2. Results
Saccharobisindole (1) was obtained as a pale, yellow oil. The molecular formula of this compound (C26H28N2O3) was deduced from high-resolution fast atom bombardment mass spectrometry (HRFABMS) coupled with the detection of an ion at m/z 417.2181 [M+H]+ (calcd for C26H29N2O3, 417.2178) indicating 14 degrees of unsaturation. The infrared (IR) spectrum of this compound indicated the presence of a hydroxy group at 3431 cm−1, a carbonyl group at 1676 cm−1, and a double bond at 1638 cm−1.
The
1H NMR spectrum of
1 exhibited two sets of 1,2,4-trisubstituted aromatic protons H-6 (
δH 6.55, 1H, s), H-8 (
δH 6.80, 1H, d,
J = 7.7 Hz), H-9 (
δH 7.35, 1H, d,
J = 7.7 Hz), H-6′ (
δH 6.53, 1H, s), H-8′ (
δH 6.45, 1H, d,
J = 7.6 Hz), and H-9′ (
δH 6.09, 1H, d,
J = 7.6 Hz), four methyl singlets H-13 (
δH 1.72, 3H, s), H-14 (
δH 1.69, 3H, s), H-13′ (
δH 1.68, 3H, s), and H-14′ (
δH 1.64, 3H, s), two triplet olefinic proton signals H-11 (
δH 5.29, 1H, t,
J = 7.3 Hz) and H-11′(
δH 5.21, 1H, t,
J = 7.5 Hz), two doublet protons H-10 (
δH 3.30, 2H, d,
J = 7.9 Hz) and H-10′ (
δH 3.19, 2H, d,
J = 7.5 Hz), and one singlet proton H-3 (
δH 3.91, 1H, s). Furthermore, three exchangeable protons at NH-1 (
δH 10.08), NH-1′ (
δH 10.22), and 3′-OH (
δH 6.40) were also observed in the
1H NMR spectrum. The analysis of
13C NMR and heteronuclear single quantum coherence (HSQC) spectroscopic data indicated two carbonyl C-2 (
δC 174.6) and C-2′ (
δC 177.3), eight sp
2 quaternary C-4 (
δC 123.2), C-5 (
δC 143.4), C-7 (
δC 141.8), C-12 (
δC 131.8), C-4′ (
δC 125.8), C-5′ (
δC 142.9), C-7′ (
δC 143.1), and C-12′ (
δC 131.9), six aromatic methine C-6 (
δC 108.8), C-8 (
δC 120.9), C-9 (
δC 126.2), C-6′ (
δC 109.4), C-8′ (
δC 120.8), and C-9′ (
δC 123.5), two olefinic C-11 (
δC 123.2) and C-11′ (
δC 122.9), two methylene C-10 (
δC 33.8) and C-10′ (
δC 33.7), and four methyl singlet carbons C-13 (
δC 25.5), C-14 (
δC 17.7), C-13′ (
δC 25.4), and C-14′ (
δC 17.6), along with one sp
3 methine group C-3 (
δC 51.1) and one sp
3 quaternary carbon C-3′ (
δC 75.4) (
Table 1).
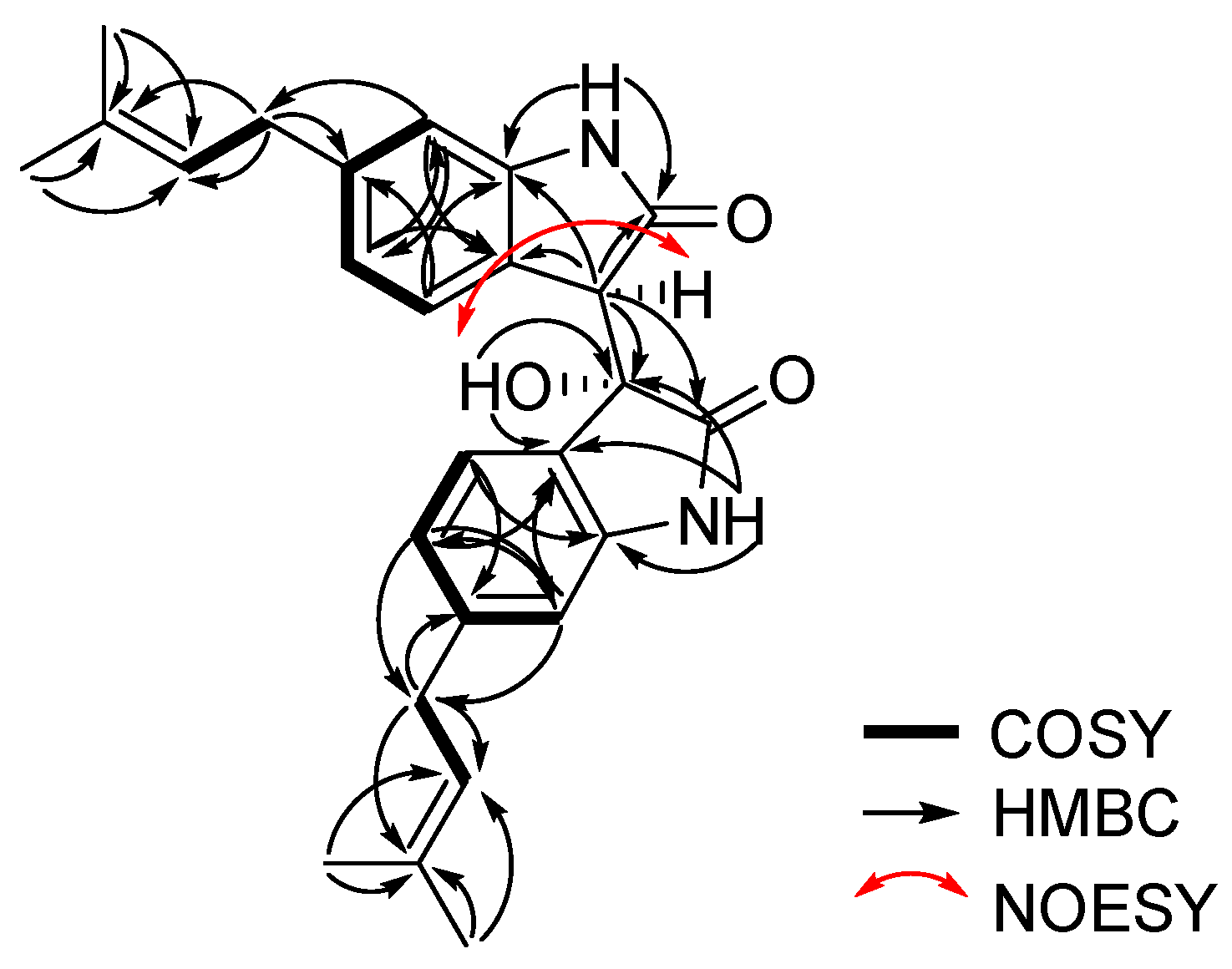
Further interpretation of 2D NMR spectroscopic data including two-bond HMBC (heteronuclear multiple bond correlation) analysis allowed for the structural construction of 1. Two sets of indolin-2-one moieties were identified by the interpretation of COSY (COrrelated SpectroscopY) and HMBC spectral data. The COSY cross peaks H-6/H-8/H-9 and H-6′/H-8′/H-9′ and the long-range HMBC correlations H-8 (δH 6.80)/ C-4 (δC 123.2), C-6 (δC 108.8); H-9 (δH 7.35)/ C-5 (δC 143.4), C-7 (δC 141.8); singlet aromatic proton H-6 (δH 6.55)/ C-4, C-8 (δC 120.9); H-8′ (δH 6.45)/ C-4′ (δC 125.8), C-6′ (δC 109.4); H-9′ (δH 6.09)/ C-5′ (δC 142.9), C-7′(δC 143.1); and H-6′ (δH 6.53)/ C-4′, C-8′(δC 120.8) confirmed the presence of two sets of 1,2,4-trisubstituted benzene moieties. Furthermore, two sets of 3-methylbut-2-enyl moieties were also identified from the interpretation of COSY and log-range HMBC analyses. COSY crosspeaks H-10/H-11 and H-10′/H-11′ and long-range HMBC correlations H-13 (δH 1.72) and H-14 (δH 1.69)/C-11 (δC 123.2), C-12 (δC 131.8); H-10 (δH 3.30)/ C-11 (δC 123.2), C-12 (δC 131.8); H-13′ (δH 1.68) and H-14′ (δH 1.64)/ C-11′ (δC 122.9), C-12′(δC 131.9); and H-10′ (δH 3.19)/ C-11′ (δC 122.9), C-12′ (δC 131.9) allowed for the assignment of two sets of 3-methylbut-2-enyl units.
One set of indolin-2-one moieties with a 3-methylbut-2-enyl group was identified via analysis of HMBC correlations (
Figure 2). HMBC correlations from H-10 to C-7 (
δC 141.8), and from H-6 (
δH 6.55) and H-8 (
δH 6.80) to C-10 (
δC 33.8) allowed for the attachment of a 3-methylbut-2-enyl group at C-7 of the benzene ring system. Further, HMBC correlations from the exchangeable NH-1 proton (
δH 10.08) to C-5 (
δC 143.4) and the carbonyl carbon C-2 (
δC 174.6), and from the methine proton H-3 (
δH 3.91) to C-2, C-4 (
δC 123.2), and C-5, allowed for the assignment of an indolin-2-one moiety with a 3-methylbut-2-enyl group. Similarly, the other indolin-2-one moiety with a 3-methylbut-2-enyl group was also assigned by interpretation of HMBC correlations. Briefly, HMBC correlations of H-10′ (
δH 3.19)/ C-7′ (
δC 143.1); H-6′ (
δH 6.53) and H-8′ (
δH 6.45)/ C-10′ (
δC 33.7); and NH-1′/ C-3′ (
δC 75.4), C-4′ (
δC 125.8), C-5′ (
δC 142.9) provided further evidence for the assignment of an indolin-2-one moiety with a 3-methylbut-2-enyl group. A hydroxy group at
δH 6.40 was attached to C-3′ based on HMBC correlations from this exchangeable proton to C-3′ and C-4′, as well as the consideration of the carbon chemical shift of C-3′ (
δC 75.4). Lastly, the connection between C-3 and C-3′ of two indolin-2-one moieties was secured from the observation of HMBC correlations from H-3 to C-3′, thus completing the assignment of
1 (
Figure 1). The relative configurations of
1 were deduced from the interpretation of nuclear Overhauser effect spectroscopy (NOESY) data. The NOESY correlation of
1 between H-3 and the 3′-OH protons indicated that the proton and hydroxy group should be placed at the same side of the rings (
Figure 2). The absolute configuration of
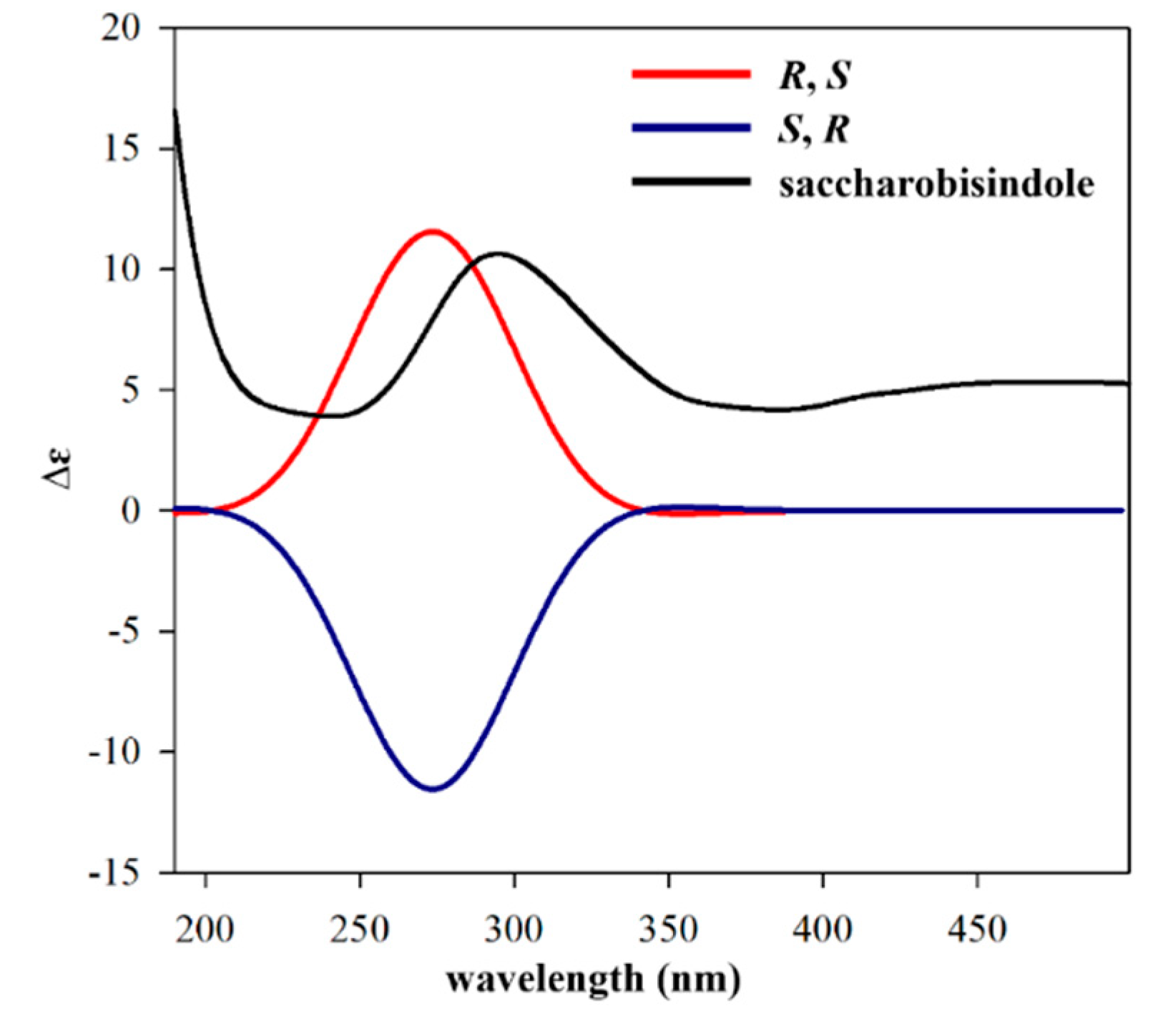
1 was determined by calculating the experimental ECD data of
1 using density functional theory (DFT) modeling. The possible enantiomers of compound
1 were selected based on NOESY NMR data. The observed ECD spectrum of compound
1 showed a positive Cotton Effect around 291 nm. Comparison of the calculated ECD data of the two enantiomers with that measured, allowed the absolute configurations of compound
1 to be assigned as
R,
S (
Figure 3). This result was also supported by comparing the experimental and calculated values of optical rotations. Compound
1 had a positive optical rotation value
= +161). The calculated values of optical rotations for four theoretical stereoisomers indicated that three stereoisomers had all negative values and only the stereoisomer with 1R,1′S configurations displayed a positive value (
= +40). Therefore,
1 was assigned as 1R,1′S configuration (
Figure S8). Compound
1 is an isatin derivative with structural differences at the side chains on two sets of indole moieties. Isatin itself was isolated from the plant
Cephalanceropsis gracilis [
13] and the deep-sea bacterium
Shewanella piezotolerans [
14].
Neoasterric methyl ester (
2) was isolated as a yellowish oil and its molecular formula was assigned as C
17H
16O
8 as determined by high-resolution fast atom bombardment mass spectrometry which illustrated a [M+H]
+ m/z value of 348.0849 (calcd for C
17H
16O
8, 348.0845), (HRFABMS). The IR absorptions at 3320 cm
−1 and 1660 cm
−1 indicated hydroxy and carbonyl functionalities, respectively. The
1H NMR spectrum showed bands for a 1,2,3,5-tetrasubstituted phenyl unit with two protons at H-2 (
δH 5.77, 1H, s) and H-4 (
δH 6.32, 1H, s), a 1,2,3,4-tetrasubstituted phenyl group at H-2′ (
δH 6.64, 1H, d,
J = 8.7 Hz) and H-3′ (
δH 6.87, 1H, d,
J = 8.7 Hz), two methoxy groups H-8 (
δH 3.75, 3H, s) and H-8′ (
δH 3.61, 3H, s), and one methyl singlet H-9 (
δH 2.08, 3H, s). The
1H,
13C and HSQC spectroscopic data revealed one methyl C-9 (
δC 21.4), two methoxys C-8 (
δC 51.8) and C-8′ (
δC 51.8), two carbonyls C-7 (
δC 166.9) and C-7′ (
δC 165.5), four aromatic methines C-2 (
δC 105.1), C-4 (
δC 109.7), C-2′ (
δC 113.3), and C-3′ (
δC 118.9), and eight quaternary carbons C-1 (
δC 156.3), C-3 (
δC 141.1), C-5 (
δC 156.0), C-6 (
δC 107.2), C-1′ (
δC 142.2), C-4′ (
δC 138.0), C-5′ (
δC 147.7), and C-6′ (
δC 116.6) (
Table 1).
Analysis of COSY and HMBC spectroscopic data allowed the chemical structure of
2 to be assigned (
Figure 4). HMBC correlations from H-2 (
δH 5.77) to C-1 (
δC 156.3), C-4 (
δC 109.7), and C-6 (
δC 107.2) and from H-4 (
δH 6.32) to C-1 and C-2 (
δC 105.1) supported the presence of a 1,2,3,5 substituted benzene moiety. Furthermore, the observation of HMBC correlations from the exchangeable proton 5-OH (
δH 10.06) to C-4, C-5 (
δC 156.0), and C-6 permitted the C-5/5-OH connectivity. HMBC correlations from the methyl singlet protons H-9 (
δH 2.08) to C-2, C-3 (
δC 141.1) and C-4 indicated that a methyl group was positioned at C-3. A methoxy group was then assigned as part of a methyl ester located at C-7 (
δC 166.9) according to analysis of the three-bond HMBC correlation from H-8 (
δH 3.75) to C-7. The long-range HMBC correlation from H-4 to C-6 (
δC 107.2) and C-7 (
δC 166.9) with the typical carbon chemical shift of C-6 (
δC 107.2) suggested the attachment of C-6 to C-7. The ortho-coupled protons (H-2′ and H-3′,
J = 8.7 Hz) displayed the COSY cross peaks H-2′/H-3′ and the HMBC correlations with C-1′ (
δC 142.2), C-5′ (
δC 147.7), and C-6′ (
δC 116.0) and C-1′, C-4′ (
δC 138.1), and C-5′, respectively. Based on these correlations, a 1,2,3,4 substituted aromatic moiety was established. The two exchangeable protons of 1′-OH (
δH 9.56) and 4′-OH (
δH 9.70) were placed at C-1′ and C-4′, respectively, based on the observation of HMBC correlations from 1′-OH to C-1′, C-2′ (
δC 113.3), and C-6′ and from the 4′-OH to C-3′ (
δC 118.9), C-4′, and C-5′. The HMBC correlation from the methoxy protons H-8′ to C-7′ (
δC 165.5) suggested the presence of a methyl ester moiety and this functional group was placed at C-6′ (
δC 116.6) based on the identification of a long-range HMBC correlation from H-2′ (
δH 6.64) to C-6′. Lastly, the linkage of two aromatic rings through an oxygen atom (C-5′/O/C-1) was deduced from the typical oxygenated carbon chemical shifts of C-1 (
δC 156.3) and C-5′ (
δC 147.7). Compound
2 is an analog of asterric acid. Methyl asterrate, an asterric acid derivative, is most structurally similar to
2 except for the hydroxy group at C-4′ and the
ortho-coupled protons (H-2′ and H-3′,
J = 8.7 Hz) [
15]. Methyl asterrate, also reported as trimethyllosoic acid, was isolated from fungi strains such as
Aspergillus sp. [
15],
Oospora sp. [
16],
Dothideomycete sp. [
17],
Phoma sp. [
18],
Pleosporales sp. [
19],
Preussia sp. [
20], and
Geomyces sp. [
21]. Methyl asterrate was found to display low cytotoxic activity in the HepG2 cell line with 22% growth inhibition at a 100 μg/mL concentration [
17].
7-Chloro-4(
1H)-quinolone (
3) was isolated as a white amorphous powder and its chemical formula was assigned as C
9H
635ClNO based on the observation of a protonated molecular ion peak at
m/z 180.0218 [M+H]
+ (calcd for C
9H
635ClNO, 180.0216) in the HRFAB mass spectrum. The
1H NMR spectrum of
3 displayed two deshielded olefinic protons H-2 (
δH 8.01, 1H, d,
J = 7.3 Hz) and H-3 (
δH 6.37, 1H, d,
J = 7.3 Hz), and a 1,3,4-trisubstituted benzene ring with protons at H-5 (
δH 8.24, 1H, d,
J = 8.8 Hz) and H-6 (
δH 7.42, 1H, dd,
J = 8.8, 1.9 Hz), and H-8 (
δH 7.63, 1H, d,
J = 1.9 Hz). The
13C NMR spectrum exhibited nine carbons C-2 (
δC 142.2), C-3 (
δC 110.3), C-4 (
δC 179.8), C-4a (
δC 125.1), C-5 (
δC 128.3), C-6 (
δC 126.1), C-7 (
δC 142.2), C-8 (
δC 118.9), C-8a (
δC 139.8). A quinolone moiety was identified based on the interpretation of COSY and HMBC spectroscopic data. The COSY correlation of H-2 (
δH 8.01) with H-3 (
δH 6.37) and long-range HMBC correlations from H-2 to C-4 (
δC 179.8) indicated the presence of an α,β-unsaturated carbonyl group. Furthermore, three-bond HMBC correlations from H-2 to C-8a and from H-3 to C-4a, along with the carbon chemical shifts of C-2 (
δC 142.2) and C-8a (
δC 142.2), and correlations from H-6 to C-4a (
δC 125.1) and C-8 (
δC 118.9), and from H-5 to C-4, C-7 (
δC 142.2) and C-8a (
δC 139.8) allowed for the construction of the quinolone moiety. The carbon chemical shift at
δC 142.2 and the isotope ratio (3:1) of two pseudomolecular ion peaks [M + H]
+ and [M + H + 2]
+ in the LR-ESI-MS spectroscopic data (
Figure S19), allowed the attachment of a chlorine atom at C-7. Compound
3 was assigned as 7-chloro-4(
1H)-quinolone (
Figure 4). The structural assignment of
3 was completed by comparing the NMR data with the data from the literature [
22].
Compound
3 has been reported as a synthesized product [
22] but this is the first report from a natural source (i.e., a bacterium). Compound
3 was approved as an antitumor drug for the treatment of late mammary cancer and non-small cell lung cancer by the SFDA (State Food and Drug Administration of China) due to its capacity to damage DNA and block DNA synthesis in tumor cells [
23]. Furthermore,
3 was found to exhibit weak inhibitory activity on cysteine protease [
24].
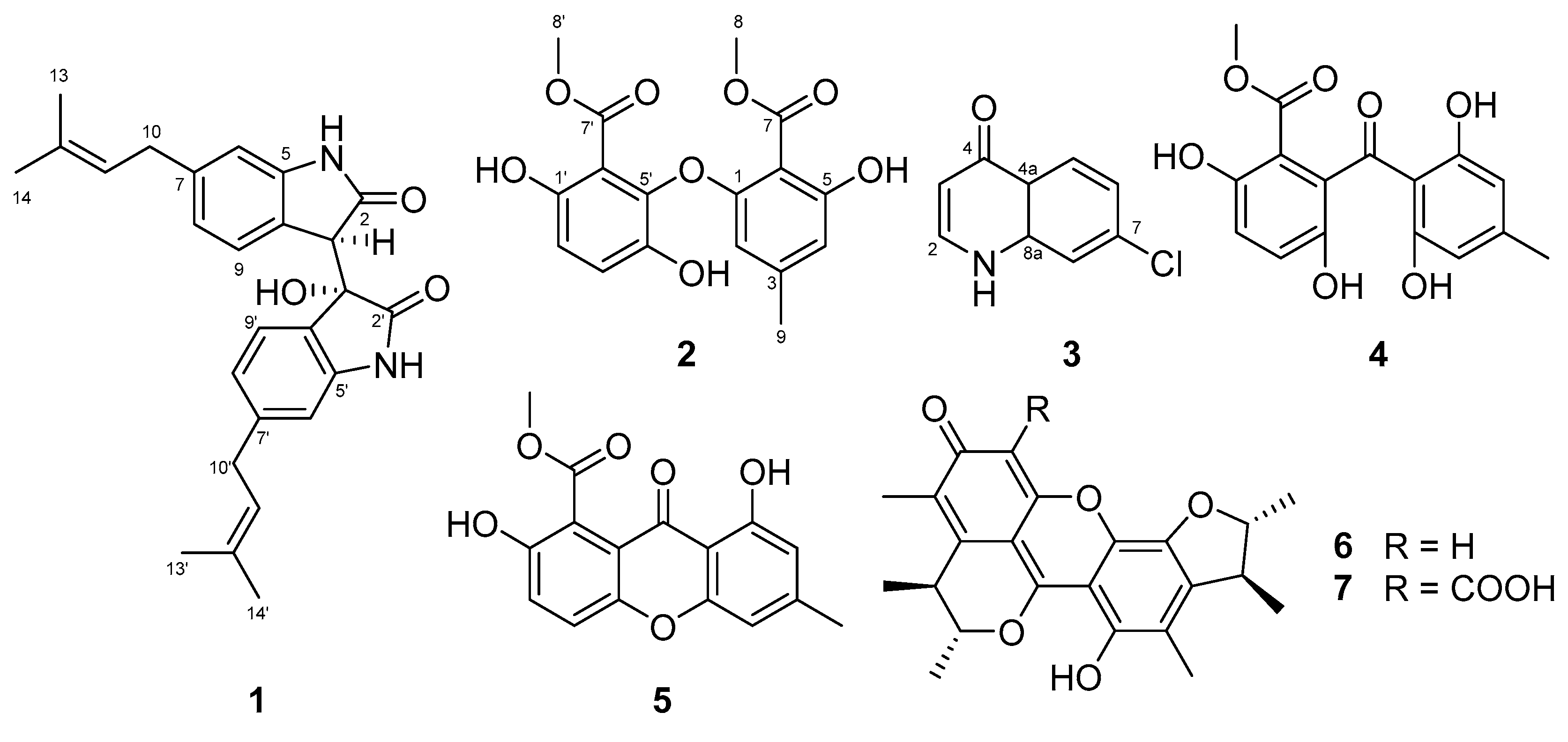
In addition to
1–
3, four known natural products were isolated and identified as acremonidin E (
4), pinselin (
5), penicitrinone A (
6), and penicitrinone E (
7) by comparing their NMR and MS spectroscopic data with those of previously reported compounds. Interestingly, the known compounds were originally isolated from fungi and had never been reported to be produced by actinomycetes. Acremonidine E (
4), penicitrinon A (
6), and penicitrinon E (
7) were isolated from
Penicillium sp. [
25,
26,
27], whereas pinselin (
5) was previously identified in the endophytic fungus
Phomopsis sp. [
28], the marine fungus
Scopuariopsis sp. [
29], and the soil fungus
Penicillium sp. [
30]. In this study, a large-scale regrowth of the strain CNQ-490 allowed us to identify the minor compounds (
1–
7) produced by this strain as well as to rediscover the major compounds such as lodopyridones A–C, sacchromonopyrones A–C, and saccharoquinoline [
7,
8,
9,
10]. These data suggest that strain CNQ-490 has very good potential in producing biosynthetically-diverse secondary metabolites.
Compounds
1–7 were tested for their antibacterial activities against three Gram-positive bacteria (
Bacillus subtilis KCTC1021,
Kocuria rhizophila KCTC1915, and
Staphylococcus aureus KCTC1927), and three Gram-negative bacteria (
Escherichia coli KCTC2441,
Salmonella typhimurium KCTC2515, and
Klebsiella pneumoniae KCTC2690) (
Table 2). In addition, quorum sensing (QS) inhibitory activity was evaluated with compounds
2, and
4–7 against six pathogenic bacteria (
Cobetia marina strain JEA023,
S. aureus KCTC1927,
Micrococcus luteus SCO560,
Pseudomonas aeruginosa SNC165,
Pseudomonas fluorescens SND204, and
Agrobacterium tumefaciens SND195) (
Table 3). The QS inhibitory activities of compounds
1 and
3 were not investigated, as these compounds could only be recovered in limited amounts. Compound
7 displayed strong antibacterial activity and weak QS inhibitory effect against
S. aureus KCTC1927 with the values of 2.0 μg/mL and 64 μg/mL, respectively. Compound
2 performed weak inhibitory activities against
B. subtilis KCTC1021 and
S. aureus KCTC1927, with minimum inhibitory concentration (MIC) values of 32 μg/mL and 64 μg/mL, respectively. Furthermore, Compounds
5 and
6 exhibited strong QS blocking activities against
C. marina JEA023 with the value 0.5 μg/mL and 1.0 μg/mL, respectively. Meanwhile, compounds
2,
4, and
6 exhibited QS blocking activities against
M. luteus SCO560 at a 1–64 μg/mL range. None of the evaluated compounds exhibited QS inhibitory effects against
P. aeruginosa SNC165,
P. fluorescens SND204, and
A. tumefaciens SND195.
3. Materials and Methods
3.1. General Experimental Procedures
UV spectra were recorded in MeOH on a Scinco UVS2100 spectrophotometer. IR spectra were collected using a Nicolet iS10 FT-IR spectrometer (Thermo Scientific Inc., Waltham, MA, USA). NMR spectra were obtained using an Agilent NMR spectrometer (Agilent, Santa Clara, CA, USA, at 400 for 1H and at 100 MHz for 13C) and a Bruker NMR spectrometer (Bruker, Middlesex, MA, USA, at 300 for 1H and and 75 MHz for 13C) using the signals of the residual solvent as internal references (δH 2.50 ppm and δC 39.5 ppm for dimethyl sulfoxide-d6 (DMSO-d6) and δH 4.87 and 3.31 ppm and δC 49.1 ppm for deuterated methanol (CD3OD). Low-resolution LC/MS measurements were performed using the Agilent Technologies 1260 quadrupole and Waters Micromass ZQ LC/MS system using a reversed-phase column (Phenomenex Luna C18 (2) 100 Å, 50 mm × 4.6 mm, 5 μm) at a flow rate of 1.0 mL/min at the National Research Facilities and Equipment Center (NanoBioEnergy Materials Center) at Ewha Womans University. Open column chromatography was performed using silica (40–63 μm, Merck silica gel 60) eluting with a gradient solvent of dichloromethane (CH2Cl2) and methanol (MeOH). The fractions were purified via semi-preparative HPLC using a Waters 996 Photodiode Array Detected HPLC coupled with a reversed-phase Phenomenex Luna C18 (2) (100 Å, 250 nm × 10 mm, 5µm) column at a 2.0 mL/min flow rate. High-resolution mass spectra were recorded on a JMS-700 mass spectrometer (JEOL Ltd., Tokyo, Japan) at Seoul National University.
3.2. Strain Isolation and Fermentation
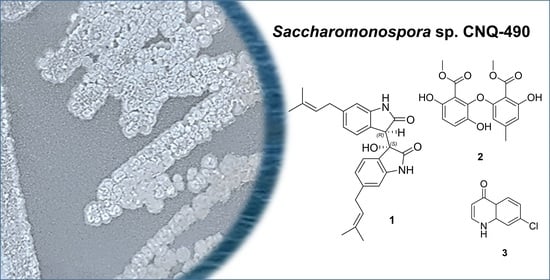
Strain CNQ-490 is an actinomycete that was isolated from a marine sediment sample obtained at a depth of 45 m from a submarine canyon in La Jolla, CA. Strain CNQ-490 was assigned to the genus Saccharomonospora based on 16S ribosomal DNA gene sequence similarity analyses and BLAST searches (GeneBank accession number EU214929). A total of 80 L of CNQ-490 was cultured in 20 × 2.5 L Ultra-Yield flasks (Thomson Scientific, Oceanside, CA, USA) each containing 1 L of the medium (10 g/L soluble starch, 2 g/L yeast, 4 g/L peptone, 10 g/L CaCO3, 20 g/L KBr, 8 g/L Fe2(SO4)3·4H2O dissolved in 750 mL natural seawater and 250 mL of distilled water) at 27 °C with constant shaking at 150 rpm. After 7 days of cultivation, the broth was extracted with ethyl acetate (EtOAc, 80 L overall) and the EtOAc-soluble fraction was dried in vacuo to obtain 6.5 g of organic extract.
3.3. Purification
The organic extract of CNQ-490 was fractionated by reversed-phase C-18 flash vacuum chromatography eluting with a step gradient from 0 to 100% methanol in water, which resulted in 11 fractions. Both fractions 3 and 4 (eluted with 9% and 10% MeOH in water, respectively) were further purified by HPLC (Phenomenex 100 Å, 250 nm × 10 mm, 5 µm, UV = 285 nm), with 60% of acetonitrile in H2O at a flow rate of 2.0 mL/min to yield 1 (1.2 mg, tR 33.7 min), 45% of acetonitrile in H2O at a flow rate 2.0 mL/min to yield 2 (4.1 mg, tR 27.5 min), 4 (40.3 mg, tR 17.0 min), 5 (6.0 mg, tR 40.0 min), 6 (4.8 mg, tR 18.0 min), and 7 (6.7 mg, tR 35.5 min), and 25% of acetonitrile in H2O at flow rate 2.0 mL/min to yield 3 (1.7 mg, tR 33.6 min).
Saccharobisindole (
1): Pale yellow oil,
= +161 (
c 0.29, MeOH), UV (MeOH) λ
max (log ε) 202 (2.93), 253 (2.64), 271 (2.08) nm; IR (KBr) ν
max 3431, 2935, 1676, 1638, 1444, 1196, 994 cm
−1,
1H and
13C NMR data:
Table 1 and
Figures S1–S5; HR-FAB-MS
m/z 417.2181 [M+H]
+ (calculated for C
26H
29N
2O
3, 417.2178).
Neoasterric methyl ester (
2): Yellowish oil, UV (MeOH) λ
max (log ε) 216 (2.42), 262 (1.93), 397 (1.65) nm; IR (KBr) ν
max 3320, 1660, 1616, 1439, 1314, 1201, 1077 cm
−1,
1H and
13C NMR data:
Table 1 and
Figures S8–S12; HR-FA-BMS
m/z 348.0849 [M+H]
+ (calculated for C
17H
16O
8, 348.0845).
7-Chloro-4(1H)-quinolone (
3): White amorphous powder, UV (MeOH) λ
max (log ε) 218 (2.58), 278 (1.87), 279 (1.77) nm; IR (KBr) ν
max 3326, 1729, 1457, 802 cm
−1,
1H and
13C NMR data:
Table 1 and
Figures S13–S17; HR-FAB-MS m/z 180.0218 [M+H]
+ (calculated for C
9H
635ClNO, 180.0216).
Acremonidine E (4): 1H NMR: (400 MHz, DMSO-d6); δH 11.36 (s, 2-OH), 11.36 (s, 6-OH), 9.70 (s, 1′-OH), 9.18 (s, 4′-OH), 6.94 (d, J = 8.8 Hz, H-5′), 6.80 (d, J = 8.8 Hz, H-6′), 6.10 (s, H-5), 6.10 (s, H-6), 3.54 (s, H-8′), 2.16 (s, H-7), 13C (100 MHz, DMSO-d6); δC 180.2 (C-8), 167.9 (C-7′), 161.5 (C-2), 161.5 (C-6), 151.1 (C-1′). 147.7 (C-4), 145.6(C-4′), 131.2 (C-3′), 122.1 (C-5′), 117.4 (C-6′), 112.3 (C-2′), 108.9 (C-1), 107,4 (C-3), 107.4 (C-5), 51.9 (C-8′), 21.6 (C-7), LR-ESI-MS m/z 319.1 [M+H]+.
Pinselin (5): 1H NMR:(400 MHz, DMSO-d6); δH 12.18 (s, 2-OH), 10.47 (s, 1′-OH), 7.45 (d, J = 9.1 Hz, H-5′), 7.60 (d, J = 9.1 Hz, H-6′), 6.89 (s, H-5), 6.65 (s, H-3), 3.84 (s, H-8′), 2.39 (s, H-7); 13C (100 MHz, DMSO-d6); δC 180.2 (C-8), 166.8 (C-7′), 160.4 (C-2), 155.4 (C-6), 150.7 (C-1′), 149.3 (C-4), 148.8 (C-4′), 125.3 (C-5′), 120.1 (C-6′), 117.2 (C-3′), 117.0 (C-2′), 110.7 (C-3), 107.4 (C-5), 106.0 (C-1), 52.2 (C-8′), 22.0 (C-7), LR-ESI-MS m/z 301.1 [M+H]+.
Penicitrinon A (6): 1H NMR: (400 MHz, CD3OD); δH 8.36 (s, 6′-OH), 6.68 (s, H-7), 5.38 (q, J = 6.8 Hz, H-3), 4.71 (dq, J = 6.7 and 4.0 Hz, H-2′), 3.48 (q, J = 6.8 Hz, H-4), 3.36 (dq, J = 6.7 and 4.0 Hz, H-3′), 2.30 (s, 5′-CH3), 2.22 (s, 5-CH3), 1.45 (d, J = 6.8 Hz, 3-CH3), 1.42 (d, J = 6.7 Hz, 2′-CH3), 1.35 (d, J = 6.7 Hz, 3′-CH3), 1.34 (d, J = 6.8 Hz, 4-CH3), 13C (100 MHz, CD3OD); δC 177.8 (C-6), 167.5 (C-1), 160.3 (C-8), 150.7(C-6′), 145.6 (C-4′), 138.5 (C-7′a), 137.4 (C-5), 128.8 (C-4a), 120.1 (C-5′), 105.7 (C-7′), 102.7 (C-8a), 101.6 (C-7), 89.0 (C-2′), 86.3 (C-3), 46.3 (C-3′), 45.8 (C-4), 21.2 (2′-CH3), 19.5 (3′-CH3), 18.9 (3-CH3), 18.8 (4-CH3), 12.1(5′-CH3), 10.7 (5-CH3), LR-ESI-MS m/z 381.1 [M+H]+.
Penicitrinon E (7): 1H NMR: (400 MHz, CD3OD); δH 5.17 (q, J = 6.8 Hz, H-3), 4.77 (dq, J = 6.5 and 4.2 Hz, H-2′), 3.29 (q, J = 6.8 Hz, H-4), 3.25 (dq, J = 6.5 and 4.2 Hz, H-3′), 2.28 (s, 5′-CH3), 2.22 (s, 5-CH3), 1.49 (d, J = 6.8 Hz, 3-CH3), 1.47 (d, J = 6.7 Hz, 2′-CH3), 1.38 (d, J = 7.0 Hz, 4-CH3), 1.36 (d, J = 7.0 Hz, 3′-CH3), LR-ESI-MS m/z 425.1 [M+H]+.
3.4. Minimum Inhibitory Concentration (MIC) against Gram-Positive and Gram-Negative Bacteria
For the antibacterial susceptibility assays, compounds 1–7 were tested against three Gram-positive bacteria (B. subtilis KCTC1021, K. rhizophila KCTC1915, and S. aureus KCTC1927) and three Gram-negative bacteria (E. coli KCTC2441, S. typhimurium KCTC2515, and K. pneumoniae KCTC2690) following the recommendations of previous studies. The bacterial inoculum was prepared in Muller-Hinton broth at 37 °C and 225 rpm for 24 h. The stock solution of compounds 1–7 and the positive control were dissolved at a 10 mg/mL concentration in DMSO and diluted with Muller-Hinton broth to obtain a 0.25–256 µg/mL concentration range. Bacterial inoculum (5 × 105 CFU/mL concentration) was then dispensed into each well. The resulting mixtures consisting of varying concentrations of compounds 1–7 with a 1 × 106 CFU/mL concentration of bacterial inoculum were transferred to 96 well microtiter plates and incubated at 37 °C for 24 h.
3.5. Detection of Anti-Quorum Sensing Activity
The 96-well plate method was employed to detect the anti-quorum sensing activity of different compounds against six bacterial strains (C. marina JEA023, M. luteus SCO560, S. aureus KCTC1927, P. aeruginosa SNC165, P. fluorescens SNA239, and A. tumefaciens SND195). S. aureus KCTC1927 and A. tumefaciens SND195 were cultured in tryptic soy broth (TSB), whereas P. aeruginosa SNC165 and P. fluorescens SND204 were cultured in King’s broth (KB). C. marina JEA023 and M. luteus SCO560 were cultured in marine broth maintaining a 0.5 McFarland standard turbidity (1.0 × 108 CFU/mL). Dimethyl sulfoxide (100% DMSO) was used as a negative control, and kanamycin and rifampin (10 mg/mL each) were used as a positive control. Next, 50 µL of inoculum was inoculated into each well and 10 mg/mL of compound 2 and 4–7 dissolved in DMSO was diluted with marine broth media to produce concentrations ranging from 0.25 to 256 µg/mL. The mixtures of compounds and broth were dispensed into each inoculated well. The 96-well plates were then incubated for 16–18 h at 37 °C depending on the bacterial strains.
3.6. Conformational Analysis and ECD Spectrum Calculations
A conformational analysis of saccharobisindole (
1) was carried out by MacroModel with the Merck molecular force field (gas phase), a 10 kJ/mol upper energy limit, and a 0.001 kJ (mol Å)
−1 convergence threshold on the rms gradient to minimize computational complexity and expense. The possible enantiomers of
1 were selected based on NOESY NMR data and the energy-minimized enantiomer structures were generated by Avogadro 1.2.0. Energy minimization of the two structures was performed by Turbomole X 4.3.2. The calculated ECD spectra corresponding to two enantiomer models were calculated using DFT at the functional B3LYP/DFT level and the def-SV(P) basis set. The ECD spectra were simulated by overlapping each transition, where
σ is the width of the band at 1/
e height. ∆
Ei is the excitation energies and
Ri is rotatory strengths for transition
i. In this calculation, the
σ value was at 0.10 eV. The observed ECD spectrum of compound
1 showed positive cotton effect around 291 nm. Comparing the calculated spectra of the two enantiomers with the measured ECD spectrum, the absolute configurations of compound
1 were deduced as
R,
S.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}