1. Introduction
Colorectal cancer (CRC) is by far the third most common malignant tumor, and it ranks as the second leading cause of death from cancer in the world [
1]. CRC deaths have ameliorated over the past few decades on account of headways in both early diagnosis and intervention; however, long-term declines in mortality have slowed during last five years [
2,
3]. At an early stage, the five-year survival rate of CRC is higher than 80%; nevertheless, it decreases to approximately 10% at the advanced stages [
1]. Therefore, it is urgent to develop effective anti-CRC agents with low toxicity to effectively treat and prevent the recurrence and metastasis of CRC.
The ocean is home to most of the world’s life. Diverse marine organisms live and thrive in an aquatic buffer system despite the many extreme physiologic stresses, such as high salinity, high pressure, and extreme hypoxia. Living in such a special environment, many marine organisms have evolved with special adaptions and have become an affluent source of bioactive substances with broad health benefits [
4]. Due to the specific environment in which marine organisms live, the structure of marine antitumor peptides is very different from that of terrestrial animal and plant peptides. Most of them are small molecular cyclic or linear peptides that are rich in polar amino acids, D-type amino acids, hydroxyl acids, thiophenol, and oxazole rings. Some also contain alkene and alkyne bonds, which greatly improve the biological stability and bioavailability of the peptides. Didemnin B is a phenolic peptide compound isolated from the tunicate
Trididem numsolidum from the Caribbean [
5]. It has been reported that the real source of didemnin B may be cyanobacteria symbiotic with the tunicates. Didemnin B can directly bind to palmitoyl protein thioesterase, and exhibits strong activity in multiple tumor models [
6]. Pettit et al. isolated a pentapeptide, dolastatin 10, from
Dolabella auricularia. Dolastatin 10 is a secondary metabolite of the cyanobacteria
Sym ploca, symbiotic with the sea hare [
7]. Dolastatin 10 has excellent antitumor activity and can strongly inhibit the binding of vinblastine to tubulin noncompetitively (Kd = 1.41 μM) and can greatly affect microtubule assembly and microtubule-dependent guanosine triphosphate hydrolysis [
8]. In addition, Dolastatin 15 is a cyanobacterial polypeptide isolated from the sea hare
D. Auricularia from the Indian Ocean. It is a linear peptide containing seven amino acids or hydroxyl acid residues. Unlike dolastatin 10, dolastatin 15 binds directly to the vinblastine binding site of tubulin to exhibit prominent antitumor efficacy [
9]. Moreover, natural peptides derived from marine organisms, some of which have been shown to act as signaling/regulatory molecules in various physiological processes, also exhibit potential antitumor effects [
10]. Among them, lysine-rich peptides have shown selective cytotoxicity against both microbes and cancer cells [
11]. Several studies have demonstrated that lysine-rich peptides have modes of action involving cellular uptake, mitochondrial apoptosis, and other intracellular events [
12,
13,
14]. However, the mechanisms behind the cytotoxic and antitumor effects of lysine-rich peptides remain unclear.
Arca inflata Reeve is a bivalve mollusk of the Arcidae family that prevails along the coastlines of Asia. The farming production of
A. inflata reaches up to 350,000 tons per year in China. Furthermore,
A. inflata is a traditional marine Chinese medicine that has several therapeutic functions, including in cancer treatment. Previously, we identified two antitumor polypeptides and a glucanase from
A. inflata [
15,
16,
17]. However, the antitumor mechanism in
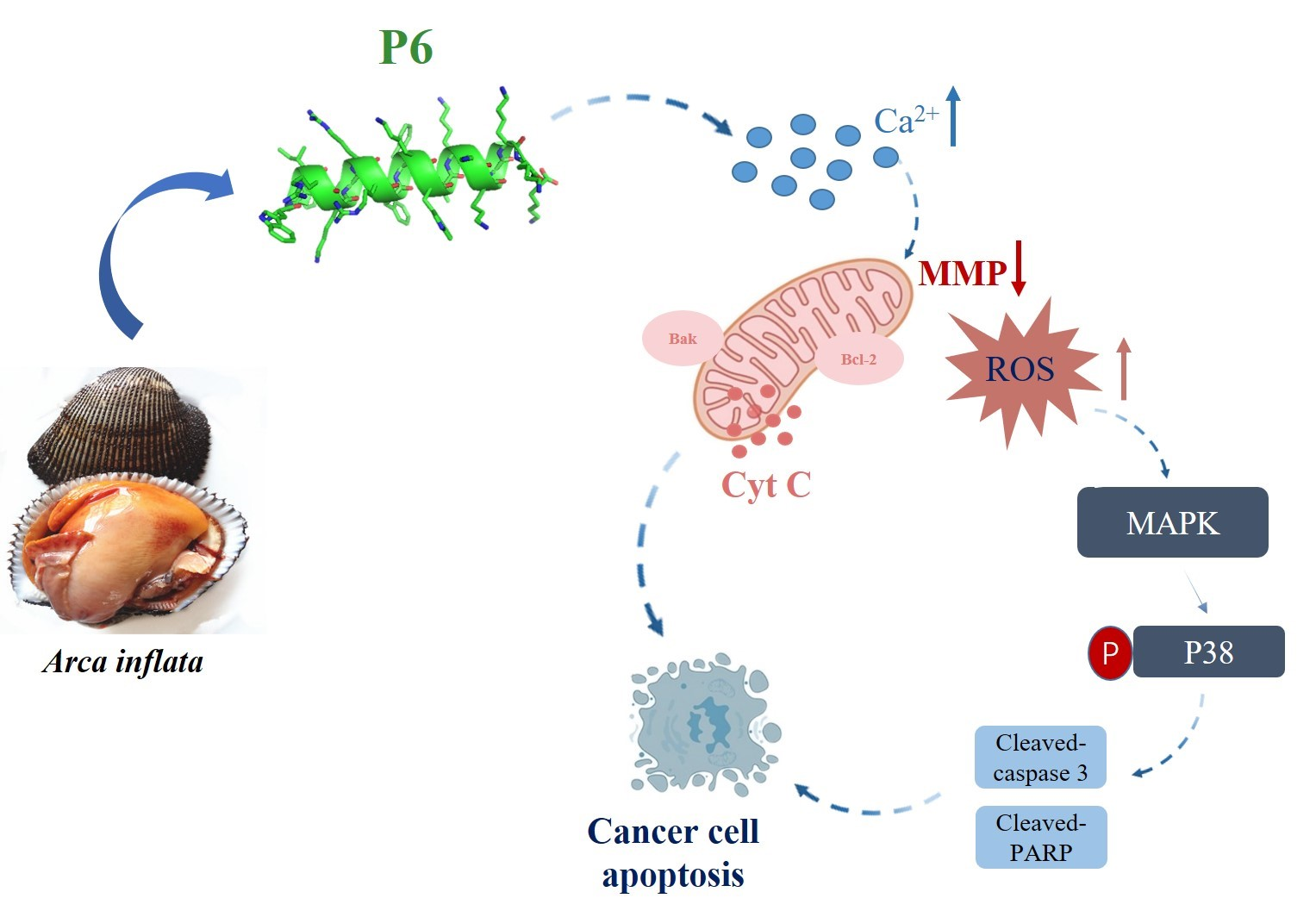
A. inflata-derived peptides remains insufficiently understood. In this study, we found that a new lysine-rich peptide, P6, displayed potent anticancer activity towards HT-29 and DLD-1 colorectal cancer cells. P6 induced Ca
2+ overload, ROS production, and mitochondrial dysfunction, thereby triggering apoptosis through the activation of the p38-MAPK signaling pathway. Moreover, P6 exerted antitumor effects in a tumor xenograft model.
3. Discussion
Colorectal cancer (CRC) kills nearly one million people each year and is becoming the world’s second most deadly cancer. As a consequence, CRC has produced a considerable disease burden and has become a major global public health problem [
24]. At the present, 5-fluorouracil and oxaliplatin are still the first choice in the clinical chemotherapy treatment of CRC, including in the National Comprehensive Cancer Network (NCCN) Guidelines of CRC (version 2020.V1) [
25]. However, the most serious treatment obstacle is the toxic side effects of 5-fluorouracil and oxaliplatin. For advanced CRC, there are only VEGFR receptor-targeted drugs (such as bevacizumab) and EGFR receptor-targeted drugs (such as cetuximab) on the market to date, which only have the effect of prolonging the approximately 3–5-months of survival for patients [
26,
27,
28]. Additionally, due to their high treatment cost and significant side effects, these targeted drugs have greatly limited clinical applications [
26,
27,
28]. Therefore, it is urgent to develop effective anti-CRC agents with low toxicity to effectively treat and prevent the recurrence and metastasis of CRC. Here, we report that P6, a novel peptide identified from the marine mollusk
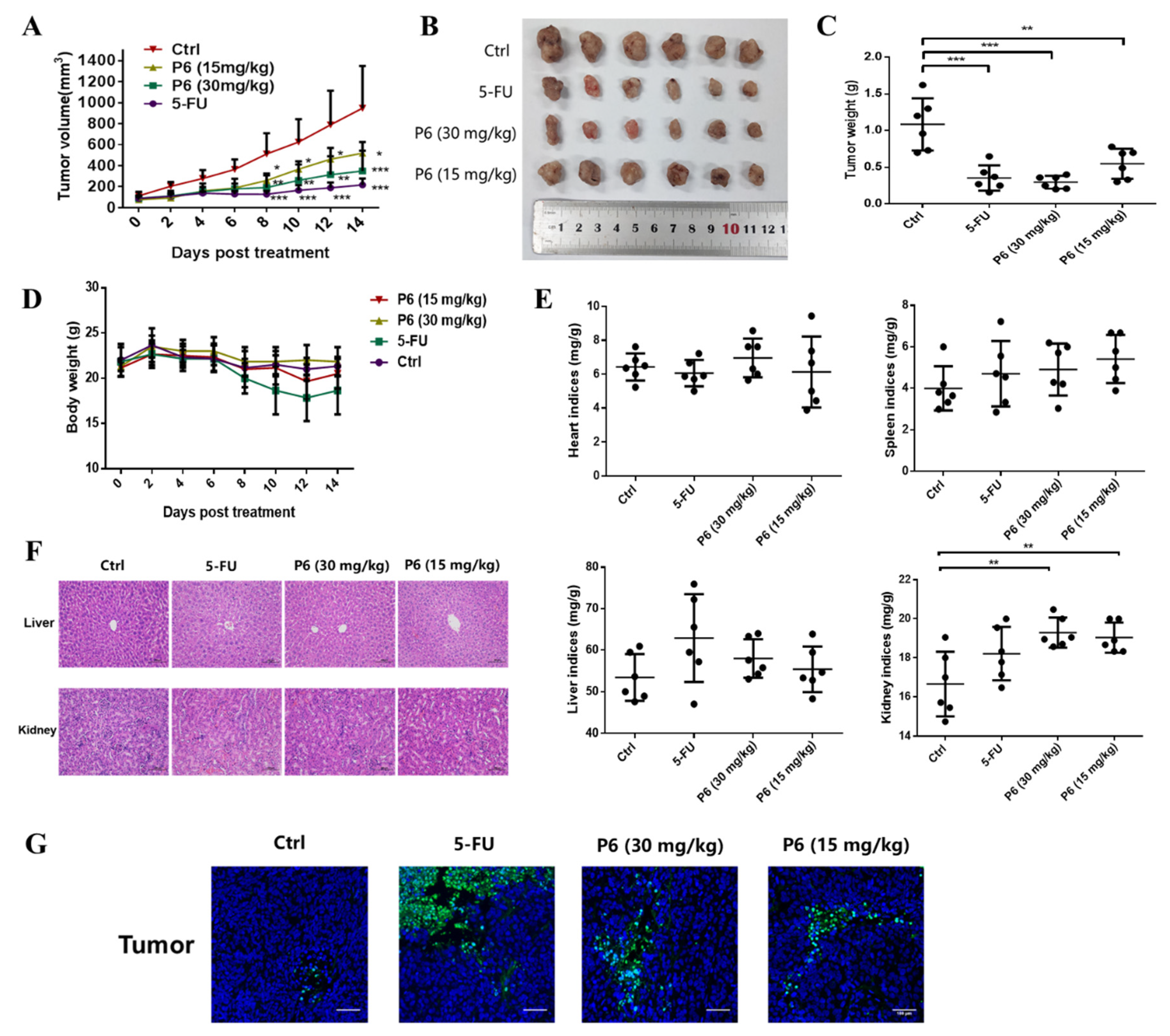
Arca inflata, was effective at suppressing CRC proliferation by inducing mitochondrial apoptosis (
Figure 5). Moreover, P6 exhibited a prominent antitumor effect in the tumor xenograft model (
Figure 7). Our study provides preliminary proof that P6 could be developed as a drug candidate for treating CRC.
Apoptosis, also known as programmed cell death, is the main mechanism for tumor cell death under the action of chemotherapeutic drugs [
29]. An abnormal apoptosis signaling pathway, the loss of apoptosis signals, or the enhancement of anti-apoptosis signals can trigger a variety of pathological changes, leading to cancer or chemotherapy failure [
30]. Under normal circumstances, apoptosis cannot trigger inflammation and the immune response; thus, promoting apoptosis has become an important strategy for tumor treatment [
31]. There are two main apoptotic pathways in mammals, the death receptor pathway [
32,
33,
34,
35] and the mitochondrial pathway [
36,
37]. There is a cross between the two pathways, and certain signaling molecules can participate in different apoptotic pathways. In recent years, nearly a hundred peptides with antitumor activity have been discovered in marine organisms, of which more than 90% trigger apoptosis through targeted apoptotic mechanisms involving the mitochondria and death receptor pathways. Both apoptotic pathways require the mitochondrial-mediated activation of caspases. The mitochondrial pathway is the main mechanism of apoptosis induced by many antitumor drugs. The activation of mitochondrial apoptosis pathways is primarily due to several factors, including ROS production and Ca
2+ overload in the cells [
38]. When the Ca
2+ homeostasis in tumor cells is disrupted, high levels of reactive oxygen species (ROS) are produced, resulting in the activation of cell apoptosis [
38,
39]. Interestingly, P6 showed a profound antitumor effect and induced tumor cell apoptosis by promoting ROS production and intracellular Ca
2+ overload in CRC cells (
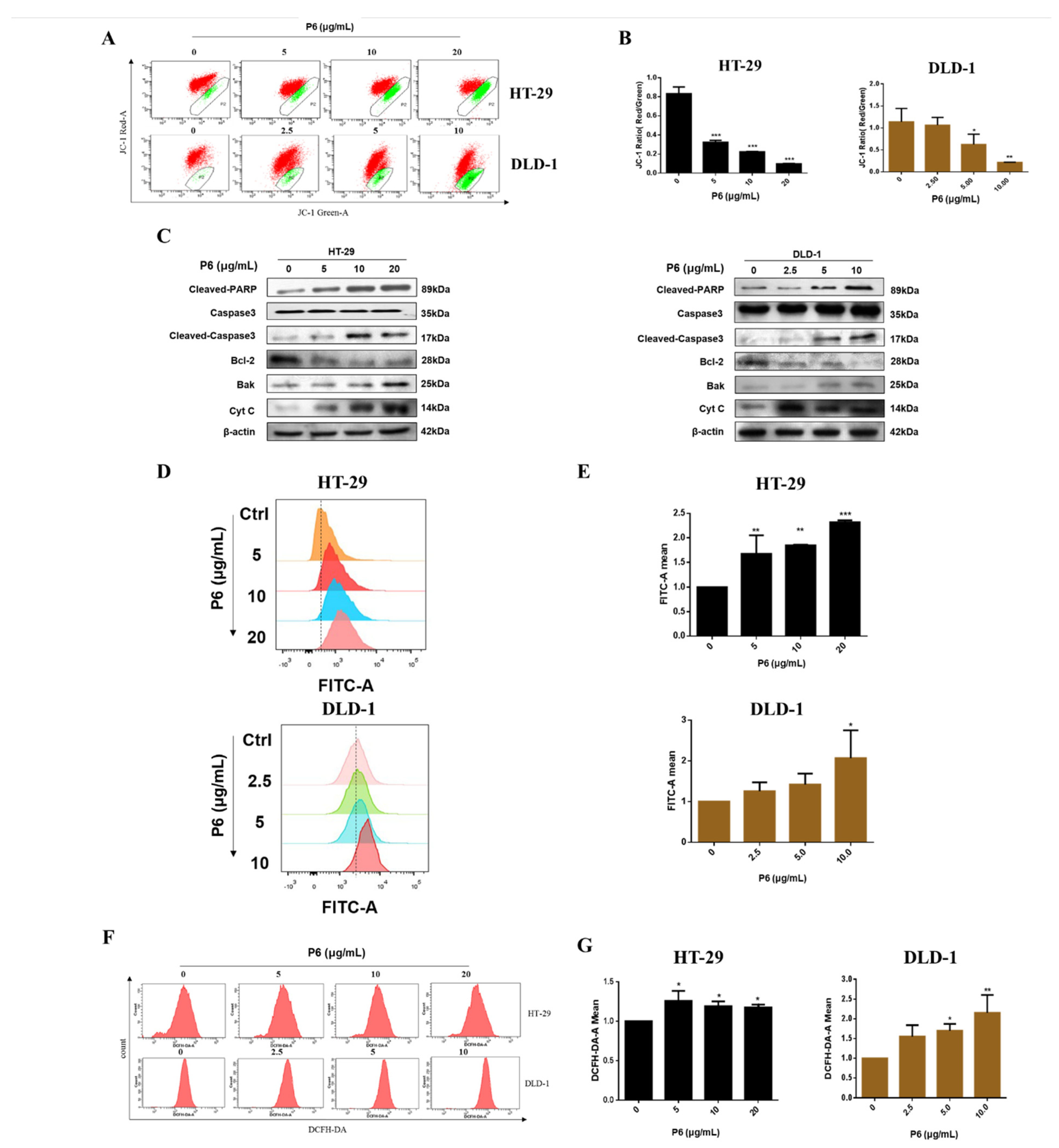
Figure 5). It has been reported that Ca
2+ overload could lead to mitochondrial dysfunction and mitochondrial ROS generation [
39]. The underlying mode of action may include Ca
2+-stimulated nitric oxide production, Ca
2+ -stimulated increase in metabolic rate, Ca
2+-induced cytochrome C (Cyt C) dissociation, Ca
2+-induced opening of the permeability transition pore with the subsequent release of Cyt C, Ca
2+-induced cardiolipin peroxidation, and Ca
2+/calmodulin dependent protein kinase activation. Several marine peptides can change the Bcl-2/Bax ratio, activate caspases, and release Cyt C to induce cell apoptosis by stimulating Ca
2+ overload and ROS production in tumor cells [
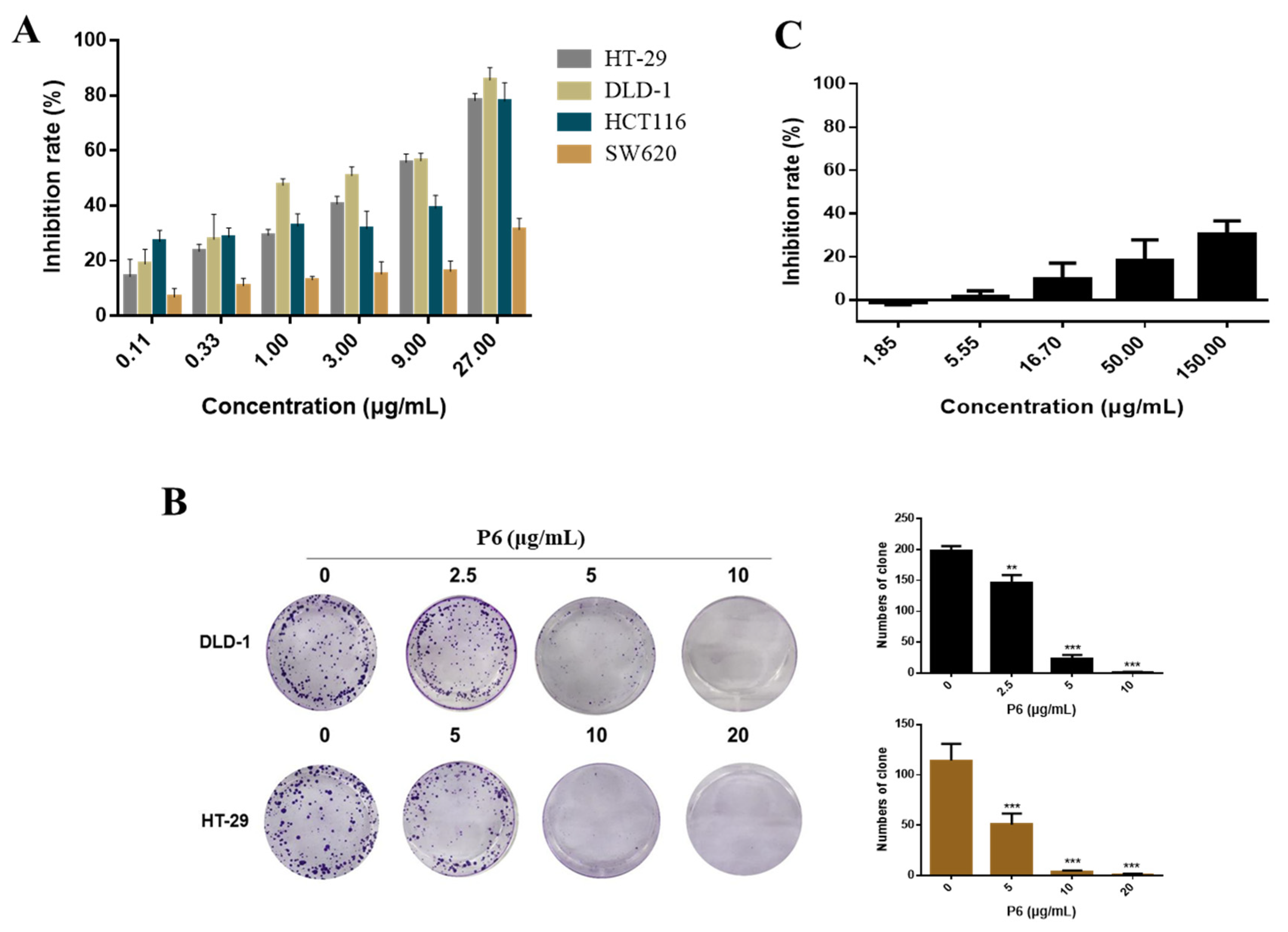
40]. In this study, we found that P6 suppressed CRC cell proliferation (
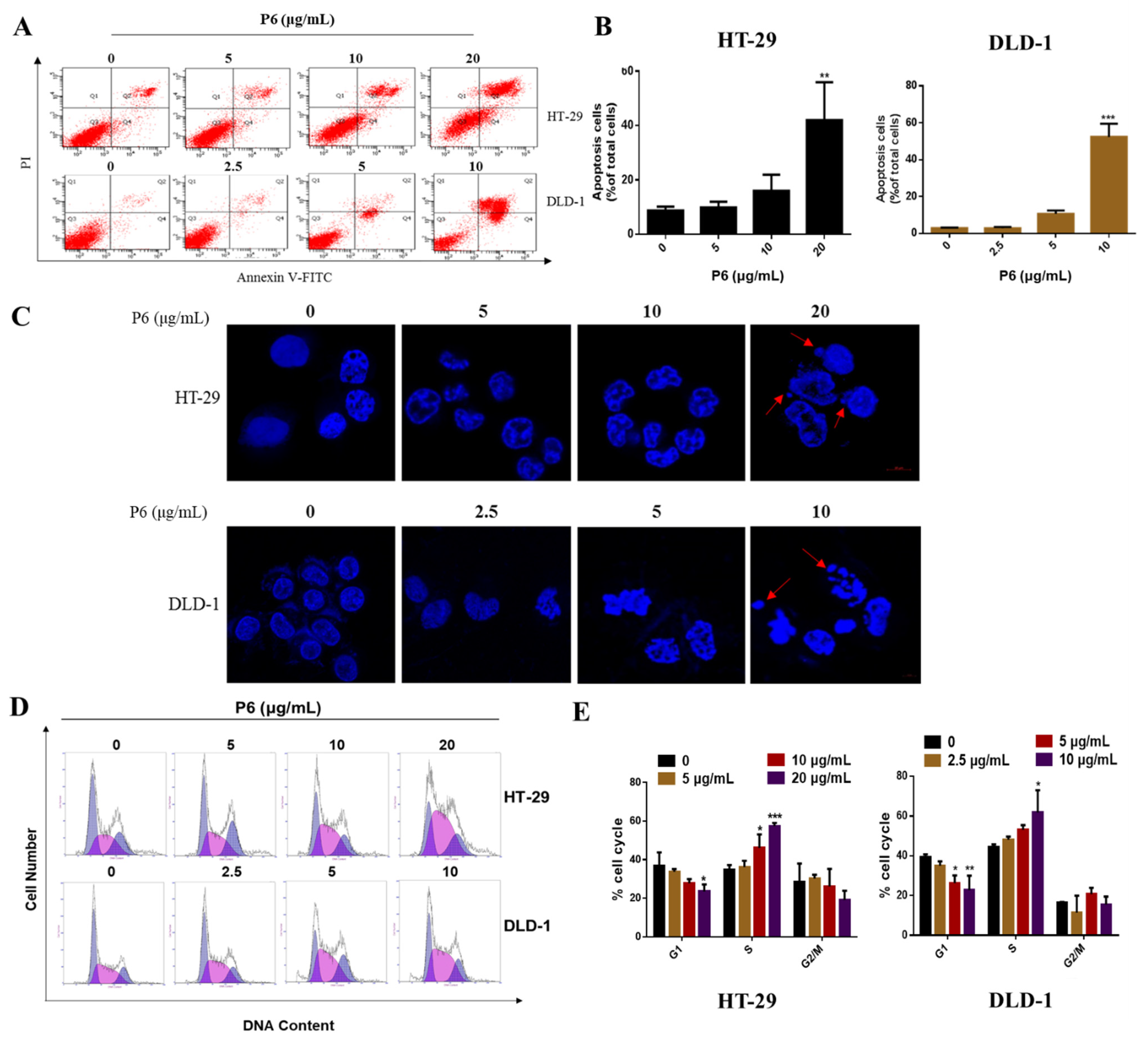
Figure 3) and induced cell apoptosis (
Figure 4) by producing a concentration-dependent increase in intracellular Ca
2+ concentration (
Figure 5D,E). Additionally, mitochondrial Ca
2+ overload induces an increase in intracellular ROS levels. Our results showed that P6 markedly reduced the mitochondrial membrane potential and increased intracellular ROS levels in both HT-29 and DLD-1 cells (
Figure 5B,F,G). The decrease in mitochondrial membrane potential indicated that the permeability of the mitochondrial membrane was increased and the internal cytochrome C was released at the same time, which induced apoptosis. Furthermore, caspases are closely related to cell apoptosis. Many marine peptides induce tumor cell apoptosis by activating caspases. Pardaxin, a peptide consisting of 33 amino acid residues, is isolated from the secretions of a fish (
Pardachirus marmoratus) found at the bottom of the Red Sea. Studies have shown that pardaxin can induce apoptosis in human fibrosarcoma HT-1080 cells, which is manifested by the increased activity of caspase-3, mitochondrial membrane potential changes, and the accumulation of reactive oxygen species (ROS) products [
40]. P6 showed a similar effect, producing an increased activity of caspase-3 and mitochondrial membrane potential changes in CRC cells (
Figure 5B,C). The subsequent Western blot results showed that P6 upregulated the expression of cytochrome C and the pro-apoptotic protein Bak (
Figure 5C). It was further confirmed that P6 exerted an apoptosis-inducing effect through the mitochondrial pathway at the molecular level.
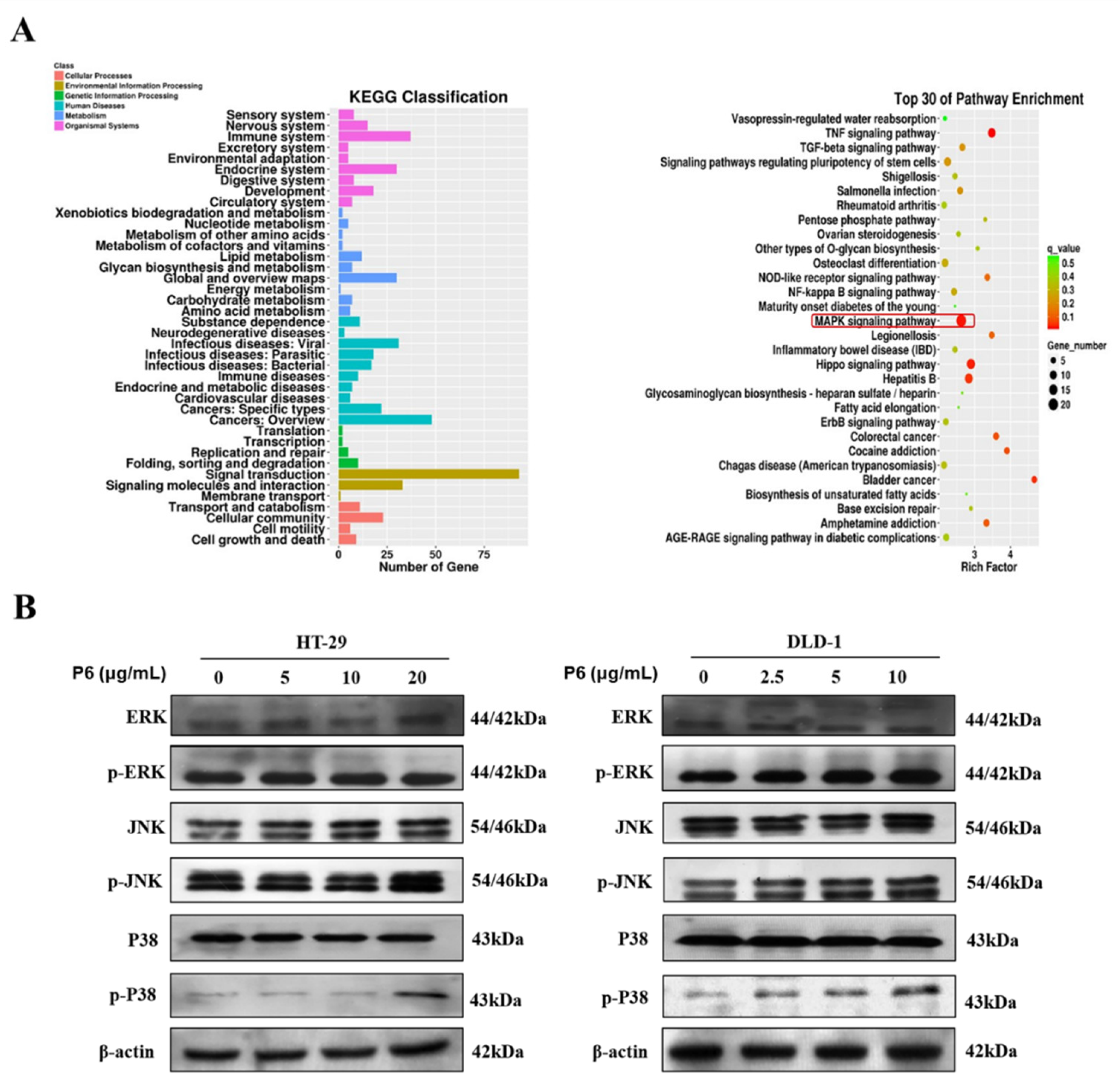
Mitogen-activated protein kinases (MAPK) form a crucial signaling pathway that regulates cell fate decisions in response to external stimuli. Among them, p38 MAPK family members play an important role in regulating cell proliferation, senescence, and tumorigenesis [
41]. Previous studies have shown that the altered expression of p38 proteins is often observed in CRC; thus, the drug-induced activation of the p38 MAPK cascade could be a scenario worth exploring for sensitizing CRC cells to apoptotic death [
41]. Herein, we hypothesized that P6 might be a promising antitumor molecule via the activation of the p38 pathway. Considering the cell proliferation-suppressing and apoptotic effects of P6 (
Figure 6A), we further investigated whether P6 influenced p38 MAPK pathways. As expected, P6 markedly increased the protein levels of the p38 proteins but showed no influence on the ERK and JNK pathways (
Figure 6B), indicating the activation of the p38 MAPK cascades. Additionally, p38 activation-mediated apoptosis is sometimes induced by secondary routes, such as through the production of ROS [
42]. In the present study, P6 suppressed proliferation and induced apoptosis in both HT-29 and DLD-1 cells. Hence, the effects of P6 on inducing mitochondrial apoptosis were partially dependent on ROS production.
4. Materials and Methods
4.1. Isolation and Identification of P6
Arca inflata Reeve materials were purchased from Qingdao of Shandong province, China; the production lot number was 20190410. The collected hemolymph of A. inflata was mixed with PBS in a ratio of 1:3 (w/v) and underwent ultrasound for 40 min at 4 °C. The supernatant was collected by 10,000 rpm centrifugation for 30 min at 4 °C. The supernatant was salted out with 100% ammonium sulfate, and the precipitate was collected by centrifugation (10,000 rpm for 30 min at 4 °C). The precipitate was dissolved in 20 mM Tris-HCl (pH 8.0) and dialyzed (molecular weight cut-off: 1 kDa) for 48 h. The samples were further separated by HPLC (Welch LP-C8, 5 μm, 4.6 × 250 mm). The sixth peak containing peptides was highlighted in red. Due to their similar hydrophobicity, the peptides in this marked peak were difficult to separate using a conventional approach. Hence, LC-MS/MS was applied to identify the peptides. The most abundant peptide fragment was named P6. According to the identified peptide sequences, the top 5 most abundant peptide fragments in this peak were synthesized by GenScript Co. (Shanghai, China) using a solid-phase peptide synthesis procedure. Their antitumor activities were tested using an MTT assay; only P6 possessed antitumor activity.
4.2. Purity of P6
High-performance liquid chromatography (HPLC) was applied to detect the purity of P6 using an Agilent series 1100 HPLC system connected to a ZORBAX®300SB-C8 column (4.6 × 150 mm, 5 µm; Agilent, Foster City, CA, USA). Water-trifluoroacetic acid (solvent A; 100:0.1, v/v) and acetonitrile-trifluoroacetic acid (solvent B; 100:0.1, v/v) were used as elution solvents. The elution procedure was set as follows: 55% solvent A and 45% solvent B with the flow rate at 1 mL/min. The wavelength of the UV detector was at 280 nm, and column temperature was 30 °C.
4.3. Molecular Weight Determination of P6
The precise molecular weight of P6 was determined using an ESI-MS spectrometer. The P6 sample was dissolved in distilled water and subsequently loaded into an API type 4000 QTRAP mass spectrometer (Applied Biosystems, Foster City, CA, USA). The mass spectrometer was used in the positive electrospray ionization (ESI + ve) mode. High-purity nitrogen gas was used for both drying (35 psi) and ESI nebulization (45 psi). The spectra were recorded over the mass/charge (m/z) range of 500–3000.
4.4. De Novo Sequencing of P6 by Tandem Mass Spectrometry (MS)
LC-MS/MS was used to determine the amino acid sequence of P6. After P6 was separated by HPLC, it was analyzed by a TripleTOF 5600 LCMS (AB SCIEX, Concord, ON, USA) connected to a nanoscale liquid-chromatography (LC) system (NanoLC-Ultra 2D; Eksigent, Dublin, CA, USA). Briefly, a reversed-phase C18 column (75 μm × 15 cm, 3 µm, 120 Å; ChromXP, Eksigent, USA) was applied for the separation of the desalted hydrolyzed peptides. Mixtures of 0.1% (v/v) formic acid in 5% acetonitrile and 0.1% (v/v) formic acid in 95% acetonitrile were used as the mobile phases A and B, respectively. The elution procedure was as follows: 5–40% B for 65 min, 40–100% B for 10 min, and maintained for 5 min. The ionization voltage and capillary temperature were set as 2.3 kV and 150 °C, respectively. The molecular masses of the purified peptides were detected in MS/MS mode. The acquired data were further analyzed by PEAKS software version 8.5 (Waterloo, ON, Canada). The peptides with de novo scores greater than 85% were selected for use in the study.
4.5. Physicochemical Characterization of P6
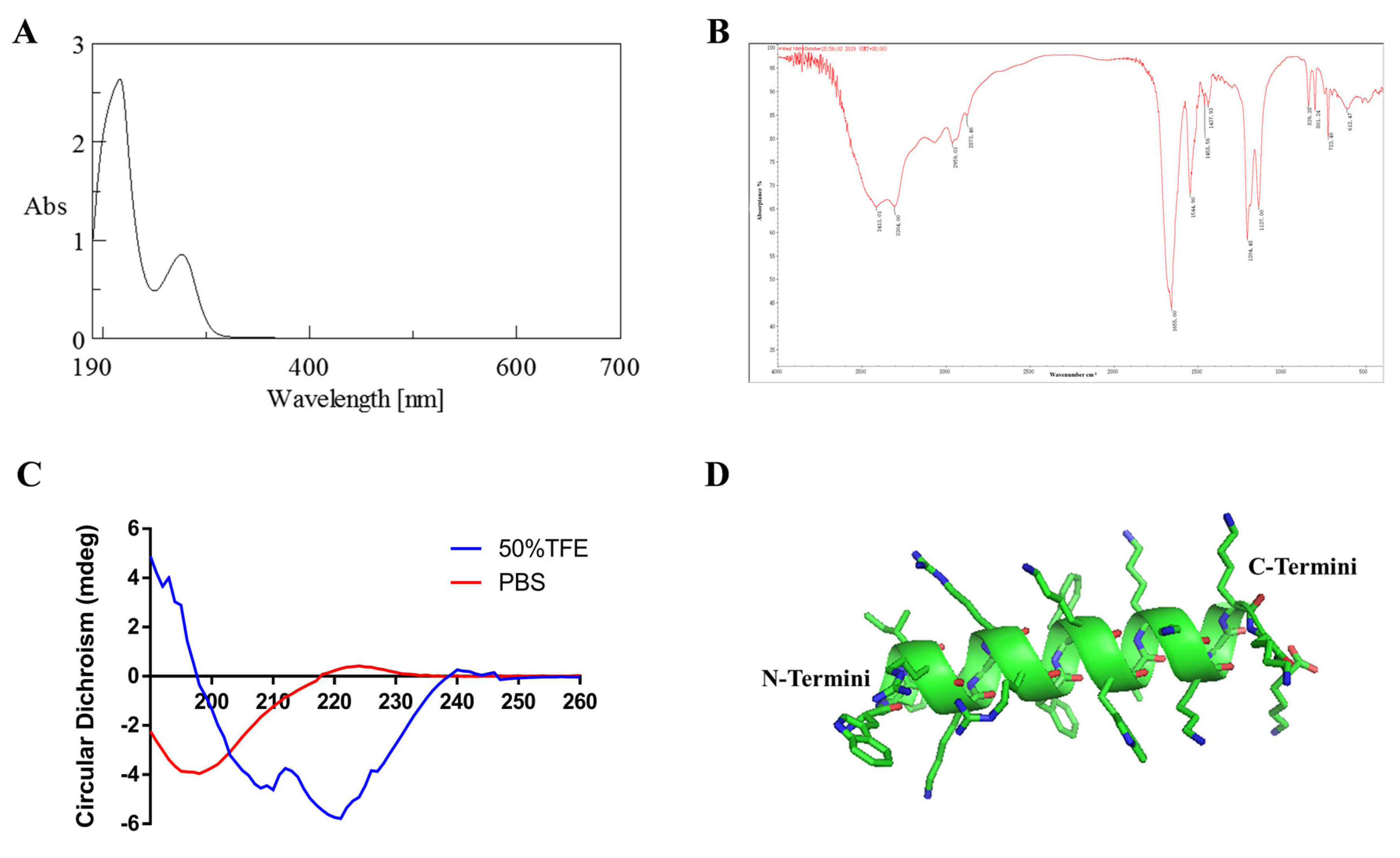
Ultraviolet (UV) spectroscopy was used to detect whether P6 contained aromatic amino acids. UV-vis absorption spectroscopy was conducted with a UV-2450 UV-vis absorption spectrophotometer (Shimadzu, Osaka, Japan) equipped with a 1.0 cm quartz cell. P6 was dissolved in distilled water to prepare a 0.05 mg/mL solution, which was scanned in the wavelength range of 190–400 nm.
Infrared spectroscopy was used to analyze the functional groups of P6. P6 and potassium bromide were fully grinded and mixed, and the mixture was put into a mold for scanning by an EQUINOX55 FT-IR spectrometer (Bruker, Bremen, Germany). The scanning range was 400–4000 cm−1.
Circular dichroism was used to analyze the secondary structure conformation of P6. P6 was dissolved in distilled water to prepare a 0.05 mg/mL solution, and CD measurements were obtained using a Jasco J-810 spectropolarimeter (Japan Spectroscopic Co., Ltd., Hachioji, Tokyo, Japan) equipped with a 0.1 cm quartz cell. The average of eight scans was used to produce the final spectrum. The spectra were all corrected for solvent contributions. The scan speed was 50 nm/min, and the scan range was 260–190 nm. The other parameters were set as follows: response time of 0.5 s, bandwidth of 2 nm, data interval of 0.2 nm, and sensitivity of 20 mdeg.
4.6. Reagents and Antibodies
N-acetyl-L-cysteine (A105420) was purchased from Aladdin (Shanghai, China). β-actin (1:1000, 3700), caspase-3 (1:1000, 9662), cleaved caspase-3 (1:1000, 9664), cleaved PARP (1:1000, 5625), Bcl-2 (1:1000, 5625), SAPK/JNK (1:1000, 9252), phospho-SAPK/JNK (1:1000, 4668), anti-rabbit IgG (1:4000, 7074S), anti-mouse IgG (1:4000, 7076S), Bak (1:1000, 12105), and cytochrome C (1:1000, 4280) were obtained from Cell Signaling Technology (Boston, Massachusetts, USA). The p38 MAPK (1:1000, AF6456), phospho-p38 MAPK (1:1000, AF3455), ERK1/2 (1:1000, AF0155), and phospho-ERK1/2 (1:1000, AF1014) were obtained from Affinity Biosciences (Pottstown, PA, USA).
4.7. Cell Culture
The human colorectal cancer cell lines (HT-29, DLD-1, HCT116, and SW620) and human normal liver cells (L02) were obtained from the Chinese Academy of Sciences Cell Bank of Type Culture Collection (Shanghai, China). The HT-29 and HCT116 cells were cultured in DMEM medium (Gibco, Gaithersburg, MD, USA) supplemented with 10% fetal bovine serum (FBS; Biological Industries, Beit Haemek, Israel), 100 units/mL penicillin, and 100 μg/mL streptomycin. The DLD-1 cells and SW620 cells were cultured in RPMI 1640 medium (Gibco, Gaithersburg, MD, USA) supplemented with 10% fetal bovine serum (FBS; Biological Industries, Beit Haemek, Israel), 100 units/mL penicillin, and 100 units/mL streptomycin. The cells were maintained in a humidified incubator with 5% CO2 at 37 °C.
4.8. Cell Viability Assay
The cytotoxicity of P6 was investigated using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. L02 cells (2.5 × 10
3 per well/100 μL), HT-29 cells, HCT116 cells, DLD-1 cells, and SW620 cells (1.5 × 10
3 per well/100 μL) were seeded into 96-well plates containing DMEM or RPIM 1640 medium, respectively. After 24 h incubation at 37 °C and 5% CO
2, the medium was replaced with various concentrations (0–27 μg/mL) of P6 for 48 h. Finally, 10 μL MTT solution (5 mg/mL) was added to each well, and the cells were incubated for 4 h. The MTT formazan product was dissolved in 200 μL DMSO and shaken for 10 min on the micro-oscillator. The optical density (OD) at 570 nm was recorded using a Synergy HT microplate reader (BioTek, Winooski, VT, USA). Each concentration was repeated at least three times. The inhibition rate was calculated as:
where D was the OD value of the experimental group, D0 was the OD value of the parallel solvent control group, and D1 was the OD value of the blank control group.
4.9. Colony Formation Assay
For the colony formation assays in monolayer cultures, the cells (1 × 103 per well) were incubated in 6-well plates for 24 h. Two milliliters of the prepared P6 solutions at different concentrations were added, and 2 mL of complete medium was added to the control group. After continuous treatment for 14 days, the medium was discarded and the cells were gently rinsed 2–3 times with PBS buffer (1 mL/well). Then, 300 μL of 4% paraformaldehyde was added to each well, incubated for 25 min, and the 4% paraformaldehyde was aspirated. Five hundred microliters of 0.1% crystal violet solution were added to each well, incubated for 25 min, and then gently rinsed with PBS buffer 2–3 times. After fixation and staining, the colonies were imaged.
4.10. Cell Morphology Analysis
Hoechst 33342 staining measurements were performed with a Hoechst 33342 staining kit (C1022, Beyotime, Shanghai, China). The cells were seeded in confocal dishes at a cell density of 3 × 104 cells/mL (200 μL/dish) and incubated at 37 °C and 5% CO2. After 24 h, the previous medium was discarded, and 200 μL of the prepared P6 solutions at different concentrations were added to the treatment group; 200 μL of complete medium was added to the control group. After 24 h of P6 treatment, the previous medium was aspirated and the cells were carefully rinsed 2–3 times. Two hundred microliters of Hoechst 33342 dye was added to each well, and after incubation in the dark for 15 min, the dye was discarded and the cells were carefully rinsed with PBS buffer 2–3 times. The photographs of the samples were recorded using a fluorescence microscope (Olympus, Tokyo, Japan).
4.11. Cell Apoptosis Assay
Cell apoptosis was detected using an Annexin V-FITC/PI staining kit (FXP018-100, 4A Biotech, Beijing, China). Cells (1 × 105 per well) were seeded into 6-well plates and incubated for 24 h with various concentrations of P6 (0, 5, 10, and 20 μg/mL for HT-29 cells or 0, 2.5, 5, and 10 μg/mL for DLD-1 cells) for 24 h. Then, the cells were washed, trypsinized, harvested, suspended in binding buffer, and stained with Annexin V-FITC and PI following the manufacturer’s instructions. The suspended cells were incubated with FITC and PI in the dark at room temperature for 15 minutes. The samples were immediately analyzed by flow cytometry (BD FACSCanto, Canto, NJ, USA).
4.12. Cell Cycle Assay
Cells (1 × 105 per well) were seeded into 6-well plates and incubated for 24 h with various concentrations of P6 (0, 5, 10, and 20 μg/mL for HT-29 cells or 0, 2.5, 5, and 10 μg/mL for DLD-1 cells) for 24 h. The cells were harvested and suspended in 70% ethanol. Then, the samples were stored at −20 °C for 2 h and stained with PI for 15 min in the dark at room temperature. The samples were immediately analyzed by flow cytometry (BD FACSCanto, Canto, NJ, USA).
4.13. Mitochondrial Membrane Potential (MMP) Assay
The mitochondrial membrane potential of colorectal cancer cells was determined using a JC-1 staining kit (C2006, Beyotime, Shanghai, China) with flow cytometry. Colorectal cancer cells were seeded into a 6-well plate at a density of 1 × 105 cells per well. After incubation overnight, the cells were treated with different concentrations of P6 (0, 5, 10, and 20 μg/mL for HT-29 or 0, 2.5, 5, and 10 μg/mL for DLD-1) for 24 h. Next, the cells were washed, trypsinized, harvested, and stained with JC-1 working buffer at 37 °C for 20 min in the dark. The samples were washed three times with PBS and immediately analyzed by flow cytometry (BD FACSCanto, Canto, NJ, USA).
4.14. Intracellular ROS Detection
A DCFH-DA staining kit (S0033, Beyotime, Shanghai, China) was used to detect intracellular ROS in CRC cells. Briefly, cells (1.5 × 105 per well) were seeded into a 6-well plate and incubated at 37 °C and 5% CO2 for 24 h. Then, each well was covered with various concentrations of P6 (0, 5, 10, and 20 μg/mL for HT-29 or 0, 2.5, 5, and 10 μg/mL for DLD-1) for 24 h. Next, the cells were washed with pre-cooled PBS, trypsinized, and harvested. Finally, the cells were stained with DCFH-DA at 37 °C for 30 min. The samples were washed three times with medium without serum and immediately analyzed by flow cytometry (BD FACSCanto, Canto, NJ, USA).
4.15. Intracellular Ca2+ Detection
Intracellular Ca2+ was detected using a Fluo-4 AM staining kit (S1060, Beyotime, Shanghai, China) and analyzed by flow cytometry. Colorectal cancer cells were seeded into a 6-well plate at a density of 1 × 105 cells per well. After incubation overnight, the cells were treated with different concentrations of P6 (0, 5, 10, and 20 μg/mL for HT-29 cells or 0, 2.5, 5, and 10 μg/mL for DLD-1 cells) for 24 h. Next, the cells were washed, trypsinized, harvested, and stained with Fluo-4 AM at 37 °C for 30 min in the dark. The samples were washed three times with PBS and immediately analyzed by flow cytometry (BD FACSCanto, Canto, NJ, USA).
4.16. Western Blotting Analysis
HT-29 and DLD-1 cells were seeded into 6-well plates (5 × 105 per well) and incubated in medium for 24 h, after which the medium was replaced with various concentrations of P6 (0, 5, 10, and 20 μg/mL for HT-29 cells or 0, 2.5, 5, and 10 μg/mL for DLD-1 cells) for 24 h. Then, the cells were washed three times with pre-cooled PBS and lysed with RIPA buffer (P0013K, Beyotime, Shanghai, China) on ice. Protein samples were collected and boiled for 15 min. The concentrations of the protein samples were quantified using a BCA protein assay kit (P0012S, Beyotime, Shanghai, China). Equal amounts of denatured protein samples were separated on 10% to 12% (w/v) sodium dodecyl sulfate (SDS)-polyacrylamide gels and transferred to polyvinylidene fluoride (PVDF) membranes (Bio-Rad, Hercules, CA, USA). The membranes were blocked in 5% BSA at room temperature for 2 h and incubated with primary antibodies overnight at 4 °C. Then, the membranes were incubated with horse radish peroxidase-linked secondary antibodies at room temperature for 2 h. After they were washed three times with TBST, the membranes were exposed to X-ray film to detect the expressions of the target proteins, which were enhanced using the ECL Kit (Tanon, Shanghai, China).
4.17. RNA-Seq Analysis
HT-29 cells in the logarithmic growth phase were collected and passaged in 100 mm Petri dishes. When the cell growth covered approximately 70% of the Petri dishes, the P6 solution or medium solution was added. After 24-hour incubation, the previous medium was discarded. The cells were carefully rinsed twice with pre-cooled PBS buffer and 3 mL of TRIZOL lysate was added to each group, lysed on ice for 5 min, and the cell lysates were collected. The purified total RNA from HT-29 cells treated with P6 or without P6 was extracted and sent to Shanghai Bohao Biotech. Co., Ltd. (SHBIO, Shanghai, China) for RNA-seq analysis using an Illumina HiSeq system after transcription. The quantity of gene expression was calculated by FPKM (fragments per kilobase of transcript per million fragments mapped). Genes with log2 (fold change) > 1 and Q < 0.001 were considered as differentially expressed genes (DEGs). Gene cluster analyses and enriched KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway analyses were performed based on the DEGs.
4.18. In Vivo Studies
All animal experiments were approved by the Medical Laboratory Animal Center of Jinan University and followed the ethical requirements for laboratory animals. Male BALB/c-nude mice were obtained from Beijing Huafukang Biotechnology Co. Ltd. These mice were fed breeding feed and water in an SPF laboratory at room temperature. HT-29 cells (1.5 × 106 cells in 0.1 mL phosphate-buffered saline) were subcutaneously injected into the left armpit of 6-week-old male BALB/c nude mice. Tumor volume and body weight were measured every 2 days, and tumor volume was calculated using the following formula: (short diameter)2 × (long diameter)/2. When the tumor volume reached about 100 mm3, these mice were randomly divided into four groups (n = 6 in each group): the model group, 5-FU treatment group, P6 (30 mg/kg) treatment group, and P6 (15 mg/kg) treatment group. The reference drug (5-FU) and P6 were dissolved in normal saline and administered by subcutaneous injection at a volume of 0.1 mL/10g. After 14 days of administration, all mice were anesthetized by intraperitoneal injection of sodium pentobarbital (1 mg/20 g mouse) and euthanized. The organs were obtained for immunohistochemical experiments.
4.19. Histopathologic Examination and TUNEL Staining
The organs were fixed in 10% buffered paraformaldehyde solution and sectioned at 4 μm thicknesses. Hematoxylin and eosin (H&E) was used to stain the specimens. An optical microscope (Nikon, Tokyo, Japan) was used to observe the histopathological changes in the specimens.
A TUNEL assay was performed according to the manufacturer’s instructions using a one-step TUNEL apoptosis assay kit (C1086, Beyotime, Shanghai, China). TUNEL-positive tissues or cells were imaged under a fluorescence microscope (Olympus, Tokyo, Japan).
4.20. Statistical Analysis
The differences between two groups were analyzed by the two-tailed Student t-test, and comparisons of multiple groups were carried out using one-way analysis of variance (ANOVA). The data represent the mean ± standard deviation (SD) in all figures. p < 0.05 was chosen as the level of significant differences.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}