing PI3K/Akt and MAPK/Erk signaling pathways. Mol. Cell Biochem. 2017, 435, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Bi, C.L.; Zhang, Y.Q.; Li, B.; Guo, M.; Fu, Y.L. MicroRNA-520a-3p suppresses epithelial-mesenchymal transition, invasion, and migration of papillary thyroid carcinoma cells via the JAK1-mediated JAK/STAT signaling pathway. J. Cell Physiol. 2019, 234, 4054–4067. [Google Scholar] [CrossRef]
- Teeuwssen, M.; Fodde, R. Wnt Signaling in Ovarian Cancer Stemness, EMT, and Therapy Resistance. J. Clin. Med. 2019, 8, 1658. [Google Scholar] [CrossRef] [Green Version]
- Mimnaugh, E.G.; Xu, W.; Vos, M.; Yuan, X.; Isaacs, J.S.; Bisht, K.S.; Gius, D.; Neckers, L. Simultaneous inhibition of hsp 90 and the proteasome promotes protein ubiquitination, causes endoplasmic reticulum-derived cytosolic vacuolization, and enhances antitumor activity. Mol. Cancer Ther. 2004, 3, 551–566. [Google Scholar]
- Martinez Molina, D.; Jafari, R.; Ignatushchenko, M.; Seki, T.; Larsson, E.A.; Dan, C.; Sreekumar, L.; Cao, Y.; Nordlund, P. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 2013, 341, 84–87. [Google Scholar] [CrossRef]
- Sekihara, K.; Harashima, N.; Tongu, M.; Tamaki, Y.; Uchida, N.; Inomata, T.; Harada, M. Pifithrin-mu, an inhibitor of heat-shock protein 70, can increase the antitumor effects of hyperthermia against human prostate cancer cells. PLoS ONE 2013, 8, e78772. [Google Scholar] [CrossRef] [Green Version]
- Banerji, U.; O’Donnell, A.; Scurr, M.; Pacey, S.; Stapleton, S.; Asad, Y.; Simmons, L.; Maloney, A.; Raynaud, F.; Campbell, M.; et al. Phase I pharmacokinetic and pharmacodynamic study of 17-allylamino, 17-demethoxygeldanamycin in patients with advanced malignancies. J. Clin. Oncol. 2005, 23, 4152–4161. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, H.; Zhao, Q.; Xia, Y.; Hu, X.; Guo, J. PD-L1 induces epithelial-to-mesenchymal transition via activating SREBP-1c in renal cell carcinoma. Med. Oncol. 2015, 32, 212. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.Y.; Hu, D.X.; Chen, W.Q.; Chen, R.Q.; Qian, S.R.; Li, C.Y.; Li, Y.J.; Xiong, X.X.; Liu, D.; Pan, F.; et al. PD-L1 confers glioblastoma multiforme malignancy via Ras binding and Ras/Erk/EMT activation. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1754–1769. [Google Scholar] [CrossRef] [PubMed]
- Alsuliman, A.; Colak, D.; Al-Harazi, O.; Fitwi, H.; Tulbah, A.; Al-Tweigeri, T.; Al-Alwan, M.; Ghebeh, H. Bidirectional crosstalk between PD-L1 expression and epithelial to mesenchymal transition: Significance in claudin-low breast cancer cells. Mol. Cancer 2015, 14, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.H.; Kang, H.; Cho, H. Natural killer cells and tumor metastasis. Arch. Pharm. Res. 2017, 40, 1037–1049. [Google Scholar] [CrossRef]
- Tarantino, P.; Morganti, S.; Curigliano, G. Biologic therapy for advanced breast cancer: Recent advances and future directions. Expert Opin. Biol. Ther. 2020, 20, 1009–1024. [Google Scholar] [CrossRef]
- Whitesell, L.; Santagata, S.; Mendillo, M.L.; Lin, N.U.; Proia, D.A.; Lindquist, S. HSP90 empowers evolution of resistance to hormonal therapy in human breast cancer models. Proc. Natl. Acad. Sci. USA 2014, 111, 18297–18302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jhaveri, K.; Wang, R.; Teplinsky, E.; Chandarlapaty, S.; Solit, D.; Cadoo, K.; Speyer, J.; D’Andrea, G.; Adams, S.; Patil, S.; et al. A phase I trial of ganetespib in combination with paclitaxel and trastuzumab in patients with human epidermal growth factor receptor-2 (HER2)-positive metastatic breast cancer. Breast Cancer Res. 2017, 19, 89. [Google Scholar] [CrossRef]
- Zagouri, F.; Sergentanis, T.N.; Chrysikos, D.; Papadimitriou, C.A.; Dimopoulos, M.A.; Psaltopoulou, T. Hsp90 inhibitors in breast cancer: A systematic review. Breast 2013, 22, 569–578. [Google Scholar] [CrossRef]
- Allan, R.K.; Mok, D.; Ward, B.K.; Ratajczak, T. Modulation of chaperone function and cochaperone interaction by novobiocin in the C-terminal domain of Hsp90: Evidence that coumarin antibiotics disrupt Hsp90 dimerization. J. Biol. Chem. 2006, 281, 7161–7171. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, A.; Koldobskiy, M.A.; Sixt, K.M.; Juluri, K.R.; Mustafa, A.K.; Snowman, A.M.; van Rossum, D.B.; Patterson, R.L.; Snyder, S.H. HSP90 regulates cell survival via inositol hexakisphosphate kinase-2. Proc. Natl. Acad. Sci. USA 2008, 105, 1134–1139. [Google Scholar] [CrossRef] [Green Version]
- Horn, L.A.; Fousek, K.; Palena, C. Tumor Plasticity and Resistance to Immunotherapy. Trends Cancer 2020, 6, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Hughes, C.J.; Ford, H.L. Cellular Plasticity in Breast Cancer Progression and Therapy. Front. Mol. Biosci. 2020, 7, 72. [Google Scholar] [CrossRef] [Green Version]
- Chini, M.G.; Malafronte, N.; Vaccaro, M.C.; Gualtieri, M.J.; Vassallo, A.; Vasaturo, M.; Castellano, S.; Milite, C.; Leone, A.; Bifulco, G.; et al. Identification of Limonol Derivatives as Heat Shock Protein 90 (Hsp90) Inhibitors through a Multidisciplinary Approach. Chemistry 2016, 22, 13236–13250. [Google Scholar] [CrossRef]
- Takebe, N.; Warren, R.Q.; Ivy, S.P. Breast cancer growth and metastasis: Interplay between cancer stem cells, embryonic signaling pathways and epithelial-to-mesenchymal transition. Breast Cancer Res. 2011, 13, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Weinberg, R.A. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef] [Green Version]
- Sirkisoon, S.R.; Carpenter, R.L.; Rimkus, T.; Miller, L.; Metheny-Barlow, L.; Lo, H.W. EGFR and HER2 signaling in breast cancer brain metastasis. Front. Biosci. (Elite Ed.) 2016, 8, 245–263. [Google Scholar] [PubMed]
- Wikman, H.; Lamszus, K.; Detels, N.; Uslar, L.; Wrage, M.; Benner, C.; Hohensee, I.; Ylstra, B.; Eylmann, K.; Zapatka, M.; et al. Relevance of PTEN loss in brain metastasis formation in breast cancer patients. Breast Cancer Res. 2012, 14, R49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, N.; Tian, C.; Wu, H.; Yang, X.; Liu, L.; Li, J.; Xiao, H.; Gao, J.; Lu, J.; Hu, X.; et al. FGFR aberrations increase the risk of brain metastases and predict poor prognosis in metastatic breast cancer patients. Ther. Adv. Med. Oncol. 2020, 12, 1758835920915305. [Google Scholar] [CrossRef]
- Erber, R.; Rubner, M.; Davenport, S.; Hauke, S.; Beckmann, M.W.; Hartmann, A.; Haberle, L.; Gass, P.; Press, M.F.; Fasching, P.A. Impact of fibroblast growth factor receptor 1 (FGFR1) amplification on the prognosis of breast cancer patients. Breast Cancer Res. Treat. 2020, 184, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Dey, N.; Barwick, B.G.; Moreno, C.S.; Ordanic-Kodani, M.; Chen, Z.; Oprea-Ilies, G.; Tang, W.; Catzavelos, C.; Kerstann, K.F.; Sledge, G.W., Jr.; et al. Wnt signaling in triple negative breast cancer is associated with metastasis. BMC Cancer 2013, 13, 537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Sanchez-Sweatman, O.; Huang, X.; Wiltrout, R.; Khokha, R.; Zhao, Q.; Gorelik, E. Anoikis and metastatic potential of cloudman S91 melanoma cells. Cancer Res. 2001, 61, 1707–1716. [Google Scholar] [PubMed]
- Jin, Y.; Huo, B.; Fu, X.; Hao, T.; Zhang, Y.; Guo, Y.; Hu, X. LSD1 collaborates with EZH2 to regulate expression of interferon-stimulated genes. Biomed. Pharmacother. 2017, 88, 728–737. [Google Scholar] [CrossRef]
- Qu, Y.; Bi, J.Z. Killing effect of Robo1 targeted Chimeric Antigen Receptor modified NK92 cells against glioma and neuroblastoma cells. Zhonghua Yi Xue Za Zhi 2018, 98, 860–866. [Google Scholar]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, A.; Qi, X.; Du, F.; Zhang, G.; Li, D.; Li, J. PNSA, a Novel C-Terminal Inhibitor of HSP90, Reverses Epithelial–Mesenchymal Transition and Suppresses Metastasis of Breast Cancer Cells In Vitro. Mar. Drugs 2021, 19, 117. https://doi.org/10.3390/md19020117
Zhang A, Qi X, Du F, Zhang G, Li D, Li J. PNSA, a Novel C-Terminal Inhibitor of HSP90, Reverses Epithelial–Mesenchymal Transition and Suppresses Metastasis of Breast Cancer Cells In Vitro. Marine Drugs. 2021; 19(2):117. https://doi.org/10.3390/md19020117
Chicago/Turabian StyleZhang, Aotong, Xin Qi, Fu Du, Guojian Zhang, Dehai Li, and Jing Li. 2021. "PNSA, a Novel C-Terminal Inhibitor of HSP90, Reverses Epithelial–Mesenchymal Transition and Suppresses Metastasis of Breast Cancer Cells In Vitro" Marine Drugs 19, no. 2: 117. https://doi.org/10.3390/md19020117
APA StyleZhang, A., Qi, X., Du, F., Zhang, G., Li, D., & Li, J. (2021). PNSA, a Novel C-Terminal Inhibitor of HSP90, Reverses Epithelial–Mesenchymal Transition and Suppresses Metastasis of Breast Cancer Cells In Vitro. Marine Drugs, 19(2), 117. https://doi.org/10.3390/md19020117