
α-Conotoxins and α-Cobratoxin Promote, while Lipoxygenase and Cyclooxygenase Inhibitors Suppress the Proliferation of Glioma C6 Cells

 ,
,  , , and
, , and 
Abstract
:
1. Introduction
2. Results
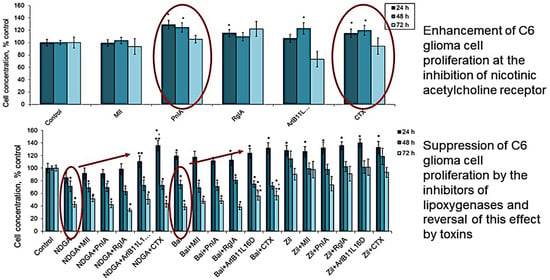
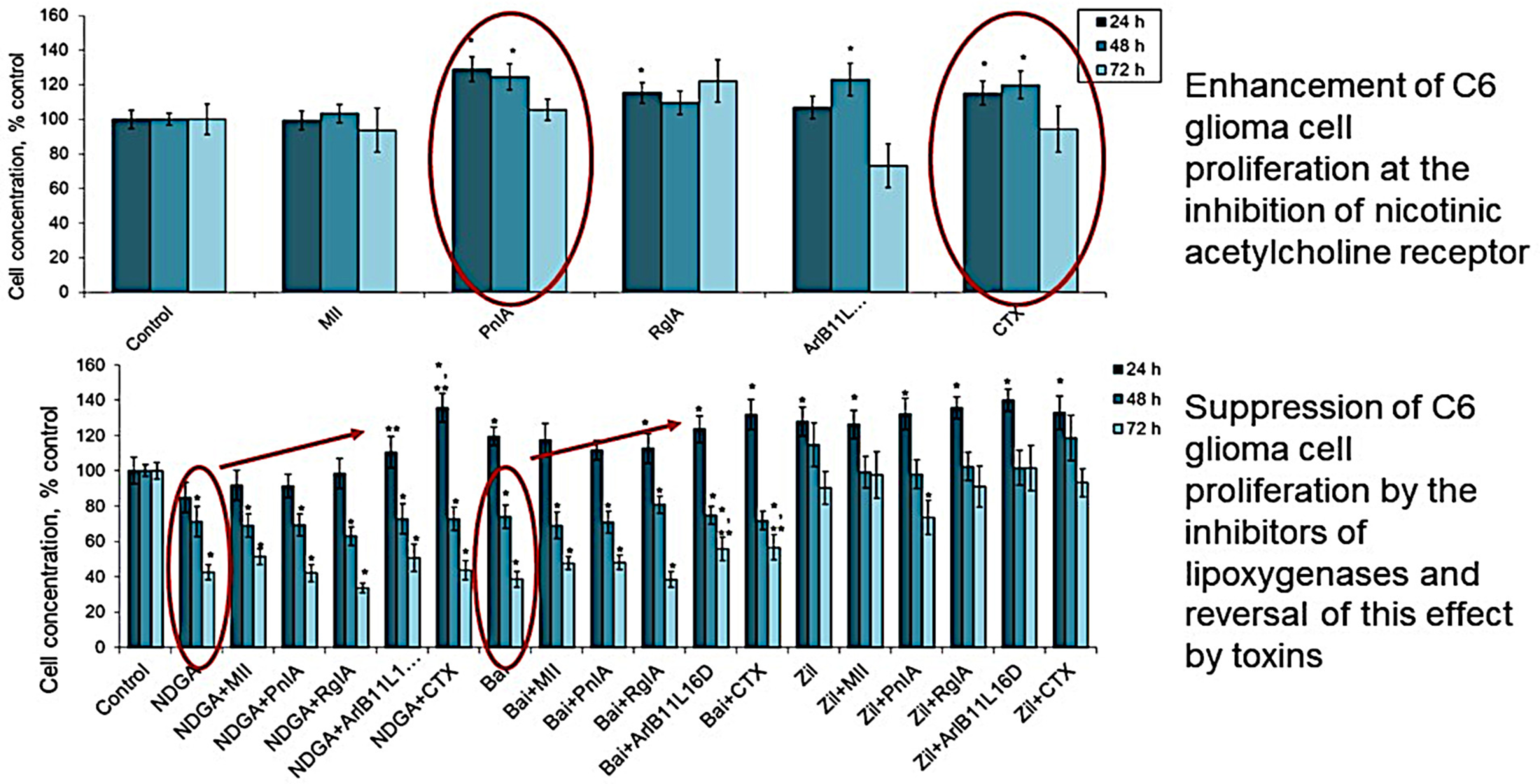
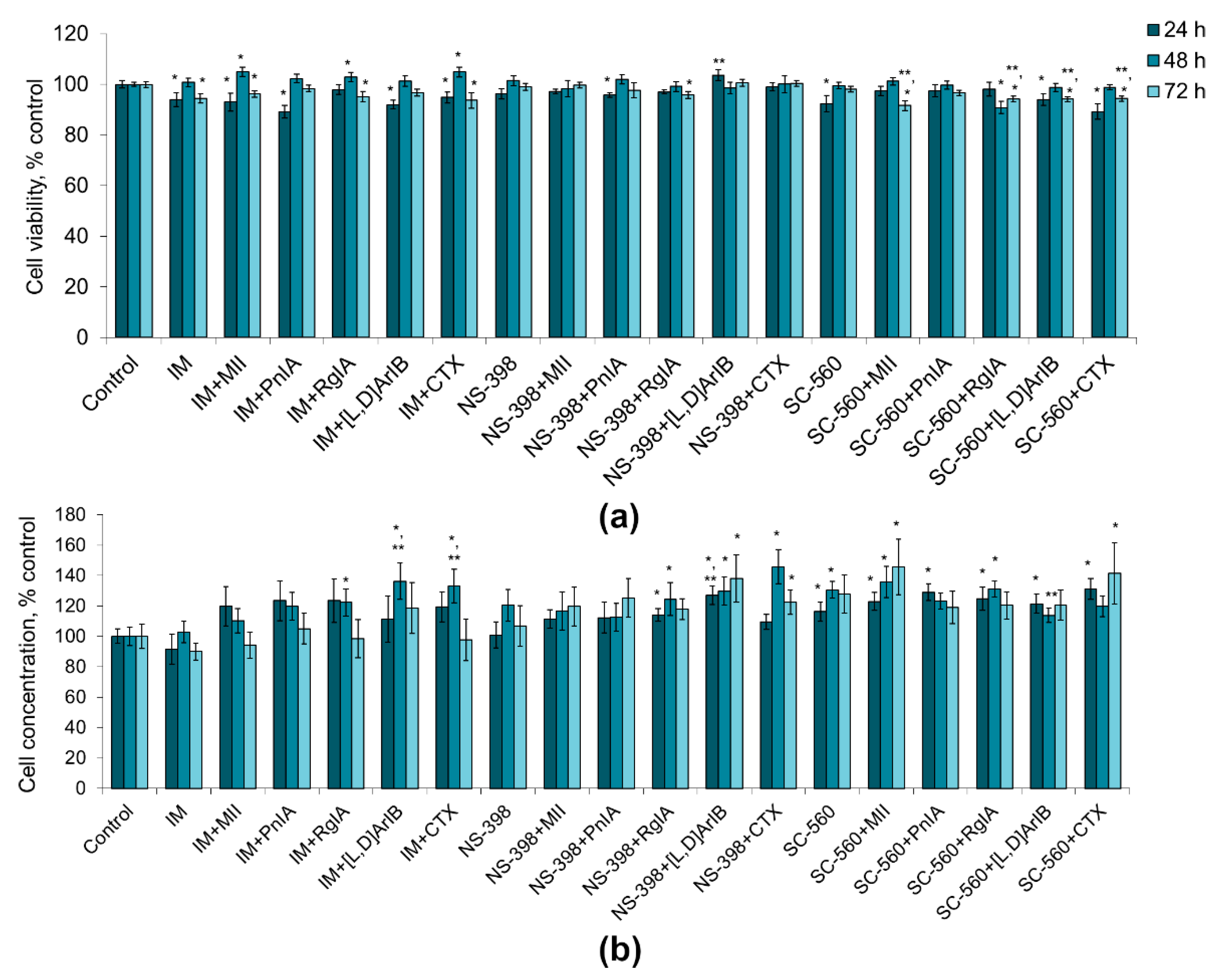
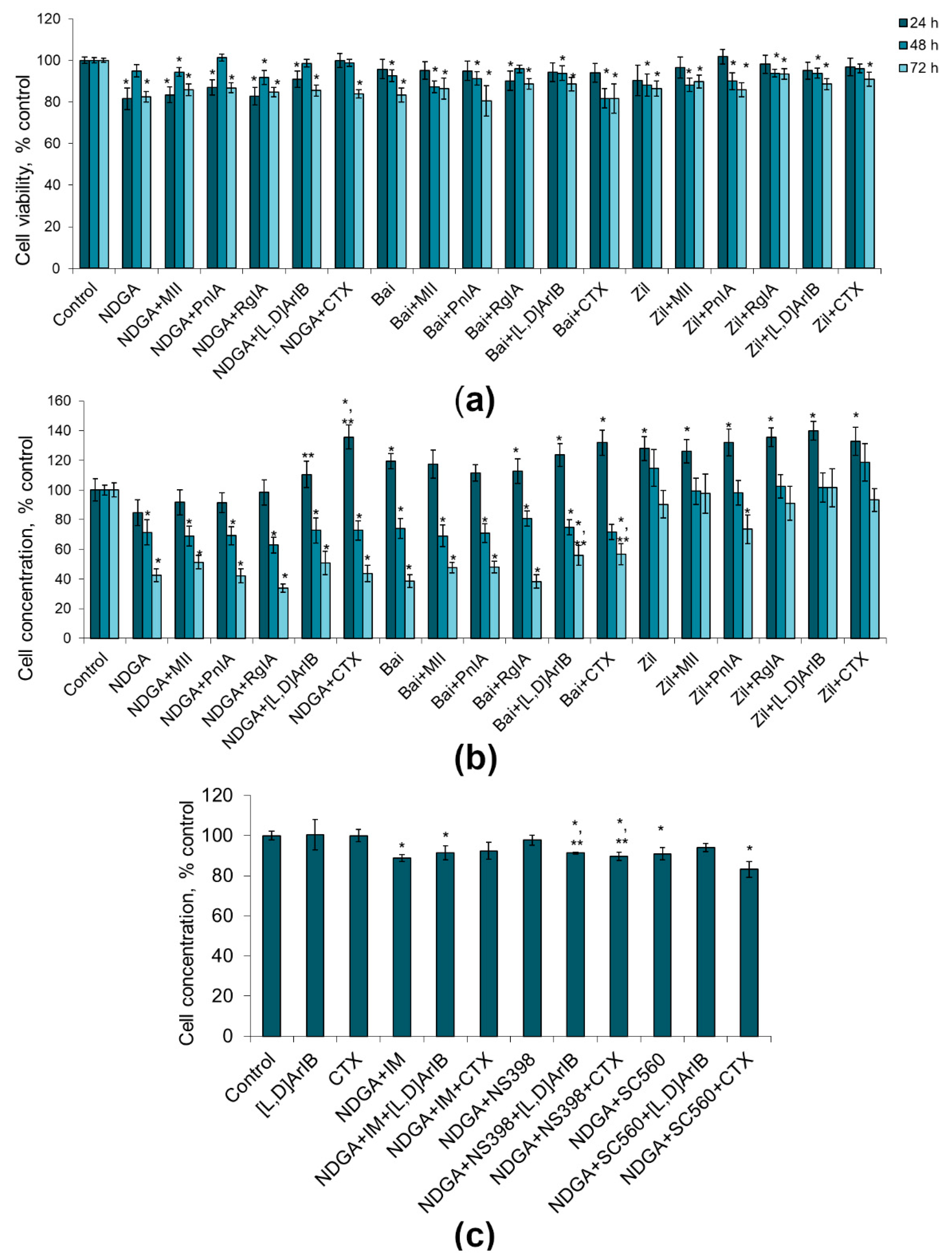
2.1. Effect of nAChR Blockade on the Viability and Proliferation of C6 Glioma Cells
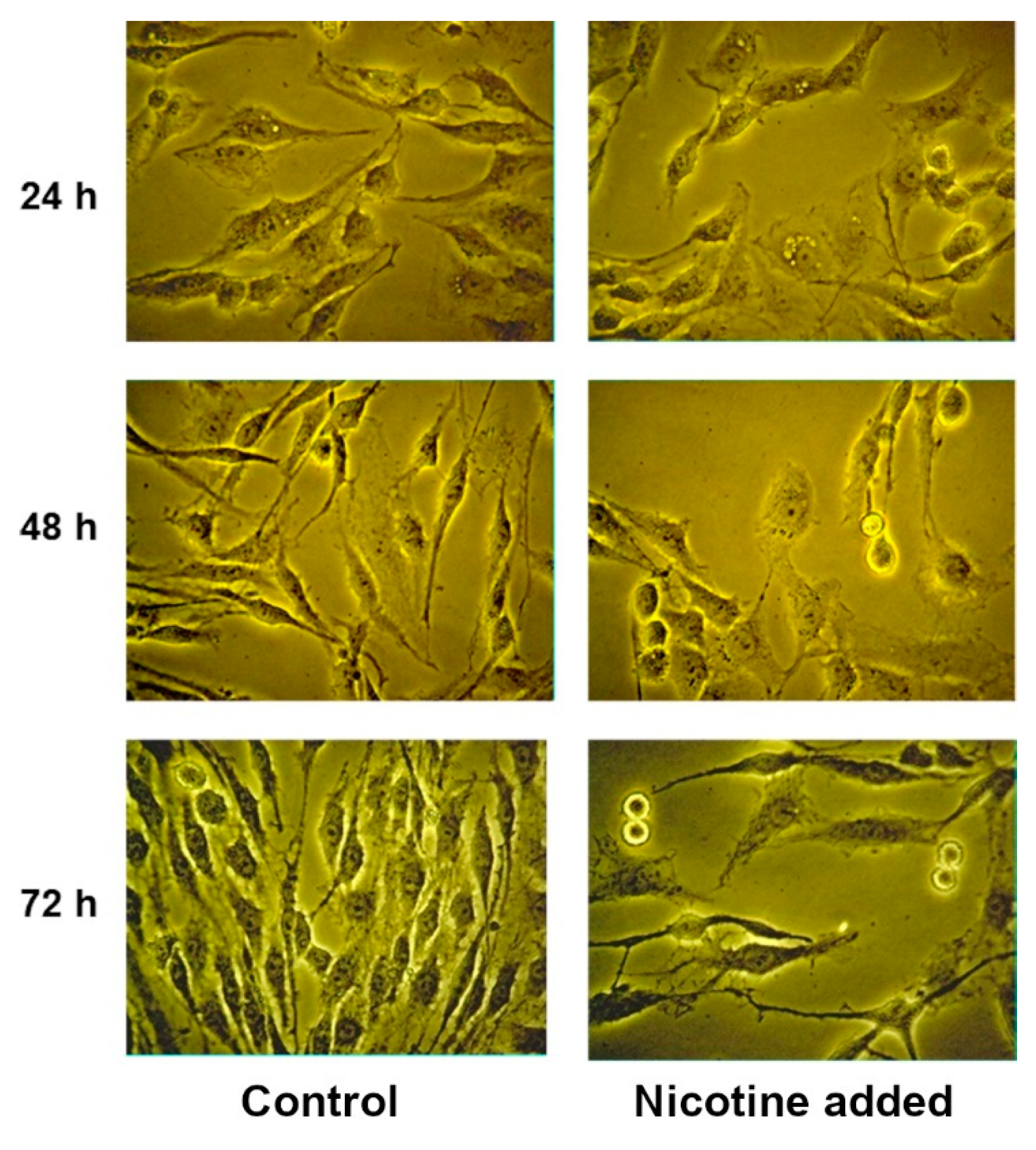
2.2. Effect of COX Inhibitors and of Their Combinations with nAChR Blockers on the Viability and Proliferation of C6 Glioma Cells
2.3. Effect of LOX Inhibitors and of Their Combinations with nAChR Blockers on the Viability and Proliferation of C6 Glioma Cells
2.4. Detection of nAChR Subunits in C6 Glioma Cells by Quantitative Real-Time PCR
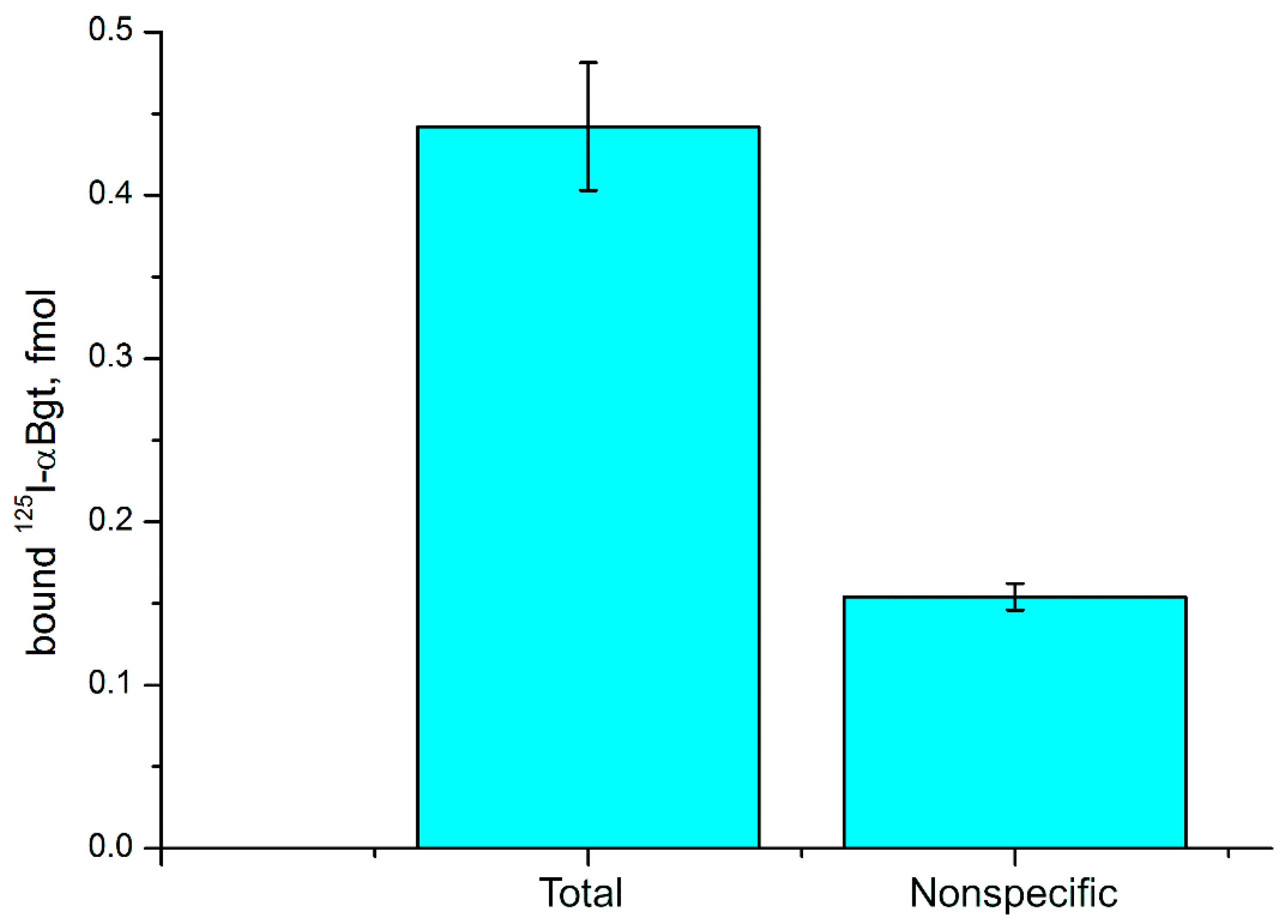
2.5. Detection of nAChR in C6 Glioma Cells by Radioligand Analysis
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Assay of the Viability and Proliferation of C6 Glioma Cells
4.3. MTT Assay
4.4. Light Microscopy
4.5. Quantitative Real-Time PCR
4.6. Toxin Binding Assay
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Friedman, J.R.; Richbart, S.D.; Merritt, J.C.; Brown, K.C.; Nolan, N.A.; Akers, A.T.; Lau, J.K.; Robateau, Z.R.; Miles, S.L.; Dasgupta, P. Acetylcholine signaling system in progression of lung cancers. Pharmacol. Ther. 2019, 194, 222–254. [Google Scholar] [CrossRef]
- Zhang, Y.; Jia, Y.; Li, P.; Li, H.; Xiao, D.; Wang, Y.; Ma, X. Reciprocal activation of α5-nAChR and STAT3 in nicotine-induced human lung cancer cell proliferation. J. Genet. Genom. 2017, 44, 355–362. [Google Scholar] [CrossRef]
- Gankhuyag, N.; Lee, K.-H.; Cho, J.-Y. The Role of Nitrosamine (NNK) in Breast Cancer Carcinogenesis. J. Mammary Gland. Biol. Neoplasia 2017, 22, 159–170. [Google Scholar] [CrossRef]
- Shin, V.Y.; Jin, H.C.; Ng, E.K.; Yu, J.; Leung, W.K.; Cho, C.H.; Sung, J.J. Nicotine and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone induce cyclooxygenase-2 activity in human gastric cancer cells: Involvement of nicotinic acetylcholine receptor (nAChR) and beta-adrenergic receptor signaling pathways. Toxicol. Appl. Pharmacol. 2008, 233, 254–261. [Google Scholar] [CrossRef]
- Shin, V.Y.; Wu, W.K.; Ye, Y.-N.; So, W.H.; Koo, M.W.; Liu, E.S.; Luo, J.-C.; Cho, C.-H. Nicotine promotes gastric tumor growth and neovascularization by activating extracellular signal-regulated kinase and cyclooxygenase-2. Carcinogenesis 2004, 25, 2487–2495. [Google Scholar] [CrossRef]
- Wang, W.; Chin-Sheng, H.; Kuo, L.-J.; Wei, P.-L.; Lien, Y.-C.; Lin, F.-Y.; Liu, H.-H.; Ho, Y.-S.; Wu, C.-H.; Chang, Y.-J. NNK Enhances Cell Migration through α7-nicotinic Acetylcholine Receptor Accompanied by Increased of Fibronectin Expression in Gastric Cancer. Ann. Surg. Oncol. 2012, 19, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Bouzat, C.; Sine, S.M. Nicotinic acetylcholine receptors at the single-channel level. Br. J. Pharmacol. 2018, 175, 1789–1804. [Google Scholar] [CrossRef]
- Zoli, M.; Pucci, S.; Vilella, A.; Gotti, C. Neuronal and Extraneuronal Nicotinic Acetylcholine Receptors. Curr. Neuropharmacol. 2018, 16, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Cetin, H.; Beeson, D.; Vincent, A.; Webster, R. The Structure, Function, and Physiology of the Fetal and Adult Acetylcholine Receptor in Muscle. Front. Mol. Neurosci. 2020, 13, 170. [Google Scholar] [CrossRef] [PubMed]
- Papke, R.L.; Lindstrom, J.M. Nicotinic acetylcholine receptors: Conventional and unconventional ligands and signaling. Neuropharmacology 2020, 168, 108021. [Google Scholar] [CrossRef]
- Arias, H.R.; Richards, V.E.; Ng, D.; Ghafoori, M.E.; Le, V.; Mousa, S.A. Role of non-neuronal nicotinic acetylcholine receptors in angiogenesis. Int. J. Biochem. Cell Biol. 2009, 41, 1441–1451. [Google Scholar] [CrossRef]
- Wang, S.; Hu, Y. α7 nicotinic acetylcholine receptors in lung cancer. Oncol. Lett. 2018, 16, 1375–1382. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.D.; Liao, Y.C.; Ho, Y.S.; Chen, L.C.; Chang, H.W.; Cheng, T.C.; Liu, D.; Lee, W.R.; Shen, S.C.; Wu, C.H.; et al. The α9 nicotinic acetylcholine receptor mediates nicotine-induced PD-L1 expression and regulates melanoma cell proliferation and migration. Cancers 2019, 11, 1991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.Y.; Liu, Y.; Ni, X.Y.; Bai, Z.H.; Chen, Q.Y.; Zhang, Y.; Gao, F.G. Nicotine promotes cell proliferation and induces resistance to cisplatin by α7 nicotinic acetylcholine receptor-mediated activation in Raw264.7 and El4 cells. Oncol. Rep. 2013, 31, 1480–1488. [Google Scholar] [CrossRef] [PubMed]
- Fararjeh, A.S.; Tu, S.H.; Chen, L.C.; Cheng, T.C.; Liu, Y.R.; Chang, H.L.; Chang, H.W.; Huang, C.C.; Wang, H.R.; Hwang-Verslues, W.W.; et al. Long-term exposure to extremely low-dose of nicotine and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) induce non-malignant breast epithelial cell transformation through activation of the a9-nicotinic acetylcholine receptor-mediated signaling pathway. Environ. Toxicol. 2019, 34, 73–82. [Google Scholar]
- Jia, Y.; Sun, H.; Wu, H.; Zhang, H.; Zhang, X.; Xiao, D.; Ma, X.; Wang, Y. Nicotine inhibits cisplatin-induced apoptosis via regu-lating α5-nAChR/AKT signaling in human gastric cancer cells. PLoS ONE 2016, 11, e0149120. [Google Scholar] [CrossRef]
- Wang, C.; Niu, W.; Chen, H.; Shi, N.; He, D.; Zhang, M.; Ge, L.; Tian, Z.; Qi, M.; Chen, T.; et al. Nicotine suppresses apoptosis by regulating α7nAChR/Prx1 axis in oral precancerous lesions. Oncotarget 2017, 8, 75065–75075. [Google Scholar] [CrossRef]
- Terpinskaya, T.I.; Osipov, A.V.; Kuznetsova, T.E.; Ryzhkovskaya, E.L.; Ulaschik, V.S.; Ivanov, I.A.; Tsetlin, V.I.; Utkin, Y.N. α-conotoxins revealed different roles of nicotinic cholinergic receptor subtypes in oncogenesis of Ehrlich tumor and in the associated inflammation. Dokl. Biochem. Biophys. 2015, 463, 216–219. [Google Scholar] [CrossRef]
- Terpinskaya, T.I.; Osipov, A.V.; Balashevich, T.V.; Yanchanka, T.L.; Tamashionik, E.A.; Tsetlin, V.I.; Utkin, Y.N. Blockers of Nicotinic Acetylcholine Receptors Delay Tumor Growth and Increase Antitumor Activity of Mouse Splenocytes. Dokl. Biochem. Biophys. 2020, 491, 89–92. [Google Scholar] [CrossRef]
- Osipov, A.V.; Terpinskaya, T.I.; Yanchanka, T.; Balashevich, T.; Zhmak, M.N.; Tsetlin, V.I.; Utkin, Y.N. α-Conotoxins Enhance both the In Vivo Suppression of Ehrlich carcinoma Growth and In Vitro Reduction in Cell Viability Elicited by Cyclooxygenase and Lipoxygenase Inhibitors. Mar. Drugs 2020, 18, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grozio, A.; Paleari, L.; Catassi, A.; Servent, D.; Cilli, M.; Piccardi, F.; Paganuzzi, M.; Cesario, A.; Granone, P.; Mourier, G.; et al. Natural agents targeting the alpha7-nicotinic-receptor in NSCLC: A promising prospective in anti-cancer drug development. Int. J. Cancer 2008, 122, 1911–1915. [Google Scholar] [CrossRef]
- Berne, S.; Čemažar, M.; Frangež, R.; Juntes, P.; Kranjc, S.; Grandič, M.; Savarin, M.; Turk, T. APS8 Delays Tumor Growth in Mice by Inducing Apoptosis of Lung Adenocarcinoma Cells Expressing High Number of α7 Nicotinic Receptors. Mar. Drugs 2018, 16, 367. [Google Scholar] [CrossRef] [Green Version]
- Lan, X.; Lederman, R.; Eng, J.M.; Shoshtari, S.S.; Saleem, M.A.; Malhotra, A.; Singhal, P.C. Nicotine induces podocyte apopto-sis through increasing oxidative stress. PLoS ONE 2016, 11, e0167071. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.B.; Kshirasagar, N.; Patibandla, S.; Puchchakayala, G.; Koka, S.; Boini, K.M. Nicotine instigates podocyte injury via NLRP3 inflammasomes activation. Aging 2019, 11, 12810–12821. [Google Scholar] [CrossRef] [PubMed]
- Marinucci, L.; Balloni, S.; Fettucciari, K.; Bodo, M.; Talesa, V.N.; Antognelli, C. Nicotine induces apoptosis in human osteo-blasts via a novel mechanism driven by H2O2 and entailing glyoxalase 1-dependent MG-H1 accumulation leading to TG2-mediated NF-kB desensitization: Implication for smokers-related osteoporosis. Free Radic. Biol. Med. 2018, 117, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.S.; Choi, J.S.; Joo, S.Y.; Bae, E.H.; Ma, S.K.; Lee, J.; Kim, S.W. Nicotine-Induced Apoptosis in Human Renal Proximal Tubular Epithelial Cells. PLoS ONE 2016, 11, e0152591. [Google Scholar] [CrossRef] [Green Version]
- Hajiasgharzadeh, K.; Somi, M.H.; Mansoori, B.; Doustvandi, M.A.; Vahidian, F.; Alizadeh, M.; Mokhtarzadeh, A.; Shanehbandi, D.; Baradaran, B. Alpha7 nicotinic acetylcholine receptor mediates nicotine-induced apoptosis and cell cycle arrest of hepato-cellular carcinoma HepG2 cells. Adv. Pharm. Bull. 2020, 10, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.L.; Urade, Y.; Jakobsson, P.-J. Enzymes of the Cyclooxygenase Pathways of Prostanoid Biosynthesis. Chem. Rev. 2011, 111, 5821–5865. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, H.; Banthiya, S.; Van Leyen, K. Mammalian lipoxygenases and their biological relevance. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2015, 1851, 308–330. [Google Scholar] [CrossRef] [Green Version]
- Mazaleuskaya, L.L.; Ricciotti, E. Druggable Prostanoid Pathway. Adv. Exp. Med. Biol. 2020, 1274, 29–54. [Google Scholar] [CrossRef]
- Ocallaghan, G.; Houston, A. Prostaglandin E2 and the EP receptors in malignancy: Possible therapeutic targets? Br. J. Pharmacol. 2015, 172, 5239–5250. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Qu, L.; Yan, S. Cyclooxygenase-2 promotes tumor growth and suppresses tumor immunity. Cancer Cell Int. 2015, 15, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kam, P.C.A.; So, A. COX-3: Uncertainties and controversies. Curr. Anaesth. Crit. Care 2009, 20, 50–53. [Google Scholar] [CrossRef]
- Oksuz, E.; Atalar, F.; Tanırverdi, G.; Bilir, A.; Shahzadi, A.; Yazici, Z. Therapeutic potential of cyclooxygenase-3 inhibitors in the management of glioblastoma. J. Neuro-Oncol. 2016, 126, 271–278. [Google Scholar] [CrossRef]
- Gautam, S.; Roy, S.; Ansari, M.N.; Saeedan, A.S.; Saraf, S.A.; Kaithwas, G. DuCLOX-2/5 inhibition: A promising target for can-cer chemoprevention. Breast Cancer 2017, 24, 180–190. [Google Scholar] [CrossRef]
- Powell, W.S.; Rokach, J. Biosynthesis, biological effects, and receptors of hydroxyeicosatetraenoic acids (HETEs) and oxoe-icosatetraenoic acids (oxo-ETEs) derived from arachidonic acid. Biochim. Biophys. Acta 2015, 1851, 340–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuncer, S.; Banerjee, S. Eicosanoid pathway in colorectal cancer: Recent updates. World J. Gastroenterol. 2015, 21, 11748–11766. [Google Scholar] [CrossRef]
- Klampfl, T.; Bogner, E.; Bednar, W.; Mager, L.; Massudom, D.; Kalny, I.; Heinzle, C.; Berger, W.; Stättner, S.; Karner, J.; et al. Up-regulation of 12(S)-lipoxygenase induces a migratory phenotype in colo-rectal cancer cells. Exp. Cell Res. 2012, 318, 768–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moris, D.; Kontos, M.; Spartalis, E.; Fentiman, I.S. The role of NSAIDs in breast cancer prevention and relapse: Current evi-dence and future perspectives. Breast Care 2016, 11, 339–344. [Google Scholar] [CrossRef] [Green Version]
- Langley, R.E.; Rothwell, P.M. Aspirin in gastrointestinal oncology: New data on an old friend. Curr. Opin. Oncol. 2014, 26, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Sahin, I.H.; Hassan, M.M.; Garrett, C.R. Impact of non-steroidal anti-inflammatory drugs on gastrointestinal cancers: Cur-rent state-of-the science. Cancer Lett. 2014, 345, 249–257. [Google Scholar] [CrossRef]
- Umar, A.; Steele, V.E.; Menter, D.G.; Hawk, E.T. Mechanisms of nonsteroidal anti-inflammatory drugs in cancer prevention. Semin. Oncol. 2016, 43, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Bie, B.; Sun, J.; Guo, Y.; Li, J.; Jiang, W.; Yang, J.; Huang, C.; Li, Z. Baicalein: A review of its anti-cancer effects and mechanisms in Hepatocellular Carcinoma. Biomed. Pharmacother. 2017, 93, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Singh, A.K.; Kumar, M.; Shekhar, S.; Rai, N.; Kaur, P.; Parshad, R.; Dey, S. Serum 5-LOX: A progressive protein marker for breast cancer and new approach for therapeutic target. Carcinogenesis 2016, 37, 912–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bush, N.A.O.; Chang, S.M.; Berger, M.S. Current and future strategies for treatment of glioma. Neurosurg. Rev. 2017, 40, 1–14. [Google Scholar] [CrossRef]
- Giakoumettis, D.; Kritis, A.; Foroglou, N. C6 cell line: The gold standard in glioma research. Hippokratia 2018, 22, 105–112. [Google Scholar]
- Thompson, E.G.; Sontheimer, H. Acetylcholine Receptor Activation as a Modulator of Glioblastoma Invasion. Cells 2019, 8, 1203. [Google Scholar] [CrossRef] [Green Version]
- Kitabatake, T.; Moaddel, R.; Cole, R.; Gandhari, M.; Frazier, C.; Hartenstein, J.; Rosenberg, A.; Bernier, M.; Wainer, I.W. Characterization of a multiple ligand-gated ion channel cellular membrane affinity chromatography column and identification of endogenously expressed receptors in astrocytoma cell lines. Anal. Chem. 2008, 80, 8673–8680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiu, J.; Nordberg, A.; Zhang, J.T.; Guan, Z.Z. Expression of nicotinic receptors on primary cultures of rat astrocytes and up-regulation of the alpha7, alpha4 and beta2 subunits in response to nanomolar concentrations of the beta-amyloid peptide (1-42). Neurochem. Int. 2005, 47, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.F.; Tang, X.C. Huperzine A protects C6 rat glioma cells against oxygen-glucose deprivation-induced injury. FEBS Lett. 2007, 581, 596–602. [Google Scholar] [CrossRef] [Green Version]
- Niranjan, R.; Nath, C.; Shukla, R. Melatonin attenuated mediators of neuroinflammation and alpha-7 nicotinic acetylcholine receptor mRNA expression in lipopolysaccharide (LPS) stimulated rat astrocytoma cells, C6. Free. Radic. Res. 2012, 46, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, S.; Nicke, A.; Tsetlin, V.I. Nicotinic acetylcholine receptor inhibitors derived from snake and snail venoms. Neuropharmacology 2017, 127, 196–223. [Google Scholar] [CrossRef] [PubMed]
- Osipov, A.V.; Kasheverov, I.E.; Makarova, Y.V.; Starkov, V.G.; Vorontsova, O.V.; Ziganshin, R.K.; Andreeva, T.V.; Serebry-akova, M.V.; Benoit, A.; Hogg, R.C.; et al. Naturally occurring disulfide-bound dimers of three-fingered toxins: A paradigm for biological activity diversification. J. Biol. Chem. 2008, 283, 14571–14580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hone, A.J.; Whiteaker, P.; Christensen, S.; Xiao, Y.; Meyer, E.L.; McIntosh, J.M. A novel fluorescent α-conotoxin for the study of α7 nicotinic acetylcholine receptors. J. Neurochem. 2009, 111, 80–89. [Google Scholar] [CrossRef] [Green Version]
- Whiteaker, P.; Christensen, S.; Yoshikami, D.; Dowell, C.; Watkins, M.; Gulyas, J.; Rivier, J.; Olivera, B.M.; McIntosh, J.M. Discovery, Synthesis, and Structure Activity of a Highly Selective α7 Nicotinic Acetylcholine Receptor Antagonist. Biochemisty 2007, 46, 6628–6638. [Google Scholar] [CrossRef]
- Cartier, G.E.; Yoshikami, D.; Gray, W.R.; Luo, S.; Olivera, B.M.; McIntosh, J.M. A new alpha-conotoxin which targets al-pha3beta2 nicotinic acetylcholine receptors. J. Biol. Chem. 1996, 271, 7522–7528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McIntosh, J.M.; Azam, L.; Staheli, S.; Dowell, C.; Lindstrom, J.M.; Kuryatov, A.; Garrett, J.E.; Marks, M.J.; Whiteaker, P. Analogs of α-Conotoxin MII Are Selective for α6-Containing Nicotinic Acetylcholine Receptors. Mol. Pharmacol. 2004, 65, 944–952. [Google Scholar] [CrossRef]
- Luo, S.; Nguyen, T.A.; Cartier, G.E.; Olivera, B.M.; Yoshikami, D.; McIntosh, J.M. Single-Residue Alteration in α-Conotoxin PnIA Switches Its nAChR Subtype Selectivity. Biochemisty 1999, 38, 14542–14548. [Google Scholar] [CrossRef]
- Hone, A.J.; Meyer, E.L.; McIntyre, M.; McIntosh, J.M. Nicotinic acetylcholine receptors in dorsal root ganglion neurons include the α6β4* subtype. FASEB J. 2012, 26, 917–926. [Google Scholar] [CrossRef] [Green Version]
- Ellison, M.; Haberlandt, C.; Gomez-Casati, M.E.; Watkins, M.; Elgoyhen, A.B.; McIntosh, J.M.; Olivera, B.M. α-RgIA: A Novel Conotoxin That Specifically and Potently Blocks the α9α10 nAChR. Biochemisty 2006, 45, 1511–1517. [Google Scholar] [CrossRef]
- Giribaldi, J.; Dutertre, S. α-Conotoxins to explore the molecular, physiological and pathophysiological functions of neuronal nicotinic acetylcholine receptors. Neurosci. Lett. 2018, 679, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Hone, A.J.; Talley, T.T.; Bobango, J.; Melo, C.H.; Hararah, F.; Gajewiak, J.; Christensen, S.; Harvey, P.J.; Craik, D.J.; McIntosh, J.M. Molecular determinants of α-conotoxin potency for inhibition of human and rat α6β4 nicotinic acetylcholine receptors. J. Biol. Chem. 2018, 293, 17838–17852. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-S.; Lee, C.-H.; Hsieh, C.-D.; Ho, C.-T.; Pan, M.-H.; Huang, C.-S.; Tu, S.-H.; Wang, Y.-J.; Chen, L.-C.; Chang, Y.-J.; et al. Nicotine-induced human breast cancer cell proliferation attenuated by garcinol through down-regulation of the nicotinic receptor and cyclin D3 proteins. Breast Cancer Res. Treat. 2010, 125, 73–87. [Google Scholar] [CrossRef]
- Hermann, P.C.; Sancho, P.; Cañamero, M.; Martinelli, P.; Madriles, F.; Michl, P.; Gress, T.; De Pascual, R.; Gandia, L.; Guerra, C.; et al. Nicotine promotes initiation and progression of KRAS-induced pancreatic cancer via Gata6-dependent dedifferentiation of acinar cells in mice. Gastroenterology 2014, 147, 1119–1133. [Google Scholar] [CrossRef]
- Dang, N.; Meng, X.; Song, H. Nicotinic acetylcholine receptors and cancer. Biomed. Rep. 2016, 4, 515–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieh, S.; Jao, S.-W.; Yang, C.-Y.; Lin, Y.-S.; Tseng, Y.-H.; Liu, C.-L.; Lee, T.-Y.; Liu, T.-Y.; Chu, Y.-H.; Chen, S.-F. Regulation of tumor progression via the Snail-RKIP signaling pathway by nicotine exposure in head and neck squamous cell carcinoma. Head Neck 2015, 37, 1712–1721. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, P.; Rizwani, W.; Pillai, S.; Kinkade, R.; Kovacs, M.; Rastogi, S.; Banerjee, S.; Carless, M.; Kim, E.; Coppola, D.; et al. Nicotine induces cell proliferation, invasion and epithelial-mesenchymal transition in a variety of human cancer cell lines. Int. J. Cancer 2008, 124, 36–45. [Google Scholar] [CrossRef] [Green Version]
- Guha, P.; Bandyopadhyaya, G.; Polumuri, S.K.; Chumsri, S.; Gade, P.; Kalvakolanu, D.V.; Ahmed, H. Nicotine promotes apoptosis resistance of breast cancer cells and enrichment of side population cells with cancer stem cell-like properties via a signaling cascade involving galectin-3, α9 nicotinic acetylcholine receptor and STAT3. Breast Cancer Res. Treat. 2014, 145, 5–22. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Qi, Y.; Avercenc-Leger, L.; Vincourt, J.-B.; Hupont, S.; Huselstein, C.; Wang, H.; Chen, L.; Magdalou, J. Effect of nicotine on the proliferation and chondrogenic differentiation of the human Wharton’s jelly mesenchymal stem cells. BioMed Mater. Eng. 2017, 28, S217–S228. [Google Scholar] [CrossRef]
- Guan, Y.-Z.; Jin, X.-D.; Guan, L.-X.; Yan, H.-C.; Wang, P.; Gong, Z.; Li, S.-J.; Cao, X.; Xing, Y.-L.; Gao, T.-M. Nicotine Inhibits Microglial Proliferation and Is Neuroprotective in Global Ischemia Rats. Mol. Neurobiol. 2015, 51, 1480–1488. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, H.-L. Restoration of miR-1305 relieves the inhibitory effect of nicotine on periodontal ligament-derived stem cell proliferation, migration, and osteogenic differentiation. J. Oral. Pathol. Med. 2016, 46, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Xu, W.; Wu, J.; Zhang, D.; Abou-Shakra, A.; Di, L.; Wang, Z.; Wang, L.; Yang, F.; Qiao, Z. Nicotine induced autophagy of Leydig cells rather than apoptosis is the major reason of the decrease of serum testosterone. Int. J. Biochem. Cell Biol. 2018, 100, 30–41. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, H.; Qi, W.; Zhang, Y.; Li, J.; Li, Z.; Lin, Y.; Bai, X.; Liu, X.; Chen, X.; et al. Nicotine promotes atherosclerosis via ROS-NLRP3-mediated endothelial cell pyroptosis. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Qu, Q.; Zhang, F.; Zhang, X.; Yin, W. Bidirectional Regulation of Mouse Embryonic Stem Cell Proliferation by Nicotine Is Mediated Through Wnt Signaling Pathway. Dose-Response 2017, 15, 1559325817739760. [Google Scholar] [CrossRef] [PubMed]
- Verbitsky, M.; Rothlin, C.V.; Katz, E.; Elgoyhen, A.B. Mixed nicotinic–muscarinic properties of the α9 nicotinic cholinergic receptor. Neuropharmacology 2000, 39, 2515–2524. [Google Scholar] [CrossRef]
- Boulin, T.; Gielen, M.; Richmond, J.E.; Williams, D.C.; Paoletti, P.; Bessereau, J.-L. Eight genes are required for functional reconstitution of the Caenorhabditis elegans levamisole-sensitive acetylcholine receptor. Proc. Natl. Acad. Sci. USA 2008, 105, 18590–18595. [Google Scholar] [CrossRef] [Green Version]
- Charvet, C.L.; Robertson, A.P.; Cabaret, J.; Martin, R.J.; Neveu, C. Selective effect of the anthelmintic bephenium on Haemonchus contortus levamisole-sensitive acetylcholine receptors. Invertebr. Neurosci. 2012, 12, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Kehoe, J.; McIntosh, J.M. Two Distinct Nicotinic Receptors, One Pharmacologically Similar to the Vertebrate α7-Containing Receptor, Mediate Cl Currents inAplysiaNeurons. J. Neurosci. 1998, 18, 8198–8213. [Google Scholar] [CrossRef] [Green Version]
- Ryoo, H.D.; Bergmann, A. The Role of Apoptosis-Induced Proliferation for Regeneration and Cancer. Cold Spring Harb. Perspect. Biol. 2012, 4, a008797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vizi, E.S.; Fekete, A.; Karoly, R.; Mike, A. Non-synaptic receptors and transporters involved in brain functions and targets of drug treatment. Br. J. Pharmacol. 2010, 160, 785–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín, A.; Szczupak, B.; Gómez-Vallejo, V.; Domercq, M.; Cano, A.; Padro, D.; Muñoz, C.; Higuchi, M.; Matute, C.; Llop, J. In VivoPET Imaging of the α4β2 Nicotinic Acetylcholine Receptor as a Marker for Brain Inflammation after Cerebral Ischemia. J. Neurosci. 2015, 35, 5998–6009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morioka, N.; Tokuhara, M.; Nakamura, Y.; Idenoshita, Y.; Harano, S.; Zhang, F.; Hisaoka-Nakashima, K.; Nakata, Y. Primary cultures of rat cortical microglia treated with nicotine increases in the expression of excitatory amino acid transporter 1 (GLAST) via the activation of the α7 nicotinic acetylcholine receptor. Neurosciences 2014, 258, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Everhart, D.; Reiller, E.; Mirzoian, A.; McIntosh, J.M.; Malhotra, A.; Luetje, C.W. Identification of residues that confer alpha-conotoxin-PnIA sensitivity on the alpha 3 subunit of neuronal nicotinic acetylcholine receptors. J. Pharmacol. Exp. Ther. 2003, 306, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, M.S.; Zwart, R.; Ursu, D.; Jensen, M.M.; Pinborg, L.H.; Gilmour, G.; Wu, J.; Sher, E.; Mikkelsen, J.D. α7 and β2 Nicotinic Acetylcholine Receptor Subunits Form Heteromeric Receptor Complexes that Are Expressed in the Human Cortex and Display Distinct Pharmacological Properties. PLoS ONE 2015, 10, e0130572. [Google Scholar] [CrossRef]
- Wu, J.; Liu, Q.; Tang, P.; Mikkelsen, J.D.; Shen, J.; Whiteaker, P.; Yakel, J.L. Heteromeric α7β2 Nicotinic Acetylcholine Receptors in the Brain. Trends Pharmacol. Sci. 2016, 37, 562–574. [Google Scholar] [CrossRef] [Green Version]
- Moretti, M.; Zoli, M.; George, A.A.; Lukas, R.J.; Pistillo, F.; Maskos, U.; Whiteaker, P.; Gotti, C. The Novel α7β2-Nicotinic Acetylcholine Receptor Subtype Is Expressed in Mouse and Human Basal Forebrain: Biochemical and Pharmacological Characterization. Mol. Pharmacol. 2014, 86, 306–317. [Google Scholar] [CrossRef] [Green Version]
- Murray, T.A.; Bertrand, D.; Papke, R.L.; George, A.A.; Pantoja, R.; Srinivasan, R.; Liu, Q.; Wu, J.; Whiteaker, P.; Lester, H.A.; et al. α7β2 nicotinic acetylcholine receptors assemble, function, and are activated primarily via their α7-α7 interfaces. Mol Pharmacol. 2012, 81, 175–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, R.; Ushiyama, N.; Fujii, T.; Kawashima, K. Nicotine-induced Ca2+ signaling and down-regulation of nicotinic acetylcholine receptor subunit expression in the CEM human leukemic T-cell line. Life Sci. 2003, 72, 2155–2158. [Google Scholar] [CrossRef]
- Jaismy Jacob, P.; Manju, S.L.; Ethiraj, K.R.; Elias, G. Safer anti-inflammatory therapy through dual COX-2/5-LOX inhibitors: A structure-based approach. Eur. J. Pharm. Sci. 2018, 121, 356–381. [Google Scholar]
- Crusz, S.M.; Balkwill, F.R. Inflammation and cancer: Advances and new agents. Nat. Rev. Clin. Oncol. 2015, 12, 584–596. [Google Scholar] [CrossRef]
- Grösch, S.; Maier, T.J.; Schiffmann, S.; Geisslinger, G. Cyclooxygenase-2 (COX-2)–Independent Anticarcinogenic Effects of Selective COX-2 Inhibitors. J. Natl. Cancer Inst. 2006, 98, 736–747. [Google Scholar] [CrossRef] [Green Version]
- Pannunzio, A.; Coluccia, M. Cyclooxygenase-1 (COX-1) and COX-1 Inhibitors in Cancer: A Review of Oncology and Medicinal Chemistry Literature. Pharmaceuticals 2018, 11, 101. [Google Scholar] [CrossRef] [Green Version]
- Sato, A.; Mizobuchi, Y.; Nakajima, K.; Shono, K.; Fujihara, T.; Kageji, T.; Kitazato, K.; Matsuzaki, K.; Mure, H.; Kuwayama, K.; et al. Blocking COX-2 induces apoptosis and inhibits cell proliferation via the Akt/survivin- and Akt/ID3 pathway in low-grade-glioma. J. Neuro-Oncol. 2017, 132, 231–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egashira, I.; Takahashi-Yanaga, F.; Nishida, R.; Arioka, M.; Igawa, K.; Tomooka, K.; Nakatsu, Y.; Tsuzuki, T.; Nakabeppu, Y.; Kitazono, T.; et al. Celecoxib and 2,5-dimethylcelecoxib inhibit intestinal cancer growth by suppressing the Wnt/β-catenin signaling pathway. Cancer Sci. 2016, 108, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Farhadi, P.; Yarani, R.; Dokaneheifard, S.; Mansouri, K. The emerging role of targeting cancer metabolism for cancer therapy. Tumor Biol. 2020, 42, 1010428320965284. [Google Scholar] [CrossRef] [PubMed]
- Neumann, S.; Shirley, S.A.; Kemp, R.A.; Hook, S.M. Improved antitumor activity of a therapeutic melanoma vaccine through the use of the dual COX-2/5-LO inhibitor licofelone. Front. Immunol. 2016, 7, 537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Dong, Y.; Gao, Y.; Du, Z.; Wang, Y.; Cheng, P.; Chen, A.; Huang, H. The Fascinating Effects of Baicalein on Cancer: A Review. Int. J. Mol. Sci. 2016, 17, 1681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surin, A.M.; Kryukova, E.V.; Strukov, A.S.; Zhmak, M.N.; Talka, R.; Tuominen, R.; Salminen, O.; Khiroug, L.S.; Kasheverov, I.E.; Tsetlin, V.I. The effect of MII α-conotoxin and its N-terminal derivatives on Ca2+- and Na+-signals induced by nicotine in SH-SY5Y neuroblastoma cells. Russ. J. Bioorg. Chem. 2012, 38, 184–191. [Google Scholar] [CrossRef]
- Kasheverov, I.E.; Zhmak, M.N.; Khruschov, A.Y.; Tsetlin, V.I. Design of New α-Conotoxins: From Computer Modeling to Synthesis of Potent Cholinergic Compounds. Mar. Drugs 2011, 9, 1698–1714. [Google Scholar] [CrossRef] [PubMed]
- Kryukova, E.V.; Ivanov, I.A.; Lebedev, D.S.; Spirova, E.N.; Egorova, N.S.; Zouridakis, M.; Kasheverov, I.E.; Tzartos, S.J.; Tsetlin, V.I. Orthosteric and/or Allosteric Binding of α-Conotoxins to Nicotinic Acetylcholine Receptors and Their Models. Mar. Drugs 2018, 16, 460. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terpinskaya, T.I.; Osipov, A.V.; Kryukova, E.V.; Kudryavtsev, D.S.; Kopylova, N.V.; Yanchanka, T.L.; Palukoshka, A.F.; Gondarenko, E.A.; Zhmak, M.N.; Tsetlin, V.I.; et al. α-Conotoxins and α-Cobratoxin Promote, while Lipoxygenase and Cyclooxygenase Inhibitors Suppress the Proliferation of Glioma C6 Cells. Mar. Drugs 2021, 19, 118. https://doi.org/10.3390/md19020118
Terpinskaya TI, Osipov AV, Kryukova EV, Kudryavtsev DS, Kopylova NV, Yanchanka TL, Palukoshka AF, Gondarenko EA, Zhmak MN, Tsetlin VI, et al. α-Conotoxins and α-Cobratoxin Promote, while Lipoxygenase and Cyclooxygenase Inhibitors Suppress the Proliferation of Glioma C6 Cells. Marine Drugs. 2021; 19(2):118. https://doi.org/10.3390/md19020118
Chicago/Turabian StyleTerpinskaya, Tatiana I., Alexey V. Osipov, Elena V. Kryukova, Denis S. Kudryavtsev, Nina V. Kopylova, Tatsiana L. Yanchanka, Alena F. Palukoshka, Elena A. Gondarenko, Maxim N. Zhmak, Victor I. Tsetlin, and et al. 2021. "α-Conotoxins and α-Cobratoxin Promote, while Lipoxygenase and Cyclooxygenase Inhibitors Suppress the Proliferation of Glioma C6 Cells" Marine Drugs 19, no. 2: 118. https://doi.org/10.3390/md19020118
APA StyleTerpinskaya, T. I., Osipov, A. V., Kryukova, E. V., Kudryavtsev, D. S., Kopylova, N. V., Yanchanka, T. L., Palukoshka, A. F., Gondarenko, E. A., Zhmak, M. N., Tsetlin, V. I., & Utkin, Y. N. (2021). α-Conotoxins and α-Cobratoxin Promote, while Lipoxygenase and Cyclooxygenase Inhibitors Suppress the Proliferation of Glioma C6 Cells. Marine Drugs, 19(2), 118. https://doi.org/10.3390/md19020118