
Antimicrobial Activity and Action Mechanisms of Arg-Rich Short Analog Peptides Designed from the C-Terminal Loop Region of American Oyster Defensin (AOD)

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Subsection
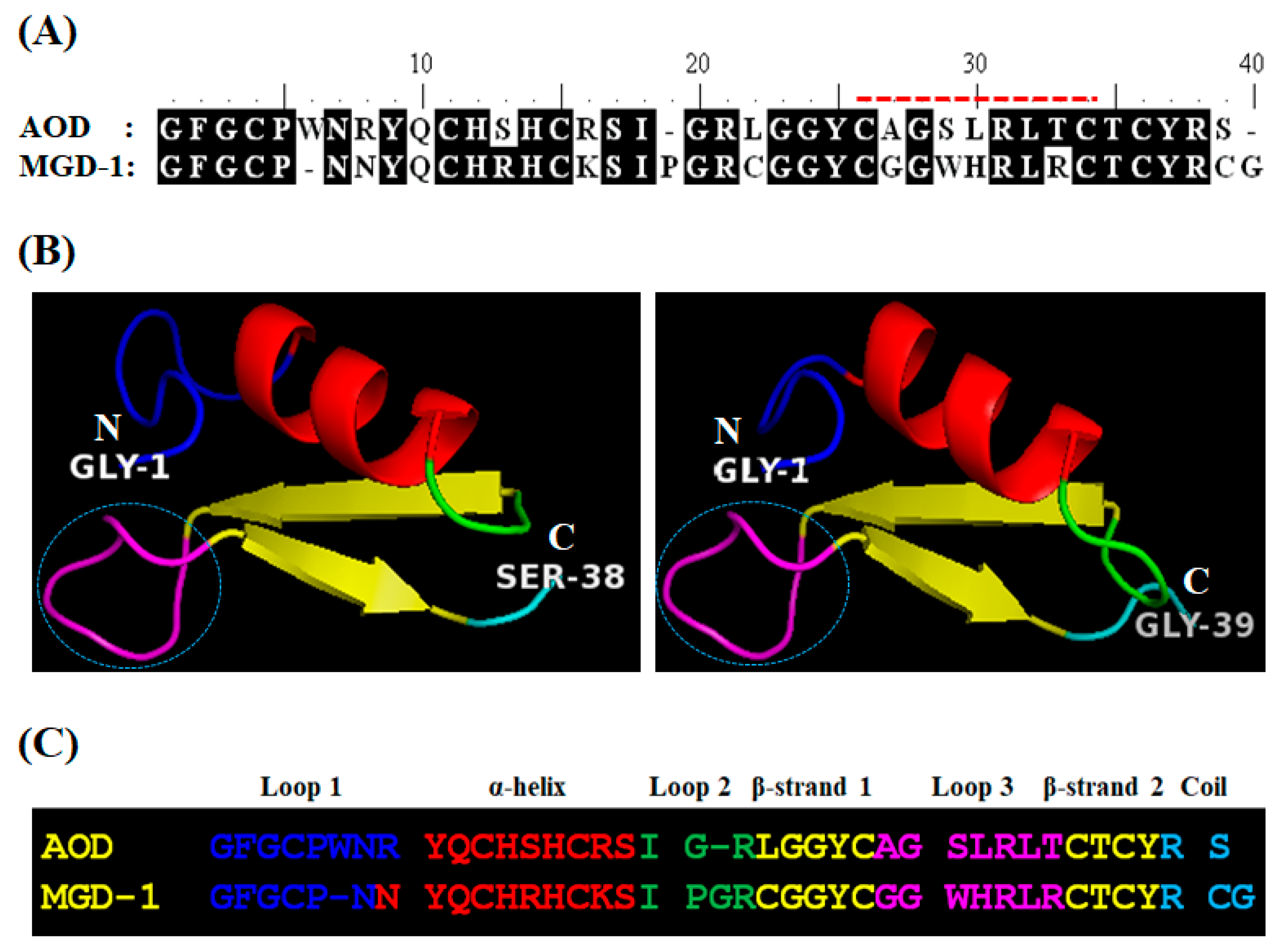
2.1.1. Analog Design and Synthesis
2.1.2. Antimicrobial Activity
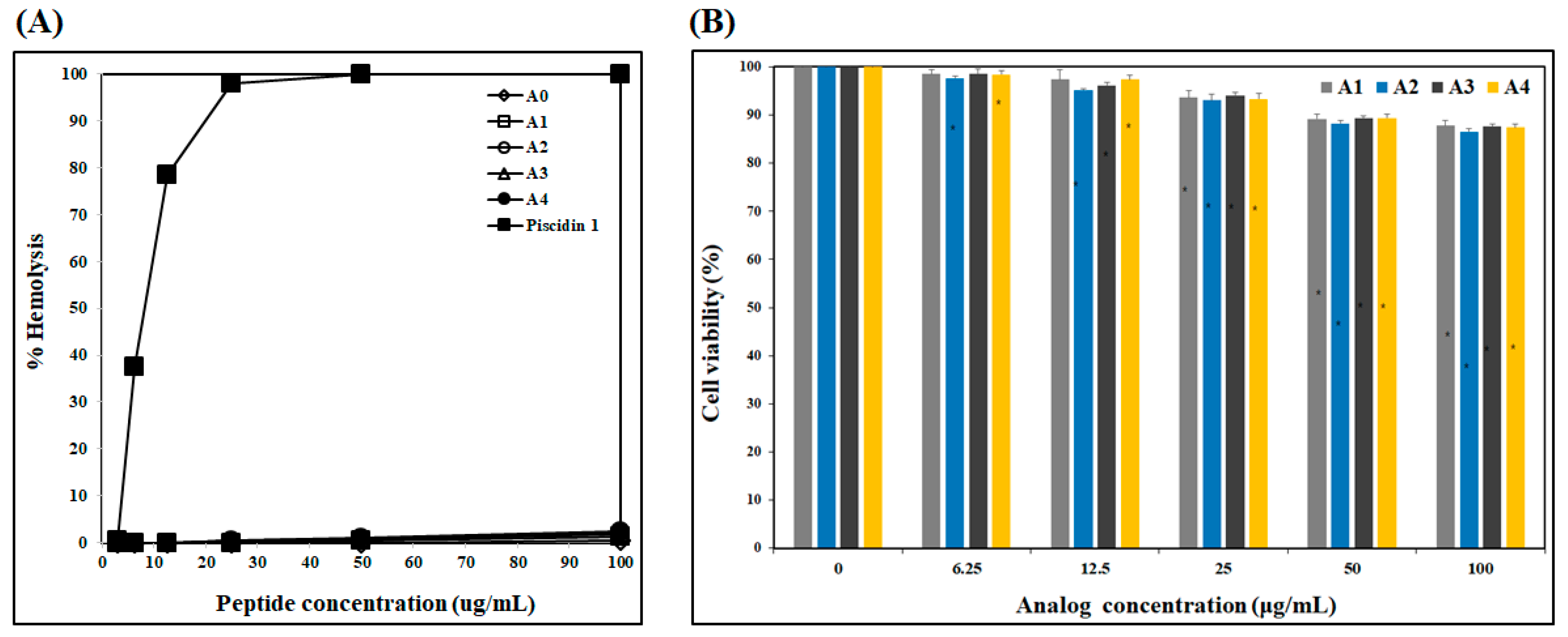
2.1.3. Cytotoxicity of Analogs
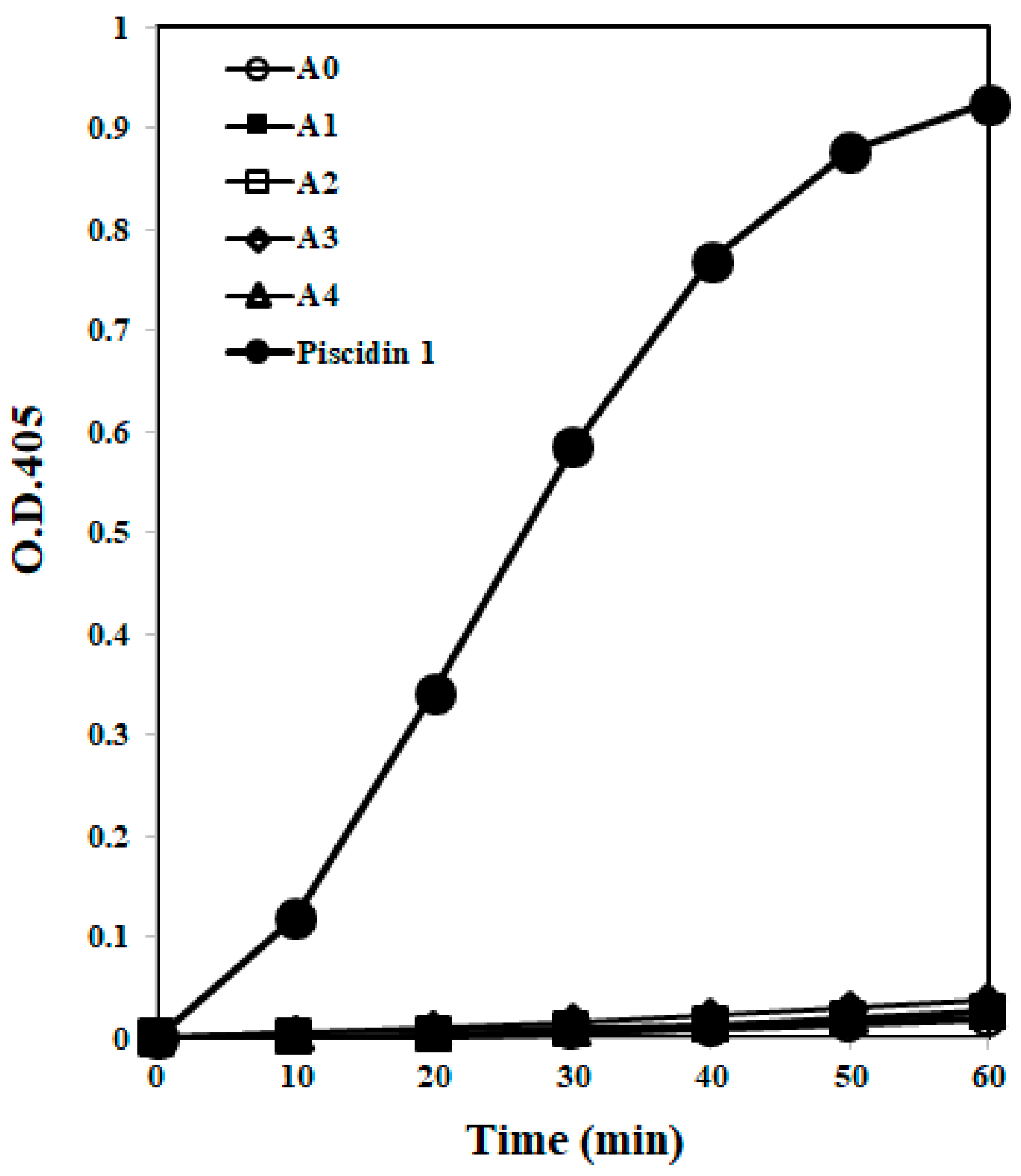
2.1.4. Membrane Permeability
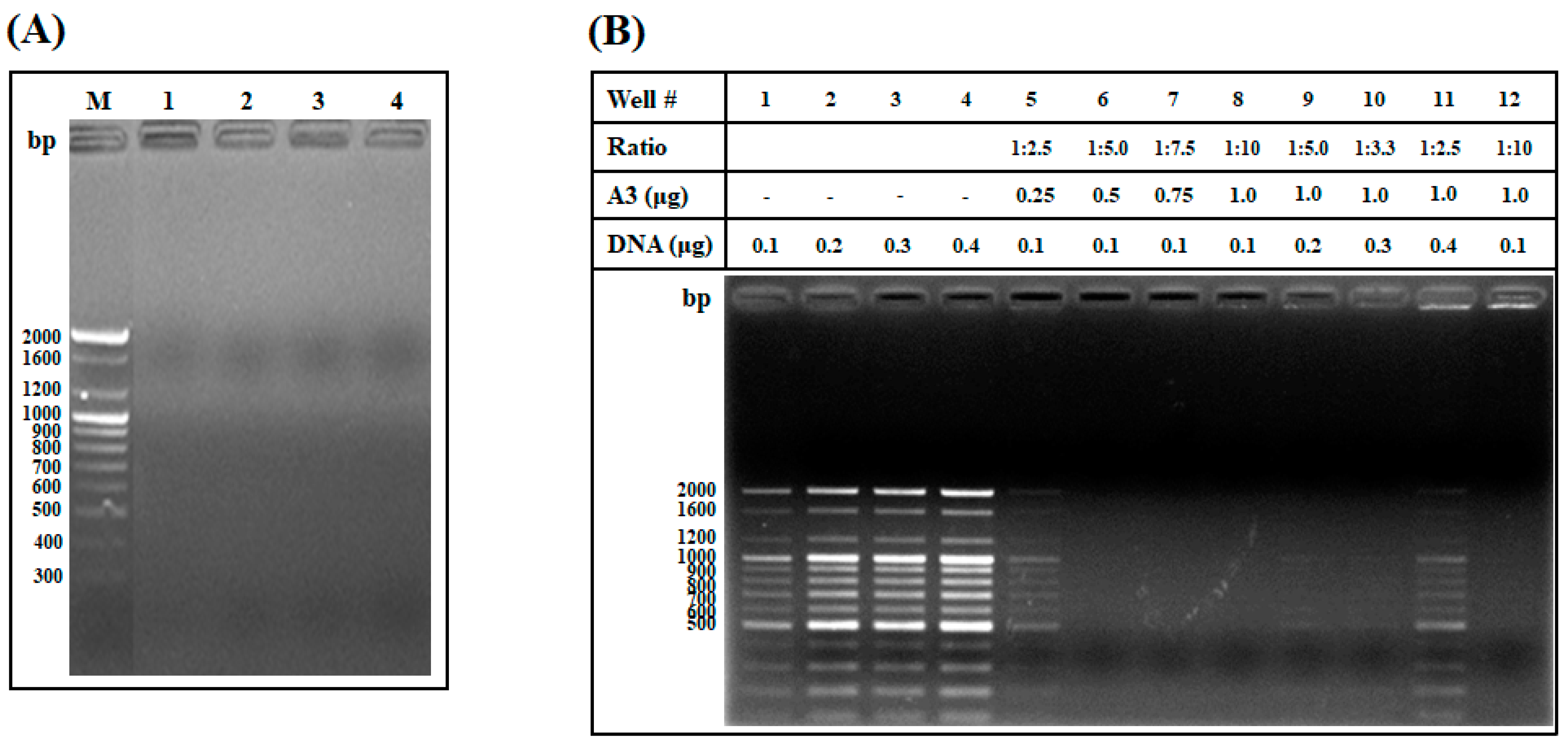
2.1.5. DNA Binding Assay and Investigation of the Weight Ratio of AOD Analog and DNA Needed for Interaction
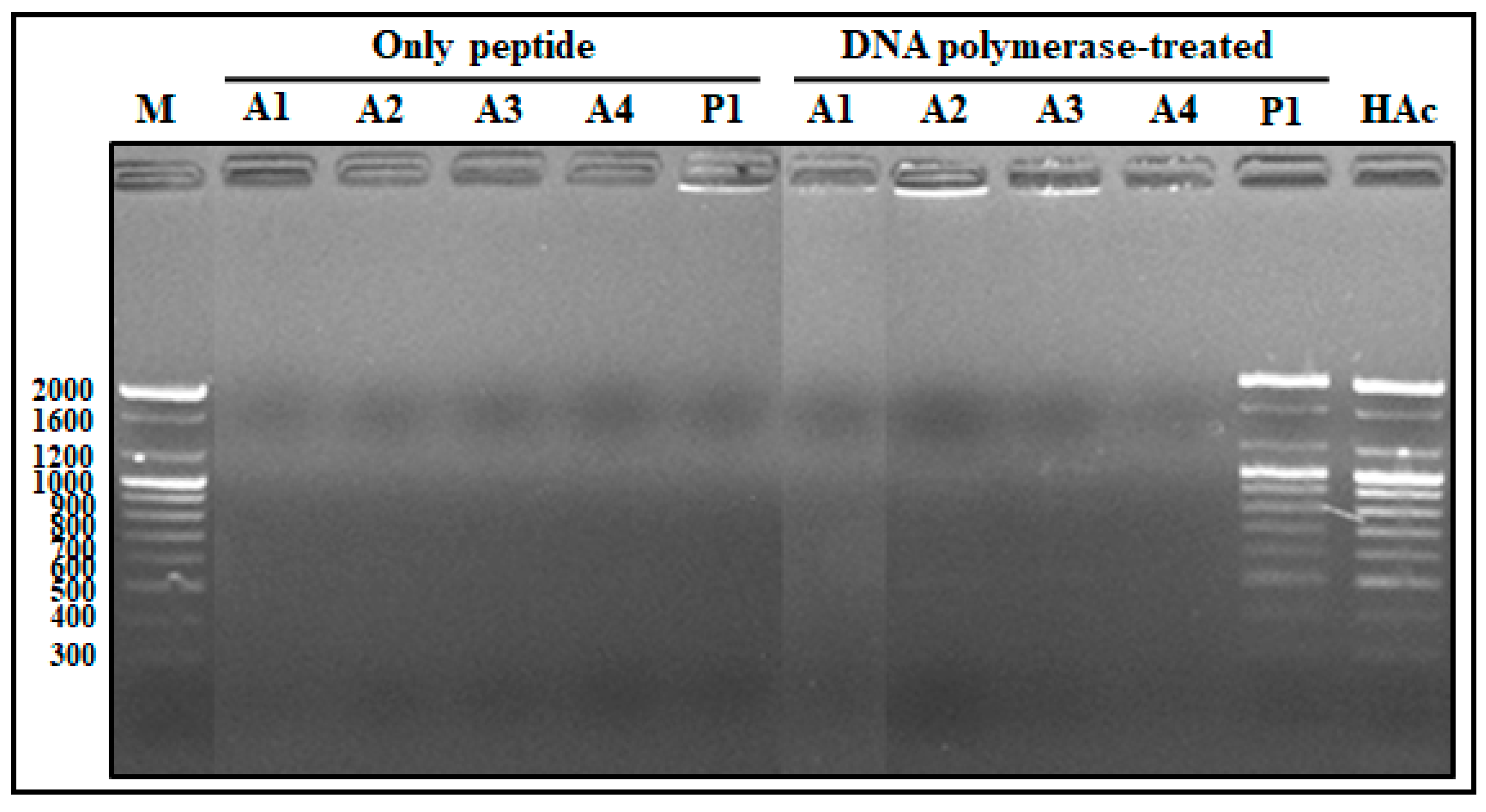
2.1.6. Competitive Binding Ability of AOD Analogs between DNA and DNA Polymerase
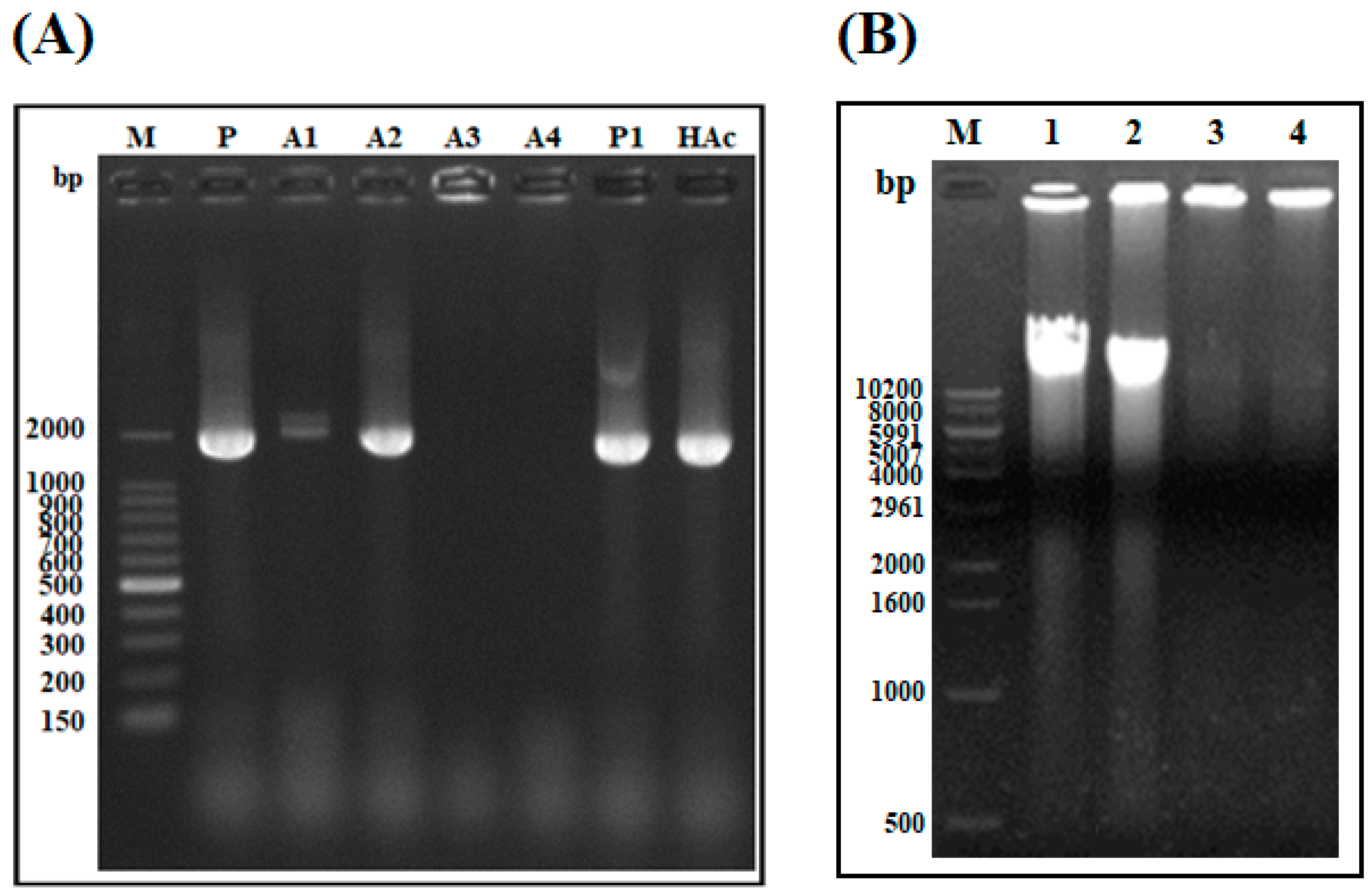
2.1.7. DNA Polymerization Inhibition Assay
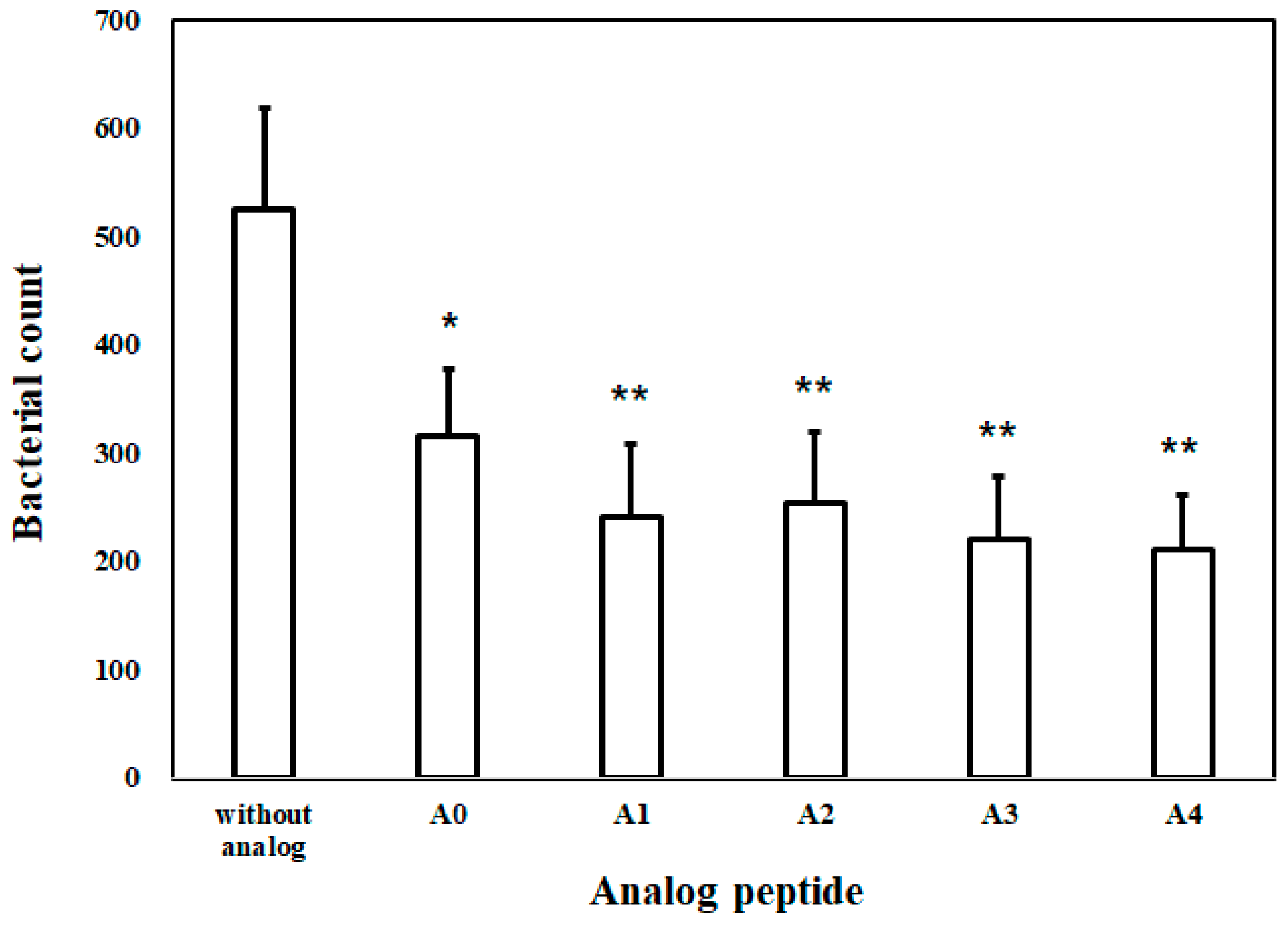
2.1.8. Bacterial Inhibition Assay (BIA) in Plasma
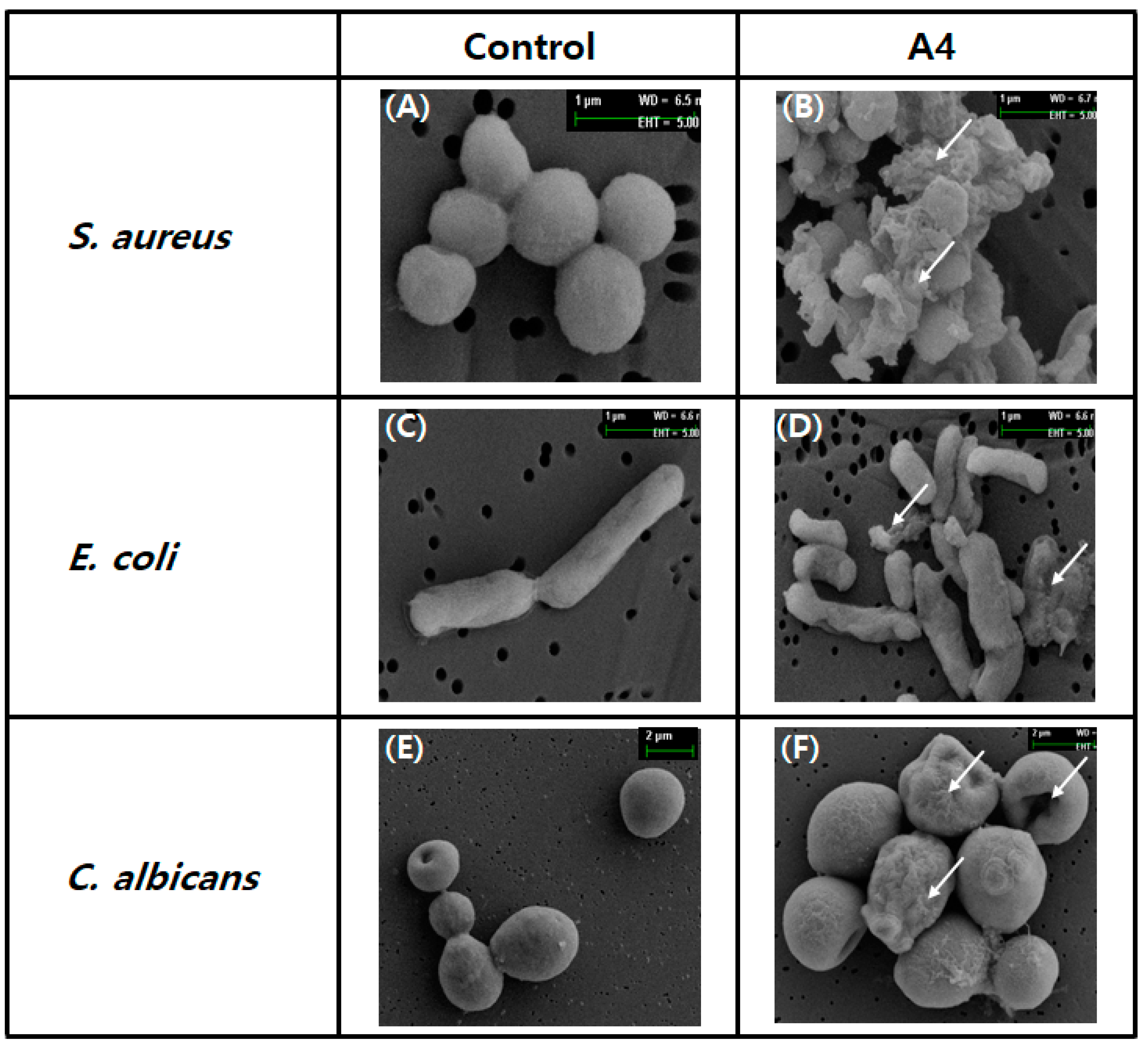
2.1.9. Damage to Microbes Observed through Electron Microscopy
3. Discussion
4. Materials and Methods
4.1. Peptide Synthesis and Purification
4.2. Ultrasensitive Radial Diffusion Assay for Antimicrobial Potency
4.3. Hemolytic Activity Assay
4.4. Effect on Human Dermal Fibroblast Cell Viability
4.5. Membrane Permeabilization
4.6. DNA Binding Assay
4.7. DNA Binding Assay and Weight Ratio of AOD Analog and DNA Needed for Interaction
4.8. Competitive Binding of Analog between DNA and DNA Polymerase
4.9. Genomic DNA Extraction and DNA Amplification Inhibition Assay
4.10. Bacterial Inhibition Assay in Mouse Plasma
4.11. Scanning Electron Microscopy
4.12. Structure Prediction and Homology Modeling
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Seo, J.K.; Go, H.J.; Moon, H.S.; Lee, M.J.; Hong, Y.K.; Jeong, H.D.; Nam, B.H.; Park, T.H.; Park, N.G. Interaction of apidaecin Ib with phospholipid bilayers and its Edwardsiella species-specific antimicrobial activity. Bull. Korean Chem. Soc. 2012, 33, 115–122. [Google Scholar] [CrossRef] [Green Version]
- Hancock, R.E.W.; Lehrer, R.I. Cationic peptides: A new source of antibiotics. Trends Biotechnol. 1998, 16, 82–88. [Google Scholar] [CrossRef]
- Hancock, R.E.W.; Chapple, D.S. Peptide antibiotics. Antimicrob. Agents Chemother. 1999, 43, 1317–1323. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.K.; Crawford, J.M.; Stone, K.L.; Noga, E.J. Purification of a novel arthropod defensin from the American oyster. Crassostrea virginica. Biochem. Biophys. Res. Comm. 2005, 338, 1998–2004. [Google Scholar] [CrossRef] [PubMed]
- Hubert, F.; Noël, T.; Roch, P. A member of the arthropod defensin family from the edible Mediterranean mussels (Mytilus galloprovincialis). Eur. J. Biochem. 1996, 240, 302–306. [Google Scholar] [CrossRef]
- Romestand, B.; Molina, F.; Richard, V.; Roch, P.; Granier, C. Key role of the loop connecting the two beta strands of mussel defensin in its antimicrobial activity. Eur. J. Biochem. 2003, 270, 2805–2813. [Google Scholar] [CrossRef] [PubMed]
- Deslouches, B.; Steckbeck, J.D.; Craigo, J.K.; Doi, Y.; Mietzner, T.A.; Montelaro, R.C. Rational Design of Engineered Cationic Antimicrobial Peptides Consisting Exclusively of Arginine and Tryptophan, and Their Activity against Multidrug-Resistant Pathogens. Antimicrob. Agents Chemother. 2013, 57, 2511–2521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, D.I.; Prenner, E.J.; Vogel, H.J. Tryptophan-and arginine rich antimicrobial peptides: Structures and mechanisms of action. BBA Biomembr. 2006, 1758, 184–202. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.H.; Chen, Y.C.; Peng, S.Y.; Tsai, A.P.; LeeT, J.; Yen, J.H.; Liou, J.W. An engineered arginine-rich alpha-helical antimicrobial peptide exhibits broad-spectrum bactericidal activity against pathogenic bacteria and reduces bacterial infections in mice. Sci. Rep. 2018, 8, 1–14. [Google Scholar]
- Walrant, A.; Bauzá, A.; Girardet, C.; Alves, I.D.; Lecomte, S.; Illien, F.; Cardon, S.; Chaianantakul, N.; Pallerla, M.; Burlina, F.; et al. Ionpair-π interactions favor cell penetration of arginine/tryptophan-rich cell-penetrating peptides. Biochim. Biophys. Acta. Biomembr. 2020, 1862, 183098. [Google Scholar] [CrossRef] [PubMed]
- Leoni, G.; Poli, A.D.; Mardirossian, M.; Gambato, S.; Florian, F.; Venier, P.; Wilson, D.N.; Tossi, A.; Pallavicini, A.; Gerdol, M. Myticalins: A novel multigenic family of linear, cationic antimicrobial peptides from marine mussels (Mytilus spp.). Mar. Drugs 2017, 15, 261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deslouches, B.; Hasek, M.L.; Craigo, J.K.; Steckbeck, J.D.; Montelaro, R.C. Comparative functional properties of engineered cationic antimicrobial peptides consisting exclusively of tryptophan and either lysine or arginine. J. Med. Microbiol. 2016, 65, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Ko, S.J.; Park, E.J.; Asandei, A.; Choi, J.Y.; Lee, S.C.; Seo, C.H.; Luchian, T.; Park, Y.K. Bee venom-derived antimicrobial peptide melectin has broad-spectrum potency, cell selectivity, and salt-resistant properties. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Skerlavaj, B.; Romeo, D.; Gennaro, R. Rapid membrane permeabilization and inhibition of vital functions of Gram-negative bacteria by bactenecins. Infect. Immun. 1990, 58, 3724–3730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrin Jr, B.S.; Tian, Y.; Fu, R.; Grant, C.V.; Chekmenev, E.Y.; Wieczorek, W.E. High-resolution structures and orientations of antimicrobial peptides piscidin 1 and piscidin 3 in fluid bilayers reveal tilting, kinking, and bilayer immersion. J. Am. Chem. Soc. 2014, 136, 3491–3504. [Google Scholar] [CrossRef]
- Han, H.; Wang, T.; Li, Z.; Teng, D.; Mao, R.; Hao, Y.; Yang, N.; Wang, X.; Wang, J. Improved Stability and Activity of a Marine Peptide-N6NH2 against Edwardsiella tarda and Its Preliminary Application in Fish. Mar. Drugs 2020, 18, 650. [Google Scholar] [CrossRef] [PubMed]
- Nam, B.H.; Shin, E.H.; Kim, Y.O.; Kim, D.G.; Kong, H.J.; Park, J.Y.; Seo, J.K. Development of novel antimicrobial peptides dericed from anti-lipopolysaccharide factor of the swimming crab, Portunus trituberculatus. Fish Shellfish. Immunol. 2019, 84, 664–672. [Google Scholar] [CrossRef]
- Cantisani, M.; Leone, M.; Mignogna, E.; Kampanaraki, K.; Falanga, A.; Morelli, G. Structure-activity relations of myxinidin, an antibacterial peptide derived from the epidermal mucus of hagfish. Antimicrob. Agents Chemother. 2013, 57, 5665–5673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huo, L.; Zhang, K.; Ling, J.; Peng, Z.; Huang, X.; Liu, H.; Gu, L. Antimicrobial and DNA-binding activity of the peptide fragments of human lactoferrin and histatin 5 against Streptococcus mutans. Arch. Oral Biol. 2011, 56, 869–876. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Chau, J.K.; Perry, N.A.; de Boer, L.; Zaat, S.A.; Vogel, H.J. Serum stabilities of short tryptophan- and arginine-rich antimicrobial peptide analogs. PLoS ONE 2010, 5, e12684. [Google Scholar] [CrossRef] [Green Version]
- Wang, G. Post-translational Modifications of Natural Antimicrobial Peptides and Strategies for Peptide Engineering. Curr. Biotechnol. 2012, 1, 72–79. [Google Scholar] [CrossRef]
- Vogel, H.J.; Schibli, D.J.; Jing, W.G.; Vogel, E.M.L.; Epand, R.F.; Epand, R.M. Towards a structure–function analysis of bovine lactoferricin and related tryptophan and arginine containing peptides. Biochem. Cell Biol. 2002, 80, 49–63. [Google Scholar] [CrossRef]
- Lee, H.; Lim, S.I.; Shin, S.H.; Lim, Y.; Koh, J.W.; Yang, S. Conjugation of Cell-Penetrating Peptides to Antimicrobial Peptides Enhances Antibacterial Activity. ACS Omega 2019, 4, 15694–15701. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, Y.; Ishibashi, J.; Yukuhiro, F.; Asaoka, A.; Taylor, D.; Yamakawa, M. Antibacterial activity and mechanism of action of tick defensin against gram-positive bacteria. Biochim. Biophys. Acta 2003, 1624, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Benfield, A.H.; Henriques, S.T. Mode-of-Action of Antimicrobial Peptides: Membrane Disruption vs. Intracellular Mechanisms. Front. Med. Technol. 2020, 2, 610997. [Google Scholar] [CrossRef]
- Lehrer, R.I.; Rosenman, M.; Harwig, S.S.I.; Jackson, R.; Eisenhaur, P. Ultrasensitive assay for endogenous antimicrobial polypeptides. J. Immunol. Meth. 1991, 137, 167–173. [Google Scholar] [CrossRef]
- Silphaduang, U.; Noga, E.J. Peptide antibiotics in mast cell of fish. Nature 2001, 414, 268–269. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.H.; Chen, C.; Jou, M.L.; Lee, A.Y.L.; Lin, Y.C.; Yu, Y.P. Structural and DNA-binding studies on the bovine antimicrobial peptide, indolicidin: Evidence for multiple conformations involved in binding to membranes and DNA. Nucleic Acids Res. 2005, 33, 4053–4064. [Google Scholar] [CrossRef] [Green Version]
- Hotta, K.; Zhu, C.B.; Phomsuwansiri, P.; Ishikawa, J.; Mizuno, S.; Hatsu, M.; Imai, S. PCR inhibition assay for DNA-targeted antibiotics. J. Antibiot. 1995, 48, 1267–1272. [Google Scholar] [CrossRef] [Green Version]
- Wilson, K. Preparation of genomic DNA from Bacteria. Curr. Protoc. Mol. Biol. 2001, 56, 2–4. [Google Scholar] [CrossRef] [PubMed]
- Dunbar, J.; Ticknor, L.O.; Kuske, C.R. Assessment of microbial diversity in four southwestern Unites States soils by 16S rRNA gene terminal restriction fragment analysis. Appled Environ. Microbiol. 2000, 66, 2943–2950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liebl, A.L.; Martin, L.B. Simple quantification of blood and plasma antimicrobial capacity using spectrophotometry. Funct. Ecol. 2009, 31, 188–201. [Google Scholar] [CrossRef]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The ClustalX windows interface: Flexible strategies for multiple alignments aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analog | Primary Structure | Length (aa) | pI a | Net C b | R c | W d | Predicted 2° Structure e |
|---|---|---|---|---|---|---|---|
| A0 | CAGSLRLTC-OH | 9 | 8.07 | +1 | 1 | 0 | e and c |
| A1 | Ac-CAGWRRLRC-NH2 | 9 | 10.4 | +3 | 3 | 1 | e and c |
| A2 | CRWRLRLRC-OH | 9 | 11.5 | +4 | 4 | 1 | e and c |
| A3 | CRRWRRRRC-OH | 9 | 12.0 | +6 | 6 | 1 | e and c |
| A4 | CRRWGWRRC-NH2 | 9 | 11.53 | +4 | 4 | 2 | e and c |
| Microbe | Gram Stain | Minimal Effective Concentration (μg/mL) a | |||||
|---|---|---|---|---|---|---|---|
| A0 | A1 | A2 | A3 | A4 | Piscidin 1 | ||
| B. subtilis KCTC1021 | + | >100.0 | 14.0 | 3.8 | 0.4 | 2.5 | 2.3 |
| C. acnes KCTC3314 | + | >100.0 | >100.0 | >100.0 | 53.7 | 27.1 | N.T. b |
| S. epidermidis KCTC1917 | + | >100.0 | >100.0 | 24.0 | 4.2 | 15.8 | N.T. |
| S. mutans KCCM40105 | + | >100.0 | 68.0 | 9.9 | 1.5 | 10.5 | 3.2 |
| A. hydrophila KCTC2358 | - | >100.0 | 17.0 | 14.0 | 3.3 | N.T. | 9.1 |
| E. coli D31 | - | >100.0 | 42.0 | 2.0 | 5.0 | 5.7 | 2.0 |
| E. coli ML35p | - | >100.0 | >100.0 | 13.0 | 6.8 | 2.2 | 2.3 |
| P. aeruginosa KCTC2004 | - | >100.0 | 15.0 | 7.6 | 3.0 | 29.5 | 8.0 |
| S. enterica KCTC2514 | - | >100.0 | 24.0 | 20.0 | 6.5 | N.T. | 4.6 |
| S. sonnei KCTC2009 | - | >100.0 | 40.0 | 18.0 | 5.5 | N.T. | 8.6 |
| C. albicans KCTC7965 | Yeast | >125.0 | >125.0 | >125.0 | 54.0 | 17.2 | 11.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seo, J.-K.; Kim, D.-G.; Lee, J.-E.; Park, K.-S.; Lee, I.-A.; Lee, K.-Y.; Kim, Y.-O.; Nam, B.-H. Antimicrobial Activity and Action Mechanisms of Arg-Rich Short Analog Peptides Designed from the C-Terminal Loop Region of American Oyster Defensin (AOD). Mar. Drugs 2021, 19, 451. https://doi.org/10.3390/md19080451
Seo J-K, Kim D-G, Lee J-E, Park K-S, Lee I-A, Lee K-Y, Kim Y-O, Nam B-H. Antimicrobial Activity and Action Mechanisms of Arg-Rich Short Analog Peptides Designed from the C-Terminal Loop Region of American Oyster Defensin (AOD). Marine Drugs. 2021; 19(8):451. https://doi.org/10.3390/md19080451
Chicago/Turabian StyleSeo, Jung-Kil, Dong-Gyun Kim, Ji-Eun Lee, Kwon-Sam Park, In-Ah Lee, Ki-Young Lee, Young-Ok Kim, and Bo-Hye Nam. 2021. "Antimicrobial Activity and Action Mechanisms of Arg-Rich Short Analog Peptides Designed from the C-Terminal Loop Region of American Oyster Defensin (AOD)" Marine Drugs 19, no. 8: 451. https://doi.org/10.3390/md19080451
APA StyleSeo, J.-K., Kim, D.-G., Lee, J.-E., Park, K.-S., Lee, I.-A., Lee, K.-Y., Kim, Y.-O., & Nam, B.-H. (2021). Antimicrobial Activity and Action Mechanisms of Arg-Rich Short Analog Peptides Designed from the C-Terminal Loop Region of American Oyster Defensin (AOD). Marine Drugs, 19(8), 451. https://doi.org/10.3390/md19080451