
Dieckol and Its Derivatives as Potential Inhibitors of SARS-CoV-2 Spike Protein (UK Strain: VUI 202012/01): A Computational Study

 ,
,  ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
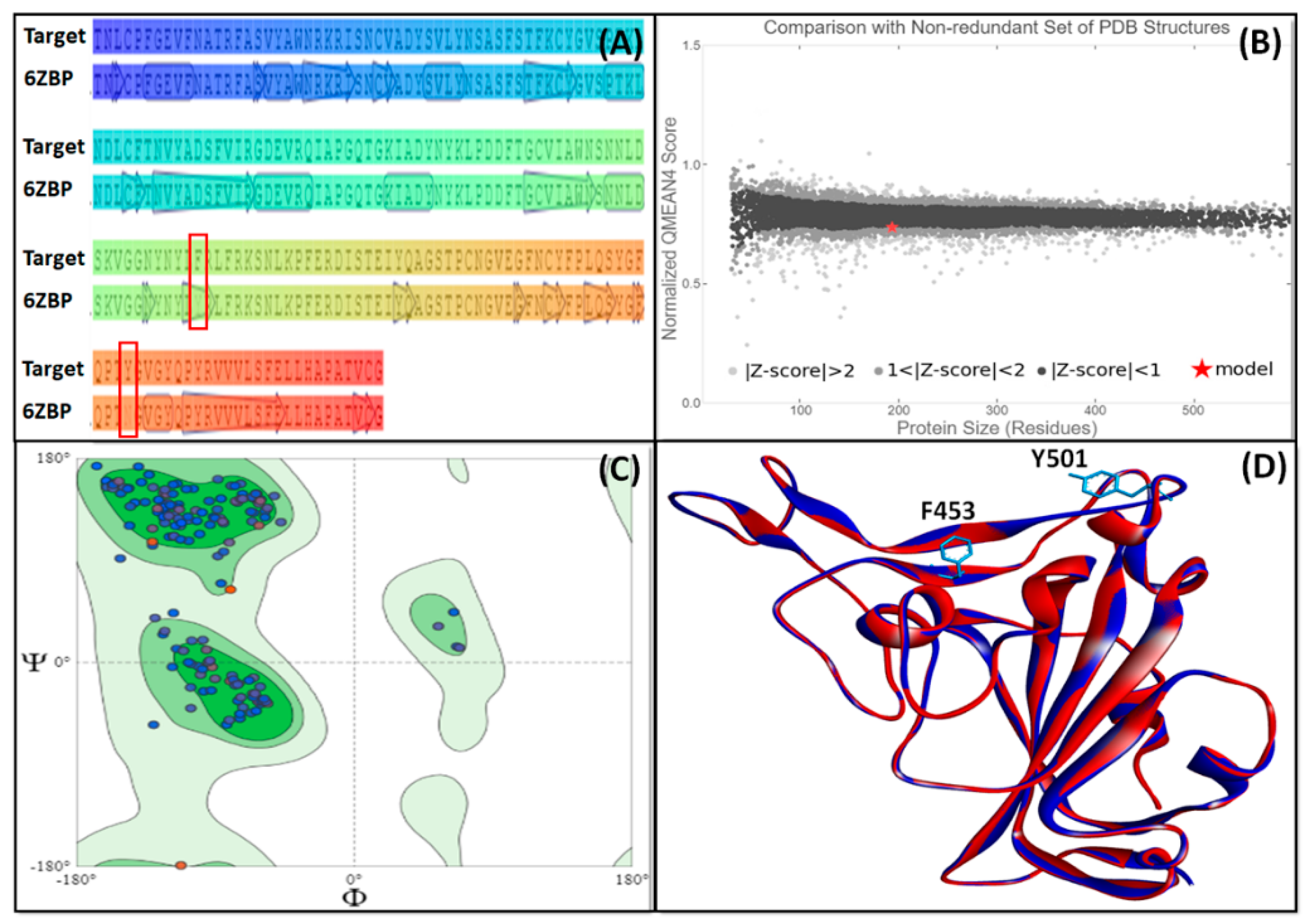
2.1. Homology Modeling and Validation of UK SARS-CoV-2 RBD
2.2. HTVS, SP, and XP Molecular Docking of Marine Seaweed Compounds against RBD of Spike Protein
2.3. Investigation of Physicochemical Pharmacokinetic, Drug-Like, and Medicinal Properties
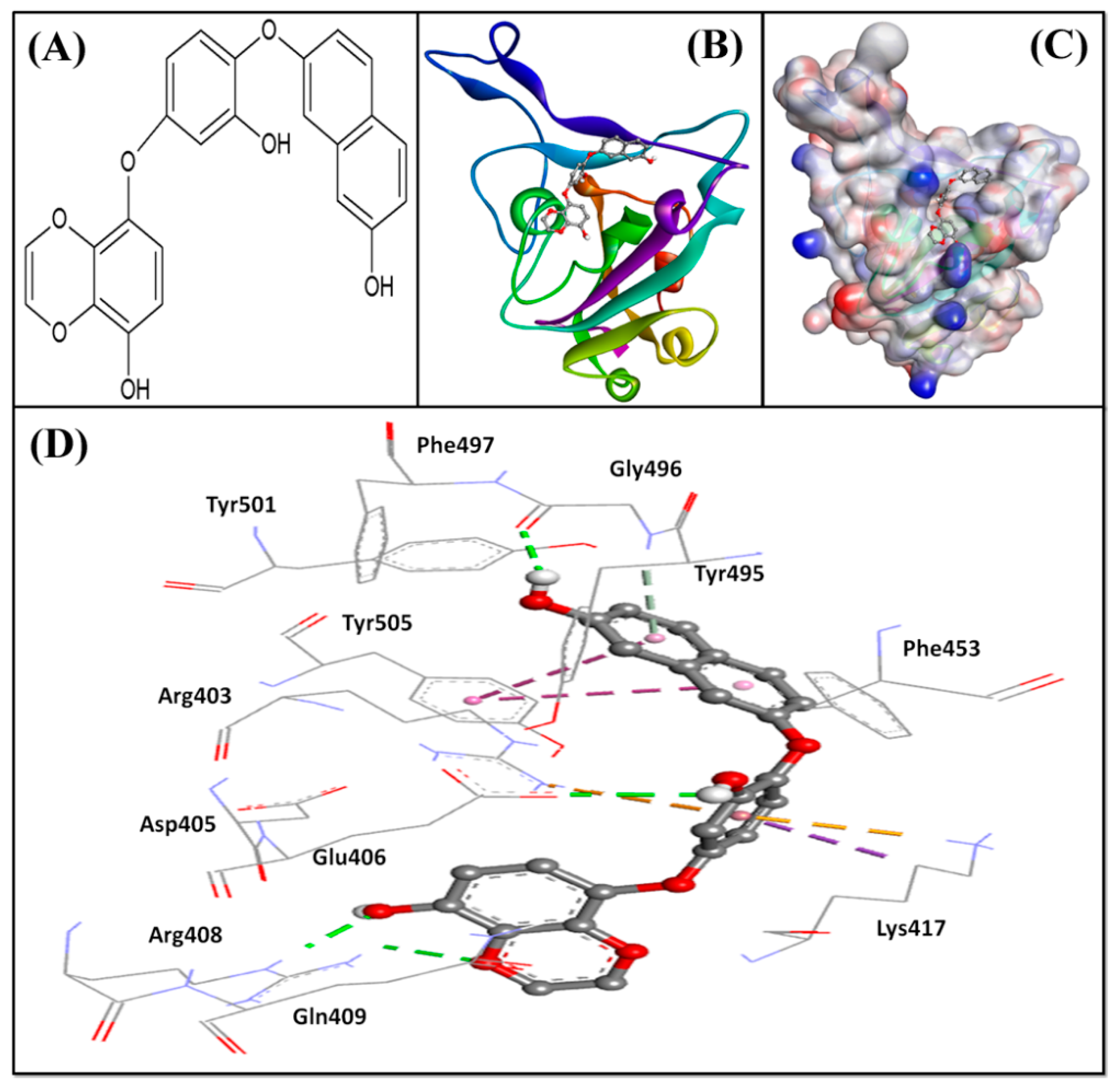
2.4. Interaction of Spike Protein RBD with ACE2 and Dieckol Derivative DK07
2.5. MD (Molecular Dynamics) Simulation Analysis
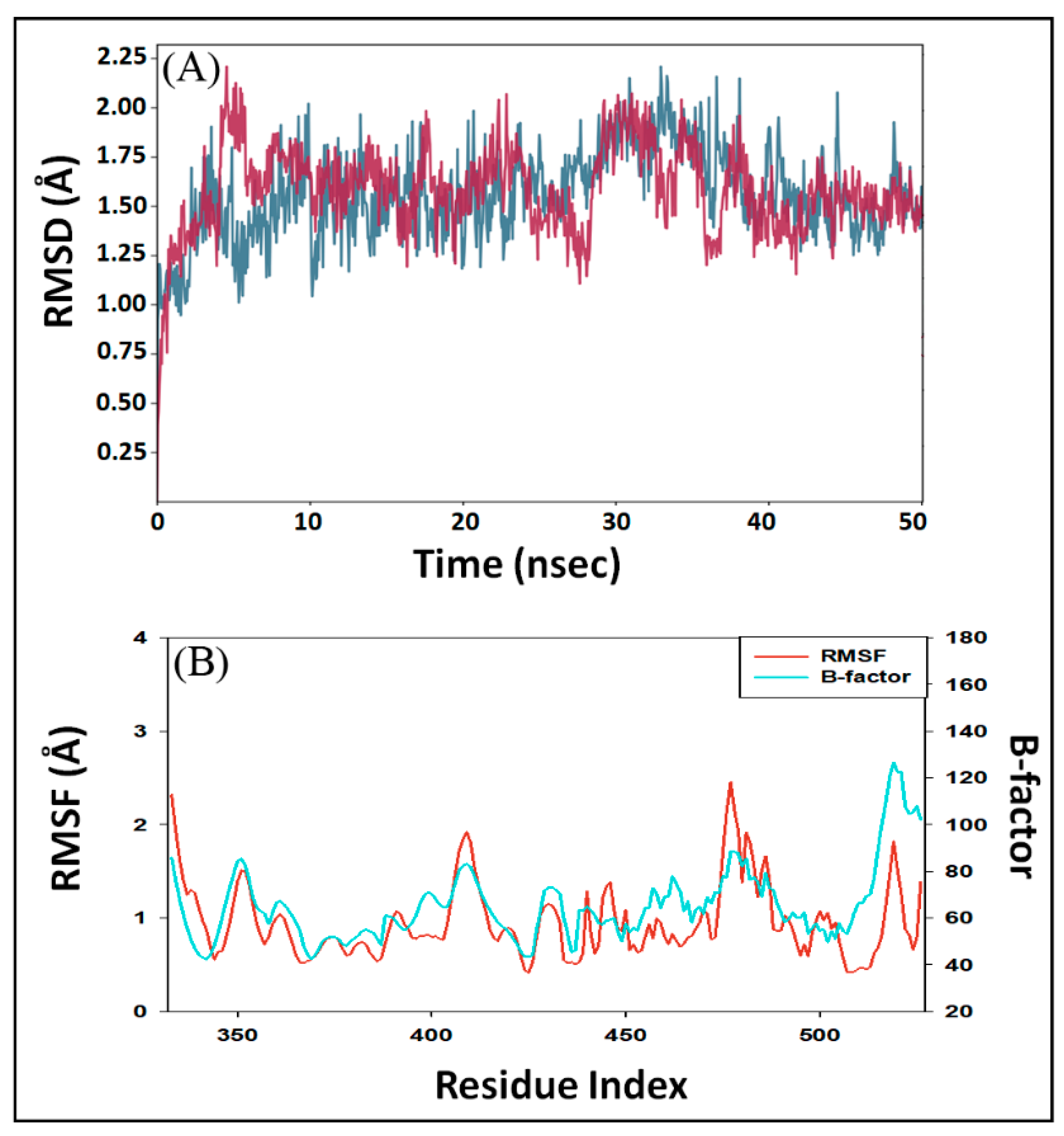
2.5.1. RMSD (Root Mean Square Deviation) Calculations
2.5.2. RMSF (Root Mean Square Fluctuation) Calculations
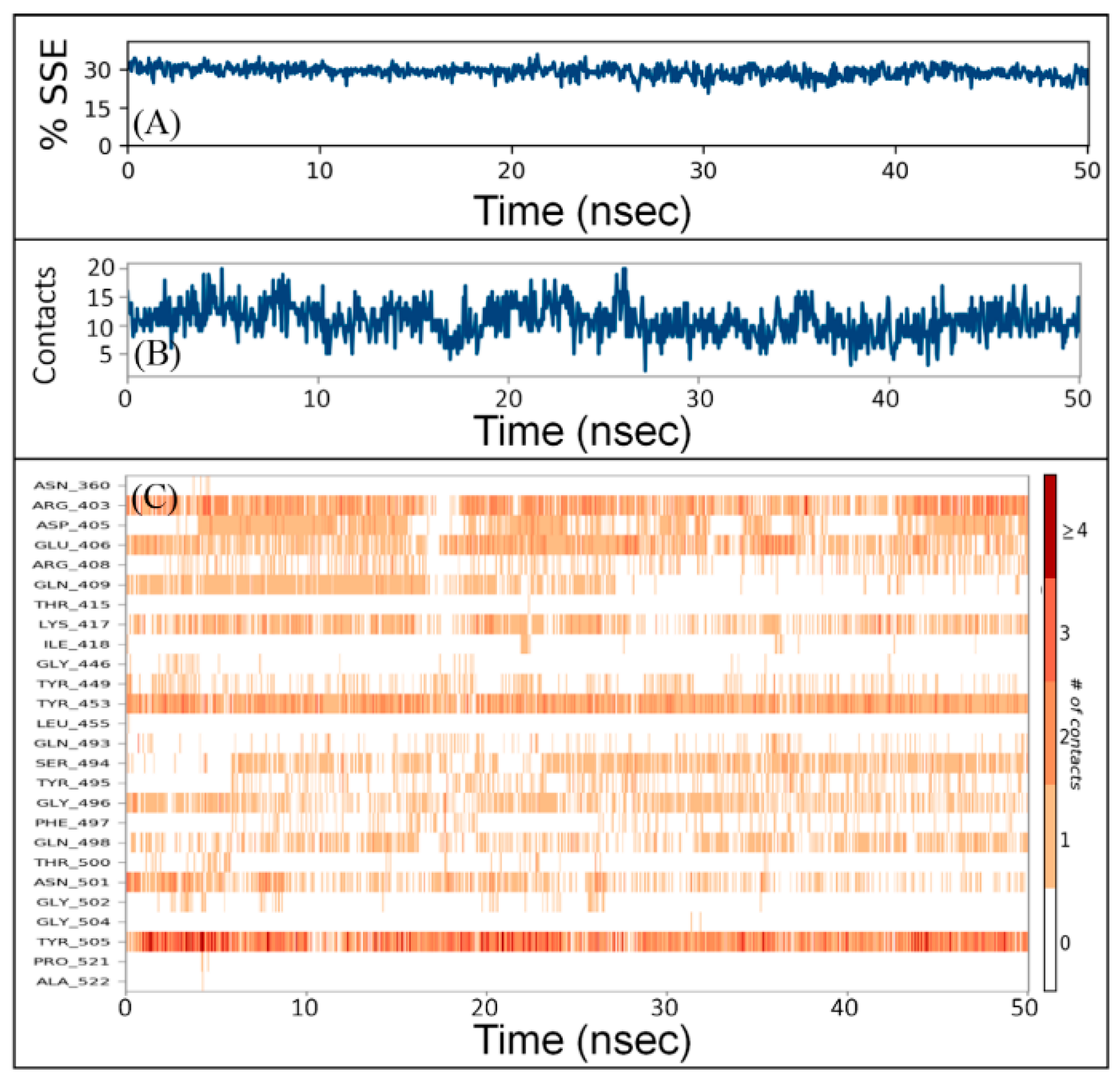
2.5.3. Secondary Structure Analysis
2.5.4. Protein–Ligand Interaction Analysis
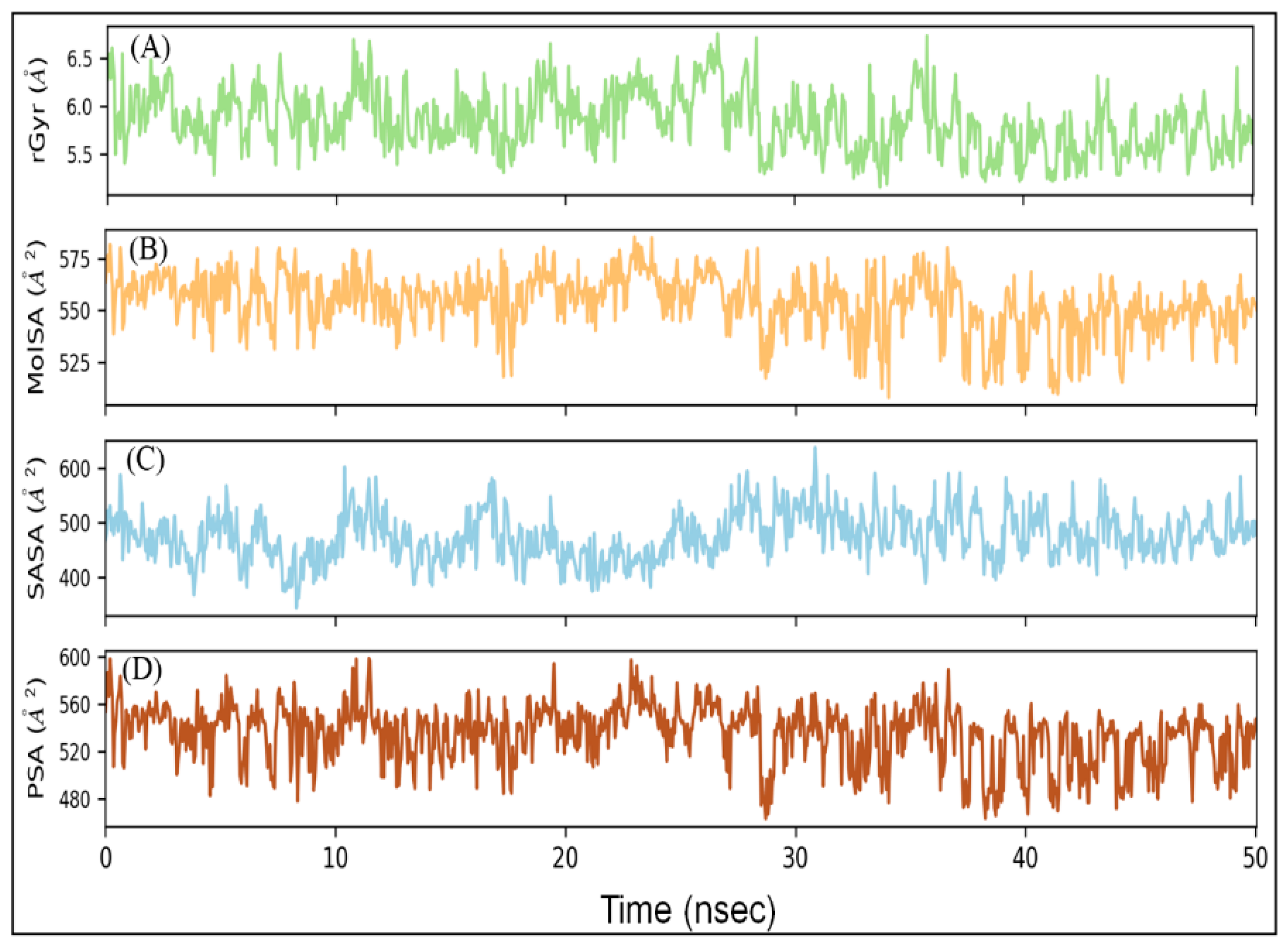
2.5.5. Radius of Gyration (rGyr) and Different Surface Area Analysis
2.6. Free Energy (Prime/MM-GBSA) Calculation of RBD–DK07 Interaction
3. Materials and Methods
3.1. Preparation of Ligands
3.2. Homology Modeling of UK SARS-CoV-2 Spike Protein RBD and Its Validation
3.3. Preparation of Target Protein (Spike Protein RBD of UK SARS-CoV-2)
3.4. Molecular Docking
3.5. Physicochemical, Drug-Like, and ADMET Properties
3.6. Molecular Dynamics (MD) Simulation
3.7. Determination of Binding Free Energy Using Prime/MM-GBSA
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cucinotta, D.; Vanelli, M. WHO declares COVID-19 a pandemic. Acta Biomed. 2020, 91, 157–160. [Google Scholar] [PubMed]
- Guo, Y.R.; Cao, Q.D.; Hong, Z.S.; Tan, Y.Y.; Chen, S.D.; Jin, H.J.; Tan, K.S.; Wang, D.Y.; Yan, Y. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak- A n update on the status. Mil. Med. Res. 2020, 7, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- She, J.; Jiang, J.; Ye, L.; Hu, L.; Bai, C.; Song, Y. 2019 novel coronavirus of pneumonia in Wuhan, China: Emerging attack and management strategies. Clin. Transl. Med. 2020, 9, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Xu, Y.; Bao, L.; Zhang, L.; Yu, P.; Qu, Y.; Zhu, H.; Zhao, W.; Han, Y.; Qin, C. From SARS to MERS, Thrusting Coronaviruses into the Spotlight. Viruses 2019, 11, 59. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef]
- Oude Munnink, B.B.; Nieuwenhuijse, D.F.; Stein, M.; O’Toole, Á.; Haverkate, M.; Mollers, M.; Kamga, S.K.; Schapendonk, C.; Pronk, M.; Lexmond, P.; et al. Rapid SARS-CoV-2 whole-genome sequencing and analysis for informed public health decision-making in the Netherlands. Nat. Med. 2020, 26, 1405–1410. [Google Scholar] [CrossRef]
- Lai, C.C.; Shih, T.P.; Ko, W.C.; Tang, H.J.; Hsueh, P.R. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): The epidemic and the challenges. Int. J. Antimicrob. Agents 2020, 55, 105924. [Google Scholar] [CrossRef]
- Palese, L.L. The Structural Landscape of SARS-CoV-2 Main Protease: Hints for Inhibitor Search. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Yang, H.; Bartlam, M.; Rao, Z. Drug Design Targeting the Main Protease, the Achilles Heel of Coronaviruses. Curr. Pharm. Des. 2006, 12, 4573–4590. [Google Scholar] [CrossRef]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Kuntz, I.D.; Blaney, J.M.; Oatley, S.J.; Langridge, R.; Ferrin, T.E. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 1982, 161, 269–288. [Google Scholar] [CrossRef]
- Faheem, M.; Rehman, M.T.; Danishuddin, M.; Khan, A.U. Biochemical Characterization of CTX-M-15 from Enterobacter cloacae and Designing a Novel Non-β-Lactam-β-Lactamase Inhibitor. PLoS ONE 2013, 8, e56926. [Google Scholar] [CrossRef]
- Li, Y.; Qian, Z.J.; Ryu, B.M.; Lee, S.H.; Kim, M.M.; Kim, S.K. Chemical components and its antioxidant properties in vitro: An edible marine brown alga, Ecklonia cava. Bioorganic Med. Chem. 2009, 17, 1963–1973. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.J.; Yoon, K.D.; Min, S.Y.; Lee, J.S.; Kim, J.H.; Kim, T.G.; Kim, S.H.; Kim, N.G.; Huh, H.; Kim, J. Inhibition of HIV-1 reverse transcriptase and protease by phlorotannins from the brown alga Ecklonia cava. Biol. Pharm. Bull. 2004, 27, 544–547. [Google Scholar] [CrossRef] [Green Version]
- Hwang, H.; Chen, T.; Nines, R.G.; Shin, H.C.; Stoner, G.D. Photochemoprevention of UVB-induced skin carcinogenesis in SKH-1 mice by brown algae polyphenols. Int. J. Cancer 2006, 119, 2742–2749. [Google Scholar] [CrossRef] [PubMed]
- Yoon, N.Y.; Kim, H.R.; Chung, H.Y.; Choi, J.S. Anti-hyperlipidemic effect of an edible brown algae, Ecklonia stolonifera, and its constituents on poloxamer 407-induced hyperlipidemic and cholesterol-fed rats. Arch. Pharmacal Res. 2008, 31, 1564–1571. [Google Scholar] [CrossRef]
- Heo, S.J.; Ko, S.C.; Cha, S.H.; Kang, D.H.; Park, H.S.; Choi, Y.U.; Kim, D.; Jung, W.K.; Jeon, Y.J. Effect of phlorotannins isolated from Ecklonia cava on melanogenesis and their protective effect against photo-oxidative stress induced by UV-B radiation. Toxicol. Vitr. 2009, 23, 1123–1130. [Google Scholar] [CrossRef]
- Lee, S.H.; Park, M.H.; Heo, S.J.; Kang, S.M.; Ko, S.C.; Han, J.S.; Jeon, Y.J. Dieckol isolated from Ecklonia cava inhibits α-glucosidase and α-amylase in vitro and alleviates postprandial hyperglycemia in streptozotocin-induced diabetic mice. Food Chem. Toxicol. 2010, 48, 2633–2637. [Google Scholar] [CrossRef]
- Moon, H.E.; Islam, M.N.; Ahn, B.R.; Chowdhury, S.S.; Sohn, H.S.; Jung, H.A.; Choi, J.S. Protein tyrosine phosphatase 1B and α-glucosidase inhibitory phlorotannins from edible brown algae, Ecklonia stolonifera and Eisenia bicyclis. Biosci. Biotechnol. Biochem. 2011, 75, 1472–1480. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.C.; Ahn, G.; Yang, X.; Kim, K.N.; Kang, S.M.; Lee, S.H.; Ko, S.C.; Ko, J.Y.; Kim, D.; Kim, Y.T.; et al. Hepatoprotective effects of dieckol-rich phlorotannins from Ecklonia cava, a brown seaweed, against ethanol induced liver damage in BALB/c mice. Food Chem. Toxicol. 2012, 50, 1986–1991. [Google Scholar] [CrossRef]
- Kim, T.H.; Ku, S.K.; Bae, J.S. Antithrombotic and profibrinolytic activities of eckol and dieckol. J. Cell. Biochem. 2012, 113, 2877–2883. [Google Scholar] [CrossRef]
- Wang, C.H.; Li, X.F.; Jin, L.F.; Zhao, Y.; Zhu, G.J.; Shen, W.Z. Dieckol inhibits non-small–cell lung cancer cell proliferation and migration by regulating the PI3K/AKT signaling pathway. J. Biochem. Mol. Toxicol. 2019, 33, e22346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.J.; Kim, Y.T.; Jeon, Y.J. Antioxidant dieckol downregulates the Rac1/ROS signaling pathway and inhibits Wiskott-Aldrich syndrome protein (WASP)-family verprolin-homologous protein 2 (WAVE2)-mediated invasive migration of B16 mouse melanoma cells. Mol. Cells 2012, 33, 363–369. [Google Scholar] [CrossRef] [Green Version]
- Park, J.Y.; Kim, J.H.; Kwon, J.M.; Kwon, H.J.; Jeong, H.J.; Kim, Y.M.; Kim, D.; Lee, W.S.; Ryu, Y.B. Dieckol, a SARS-CoV 3CLpro inhibitor, isolated from the edible brown algae Ecklonia cava. Bioorganic Med. Chem. 2013, 21, 3730–3737. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Rathi, E.; Kini, S.G. E-pharmacophore modelling, virtual screening, molecular dynamics simulations and in-silico ADME analysis for identification of potential E6 inhibitors against cervical cancer. J. Mol. Struct. 2019, 1189, 299–306. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Baig, M.S.; Alagumuthu, M.; Rajpoot, S.; Saqib, U. Identification of a Potential Peptide Inhibitor of SARS-CoV-2 Targeting its Entry into the Host Cells. Drugs R D 2020, 20, 161–169. [Google Scholar] [CrossRef]
- Rodier, F.; Bahadur, R.P.; Chakrabarti, P.; Janin, J. Hydration of protein-protein interfaces. Proteins Struct. Funct. Genet. 2005, 60, 36–45. [Google Scholar] [CrossRef]
- Gupta, P.; Khan, S.; Fakhar, Z.; Hussain, A.; Rehman, M.T.; Alajmi, M.F.; Islam, A.; Ahmad, F.; Hassan, M.I. Identification of Potential Inhibitors of Calcium/Calmodulin-Dependent Protein Kinase IV from Bioactive Phytoconstituents. Oxidative Med. Cell. Longev. 2020. [Google Scholar] [CrossRef]
- AlAjmi, M.F.; Rehman, M.T.; Hussain, A.; Rather, G.M. Pharmacoinformatics approach for the identification of Polo-like kinase-1 inhibitors from natural sources as anti-cancer agents. Int. J. Biol. Macromol. 2018, 116, 173–181. [Google Scholar] [CrossRef]
- Rehman, M.T.; Alajmi, M.F.; Hussain, A.; Rather, G.M.; Khan, M.A. High-throughput virtual screening, molecular dynamics simulation, and enzyme kinetics identified ZINC84525623 as a potential inhibitor of NDM-1. Int. J. Mol. Sci. 2019, 20, 819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unni, S.; Aouti, S.; Thiyagarajan, S.; Padmanabhan, B. Identification of a repurposed drug as an inhibitor of Spike protein of human coronavirus SARS-CoV-2 by computational methods. J. Biosci. 2020, 45, 1–20. [Google Scholar] [CrossRef]
- Sethi, A.; Sanam, S.; Munagalasetty, S.; Jayanthi, S.; Alvala, M. Understanding the role of galectin inhibitors as potential candidates for SARS-CoV-2 spike protein:: In silico studies. RSC Adv. 2020, 10, 29873–29884. [Google Scholar] [CrossRef]
- De Vita, S.; Chini, M.G.; Lauro, G.; Bifulco, G. Accelerating the repurposing of FDA-approved drugs against coronavirus disease-19 (COVID-19). RSC Adv. 2020, 10, 40867–40875. [Google Scholar] [CrossRef]
- Dicky, G.; Davis, J.; Hannah, A.; Vasanthi, R. Bioinformation Seaweed Metabolite Database (SWMD): A Database of Natural Compounds from Marine Algae; NCBI: Bethesda, MD, USA, 2011; Volume 361. [Google Scholar]
- Gallimore, W. Marine Metabolites: Oceans of Opportunity. In Pharmacognosy: Fundamentals, Applications and Strategy; Elsevier Inc.: Amsterdam, The Netherlands, 2017; pp. 377–400. ISBN 9780128020999. [Google Scholar]
- Liang, J.; Pitsillou, E.; Karagiannis, C.; Darmawan, K.K.; Ng, K.; Hung, A.; Karagiannis, T.C. Interaction of the prototypical α-ketoamide inhibitor with the SARS-CoV-2 main protease active site in silico: Molecular dynamic simulations highlight the stability of the ligand-protein complex. Comput. Biol. Chem. 2020, 87, 107292. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halgren, T.A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Rehman, M.T.; Ahmed, S.; Khan, A.U. Interaction of meropenem with ‘N’ and ‘B’ isoforms of human serum albumin: A spectroscopic and molecular docking study. J. Biomol. Struct. Dyn. 2016, 34, 1849–1864. [Google Scholar] [CrossRef]
- Rehman, M.T.; Shamsi, H.; Khan, A.U. Insight into the Binding Mechanism of Imipenem to Human Serum Albumin by Spectroscopic and Computational Approaches. Mol. Pharm. 2014, 11, 1785–1797. [Google Scholar] [CrossRef]
- Jabir, N.R.; Shakil, S.; Tabrez, S.; Khan, M.S.; Rehman, M.T.; Ahmed, B.A. In silico screening of glycogen synthase kinase-3β targeted ligands against acetylcholinesterase and its probable relevance to Alzheimer’s disease. J. Biomol. Struct. Dyn. 2020, 1–10. [Google Scholar] [CrossRef]
- Sun, H.; Li, Y.; Tian, S.; Xu, L.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 4. Accuracies of MM/PBSA and MM/GBSA methodologies evaluated by various simulation protocols using PDBbind data set. Phys. Chem. Chem. Phys. 2014, 16, 16719–16729. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Zoete, V. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [Green Version]
- Brańka, A.C. Nosé-Hoover chain method for nonequilibrium molecular dynamics simulation. Phys. Rev. E 2000, 61, 4769–4773. [Google Scholar] [CrossRef] [PubMed]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Ton, A.T.; Gentile, F.; Hsing, M.; Ban, F.; Cherkasov, A. Rapid Identification of Potential Inhibitors of SARS-CoV-2 Main Protease by Deep Docking of 1.3 Billion Compounds. Mol. Inform. 2020, 39, 200028. [Google Scholar] [CrossRef] [Green Version]
- Mittal, L.; Kumari, A.; Srivastava, M.; Singh, M.; Asthana, S. Identification of potential molecules against COVID-19 main protease through structure-guided virtual screening approach. J. Biomol. Struct. Dyn. 2020, 1–19. [Google Scholar] [CrossRef]
- Kumar, N.; Srivastava, R.; Prakash, A.; Lynn, A.M. Structure-based virtual screening, molecular dynamics simulation and MM-PBSA toward identifying the inhibitors for two-component regulatory system protein NarL of Mycobacterium Tuberculosis. J. Biomol. Struct. Dyn. 2020, 38, 3396–3410. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Mean Score | Z-Score |
|---|---|---|
| Procheck G-factor (phi/psi only) | −0.34 | −1.02 |
| Procheck G-factor (all dihedral angles) | −0.16 | −0.95 |
| Verify 3D | 0.21 | −4.01 |
| ProsaII (-ve) | 0.45 | −0.83 |
| Procheck (ϕ − ψ) | −0.34 | −1.02 |
| Procheck (all) | −0.16 | −0.95 |
| Molprobity Clash score | 1.34 | 1.30 |
| RMSD_bond length (Å) | 0.016 | - |
| RMSD_bond angle (o) | 1.9 | - |
| Close contacts (within 2.2 Å) | 0 | - |
| Ramachandran plot summary (Procheck) | ||
| 90.4 9.6 0.0 0.0 | - |
| Ramachandran plot statistics (Richardson’s lab) | ||
| 97.9 2.1 0.0 | - |
| S. No. | Compound ID | Source | Structure and Chemical Name | Docking Score (kcal mol−1) | Binding Affinity (M−1) |
|---|---|---|---|---|---|
| 1. | BE011 (Dieckol) | Ecklonia cava |  4-(4-{[6-(3,5-Dihydroxyphenoxy)-4,7,9-trihydroxyoxanthren-2-yl]oxy}-3,5-dihydroxyphenoxy)oxanthrene-1,3,6,8-tetrol | −8.326 | 1.28 × 106 |
| 2. | GA004 (Nigricanoside A) | Avrainvillea nigricans |  2,3-Dihydroxypropyl(5ξ)-6-O-[(4Z,8E,13Z)-1-carboxy-10-{[(4Z,9E)-15-carboxy-8,11-dihydroxy-4,9-pentadecadien-7-yl]oxy}-11-hydroxy-4,8,13-nonadecatrien-7-yl]-α-L-arabino-hexopyranoside | −7.952 | 6.80 × 105 |
| 3. | GA005 (Nigricanoside B) | Avrainvillea nigricans |  2,3-Dihydroxypropyl-(5ξ)-6-O-[(4Z,8E,13Z,16Z)-1-carboxy-10-{[(4Z,9E)-15-carboxy-8,11-dihydroxy-4,9-pentadecadien-7-yl]oxy}-11-hydroxy-4,8,13,16-nonadecatetraen-7-yl]-α-L-arabino-hexopyranoside | −6.962 | 1.28 × 105 |
| 4. | GA006 (Nigricanoside A dimethyl ester) | Avrainvillea nigricans |  2,3-Dihydroxypropyl-6-O-[(5Z,9E,14Z)-11-{[(4Z,9E)-8,11-dihydroxy-16-methoxy-16-oxo-4,9-hexadecadien-7-yl]oxy}-12-hydroxy-1-methoxy-1-oxo-5,9,14-icosatrien-8-yl]-β-d-galactopyranoside | −5.703 | 1.52 × 104 |
| 5. | GA007 (Nigricanoside B dimethyl ester) | Avrainvillea nigricans |  Methyl-(5Z,9E,14Z,17Z)-11-{[(4Z,9E)-8,11-dihydroxy-16-methoxy- 16-oxohexadeca-4,9-dien-7-yl]oxy}-8-{[6-(2,3-dihydroxy propoxy)-3,4,5-trihydroxyoxan-2-yl] methoxy}-12-hydroxyicosa-5,9,14,17-tetraenoate | −5.439 | 9.76 × 103 |
| Name of Compound | MW (Da) | RB | HBA | HBD | TPSA (Å2) | Lipophilicity (XlogP3) | Solubility (logS) |
|---|---|---|---|---|---|---|---|
| Dieckol | 742.55 | 6 | 18 | 11 | 287.14 | 4.87 | Poor |
| DK01 | 538.46 | 4 | 10 | 4 | 136.3 | 5.83 | Poor |
| DK02 | 528.59 | 4 | 5 | 4 | 90.15 | 7.79 | Poor |
| DK03 | 558.62 | 4 | 6 | 4 | 99.38 | 8.06 | Poor |
| DK04 | 550.51 | 4 | 9 | 5 | 138.07 | 5.5 | Poor |
| DK05 | 578.56 | 4 | 9 | 7 | 160.07 | 5.58 | Poor |
| DK06 | 472.4 | 4 | 9 | 3 | 116.07 | 4.77 | Poor |
| DK07 | 416.38 | 4 | 7 | 3 | 97.61 | 4.88 | Moderate |
| DK08 | 426.42 | 4 | 6 | 4 | 99.38 | 6.06 | Poor |
| DK09 | 410.42 | 4 | 5 | 3 | 79.15 | 6.41 | Poor |
| DK10 | 414.41 | 4 | 6 | 3 | 88.38 | 5.5 | Poor |
| Name of Compound | GIA | BBB | P-gp Substrate | Inhibitor of | logKp (cm/s) | ||||
|---|---|---|---|---|---|---|---|---|---|
| CYP1A2 | CYP2C19 | CYP2C9 | CYP2D6 | CYP3A4 | |||||
| Dieckol | Low | No | No | No | No | Yes | No | No | −7.37 |
| DK01 | Low | No | No | No | No | No | No | No | −5.45 |
| DK02 | Low | No | No | Yes | Yes | No | No | No | −3.99 |
| DK03 | Low | No | No | No | No | No | No | No | −3.98 |
| DK04 | Low | No | No | No | No | No | No | No | −5.75 |
| DK05 | Low | No | No | No | No | No | No | No | −5.87 |
| DK06 | Low | No | No | No | No | Yes | No | Yes | −5.79 |
| DK07 | High | No | No | No | Yes | Yes | Yes | Yes | −5.38 |
| DK08 | Low | No | No | No | Yes | Yes | Yes | No | −4.6 |
| DK09 | Low | No | Yes | No | Yes | Yes | Yes | No | −4.25 |
| DK10 | High | No | No | Yes | Yes | Yes | Yes | No | −4.92 |
| Name of Compound | Number of Violations in | Bioavailability Score | PAINS | Brenk Alert | Lead-likeness Alert * | Synthetic Accessibility | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Lipinski | Ghose | Veber | Egan | Muegge | Alert | |||||
| Dieckol | 3 | 4 | 1 | 2 | 5 | 0.55 | 0 | 0 | 2 | 4.68 |
| DK01 | 1 | 3 | 0 | 2 | 1 | 0.55 | 0 | 0 | 2 | 3.97 |
| DK02 | 2 | 3 | 0 | 1 | 1 | 0.17 | 0 | 0 | 2 | 4.07 |
| DK03 | 2 | 4 | 0 | 1 | 1 | 0.17 | 0 | 0 | 2 | 4.93 |
| DK04 | 1 | 3 | 0 | 2 | 1 | 0.55 | 0 | 0 | 2 | 4.59 |
| DK05 | 2 | 3 | 1 | 1 | 3 | 0.17 | 0 | 0 | 2 | 4.6 |
| DK06 | 0 | 1 | 0 | 1 | 0 | 0.55 | 0 | 0 | 2 | 4.05 |
| DK07 | 0 | 1 | 0 | 0 | 0 | 0.55 | 0 | 0 | 2 | 3.7 |
| DK08 | 0 | 1 | 0 | 1 | 1 | 0.55 | 0 | 0 | 2 | 2.92 |
| DK09 | 0 | 1 | 0 | 1 | 1 | 0.55 | 0 | 0 | 2 | 2.88 |
| DK10 | 0 | 1 | 0 | 0 | 1 | 0.55 | 0 | 0 | 2 | 3.78 |
| Donor-Acceptor Atoms * | Bond Distance (Å) | Type of Interaction | ΔG | Kd |
|---|---|---|---|---|
| (kcal mol−1) | (M−1) | |||
| ACE2 | ||||
| E:Lys417:NZ—A:Asp30:OD2 | 3 | Salt bridge | n.d. | n.d. |
| E:Lys417:NZ—A:As30:OD1 | 3.9 | Salt bridge | ||
| A:Lys31:NZ—E:Glu484:OE1 | 4.39 | Electrostatic interaction | ||
| A:Tyr41:OH—E:Thr500:OG1 | 2.7 | Conventional hydrogen bond | ||
| A:Gln42:NE2—E:Gly446:O | 3.24 | Conventional hydrogen bond | ||
| A:Gln42:NE2—E:Tyr449:OH | 2.78 | Conventional hydrogen bond | ||
| A:Tyr83:OH—E:Asn487:OD1 | 2.78 | Conventional hydrogen bond | ||
| A:Lys353:NZ—E:Gly496:O | 3.08 | Conventional hydrogen bond | ||
| E:Tyr449:OH—A:Asp38:OD2 | 2.69 | Conventional hydrogen bond | ||
| E:Asn487:ND2—A:Gln24:OE1 | 2.69 | Conventional hydrogen bond | ||
| E:Gln493:NE2:B—A:Glu35:OE1 | 3.13 | Conventional hydrogen bond | ||
| E:Gly502:N—A:Lys353:O | 2.78 | Conventional hydrogen bond | ||
| A:His34:CD2—E:Tyr453:OH | 2.86 | Carbon hydrogen bond | ||
| A:Tyr83:OH—E:Phe486 | 4.09 | Pi-donor hydrogen bond | ||
| E:Leu455:CD1—A:His34 | 3.85 | Hydrophobic (pi–sigma) | ||
| A:Tyr83—E:Phe486 | 5.14 | Hydrophobic (pi–pi stacked) | ||
| A:Lys353:C,O;Gly354:N—E:Tyr505 | 4 | Hydrophobic (amide–pi stacked) | ||
| E:Phe486—A:Met82 | 4.74 | Hydrophobic (pi–alkyl) | ||
| E:Tyr489—A:Lys31 | 4.75 | Hydrophobic (pi–alkyl) | ||
| E:Tyr505—A:Lys353 | 4.62 | Hydrophobic (pi–alkyl) | ||
| DK07 (8-{3-hydroxy-4-[(7-hydroxynaphthalen-2-yl)oxy]phenoxy}-1,4-benzodioxin-5-ol) | ||||
| Arg408:HE—UNK:O | 2.11 | Conventional hydrogen bond | −8.954 | 3.69 × 106 |
| Arg408:HH21—UNK:O | 2.41 | Conventional hydrogen bond | ||
| UNK:H—Glu406:OE2 | 2.48 | Conventional hydrogen bond | ||
| UNK:H—Gly496:O | 2.43 | Conventional hydrogen bond | ||
| Arg403:NH2—UNK | 4.1 | Pi–cation; pi–donor hydrogen bond | ||
| Lys417:HZ3—UNK | 3.15 | Pi–cation; pi–donor hydrogen bond | ||
| Gly496:HN—UNK | 3.12 | Pi–donor hydrogen bond | ||
| Lys417:CD—UNK | 3.83 | Hydrophobic (pi–sigma) | ||
| Tyr505—UNK | 5.92 | Hydrophobic (pi–pi T-shaped) | ||
| Tyr505—UNK | 5.17 | Hydrophobic (pi–pi T-shaped) | ||
| Binding Free Energy (ΔGbind) | Solvation Energy (ΔGsol) | Gas Phase Energy (ΔEgas) | Coulomb Energy (ΔEelec) | Van der Waals Energy (ΔEvdW) | Internal Energy (ΔEint) | Polar Solvation Energy (ΔGpolar) | Non-Polar Solvation Energy (ΔGnon-polar) |
|---|---|---|---|---|---|---|---|
| −52.87 | 74.88 | −127.75 | −69.99 | −58.16 | 0.40 | 78.67 | −3.79 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aatif, M.; Muteeb, G.; Alsultan, A.; Alshoaibi, A.; Khelif, B.Y. Dieckol and Its Derivatives as Potential Inhibitors of SARS-CoV-2 Spike Protein (UK Strain: VUI 202012/01): A Computational Study. Mar. Drugs 2021, 19, 242. https://doi.org/10.3390/md19050242
Aatif M, Muteeb G, Alsultan A, Alshoaibi A, Khelif BY. Dieckol and Its Derivatives as Potential Inhibitors of SARS-CoV-2 Spike Protein (UK Strain: VUI 202012/01): A Computational Study. Marine Drugs. 2021; 19(5):242. https://doi.org/10.3390/md19050242
Chicago/Turabian StyleAatif, Mohammad, Ghazala Muteeb, Abdulrahman Alsultan, Adil Alshoaibi, and Bachir Yahia Khelif. 2021. "Dieckol and Its Derivatives as Potential Inhibitors of SARS-CoV-2 Spike Protein (UK Strain: VUI 202012/01): A Computational Study" Marine Drugs 19, no. 5: 242. https://doi.org/10.3390/md19050242
APA StyleAatif, M., Muteeb, G., Alsultan, A., Alshoaibi, A., & Khelif, B. Y. (2021). Dieckol and Its Derivatives as Potential Inhibitors of SARS-CoV-2 Spike Protein (UK Strain: VUI 202012/01): A Computational Study. Marine Drugs, 19(5), 242. https://doi.org/10.3390/md19050242