
Neoechinulin A as a Promising SARS-CoV-2 Mpro Inhibitor: In Vitro and In Silico Study Showing the Ability of Simulations in Discerning Active from Inactive Enzyme Inhibitors

 ,
,  , ,
, ,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
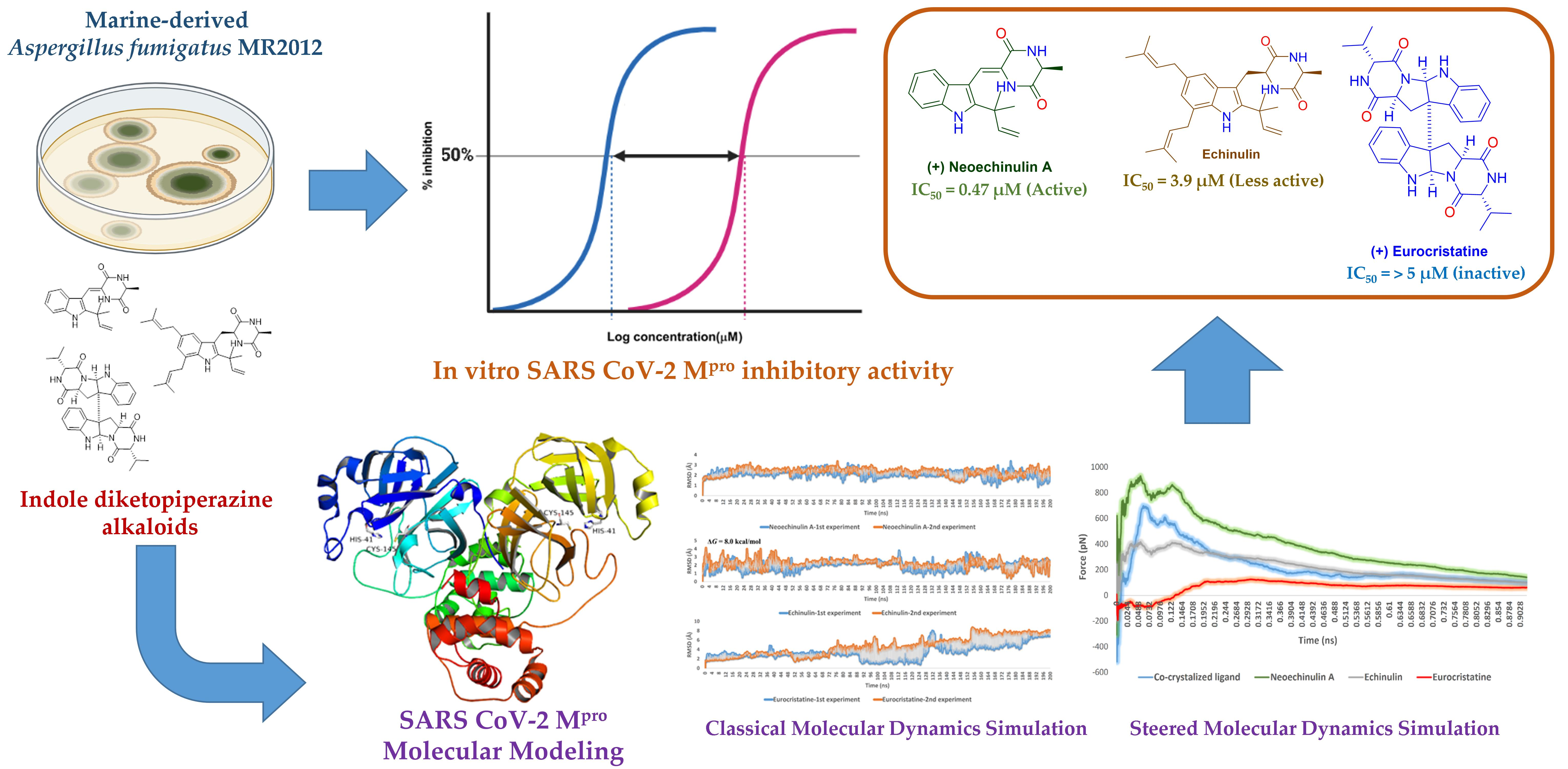
2. Results
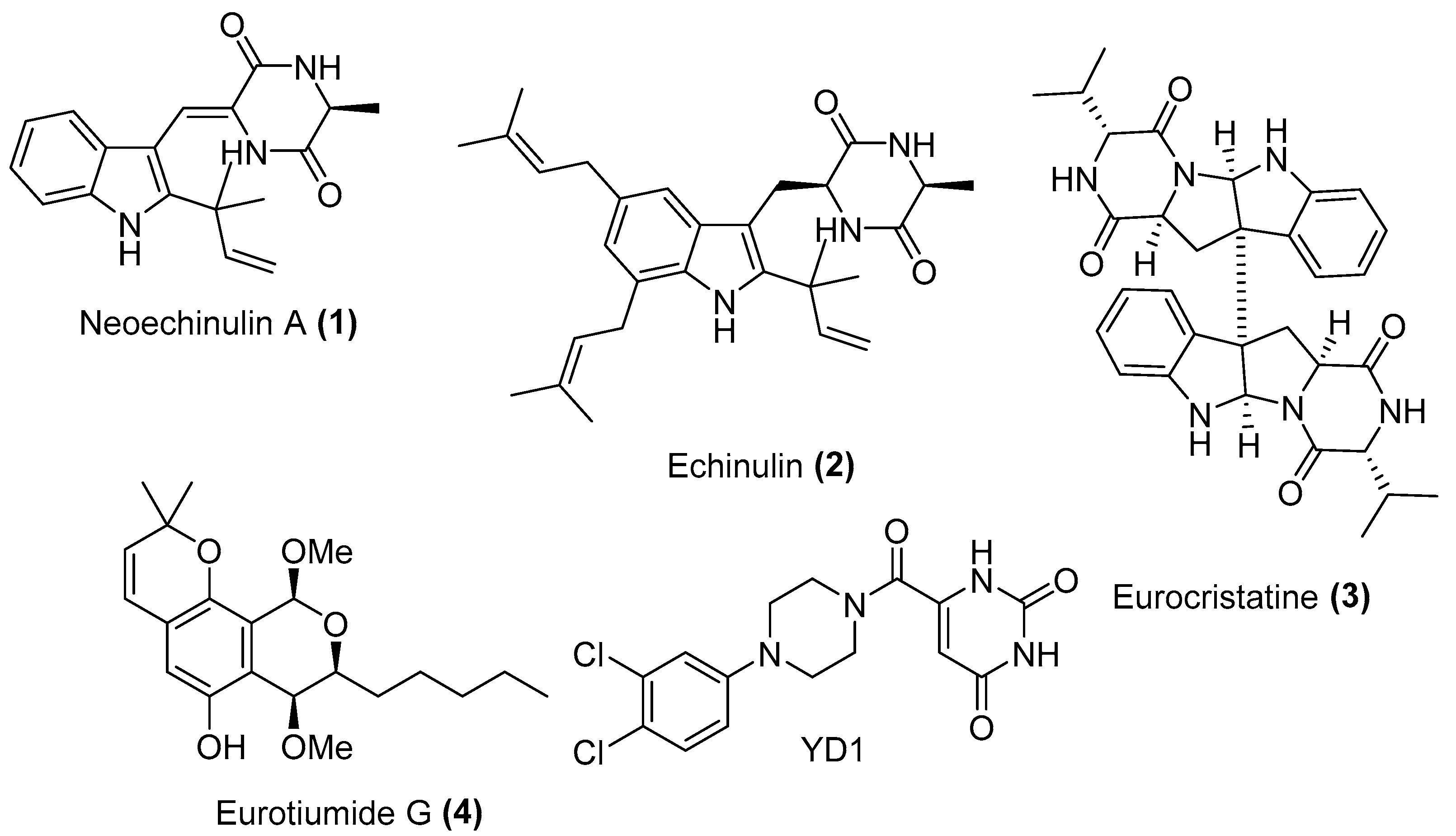
2.1. Bioguided Isolation and Structural Identification
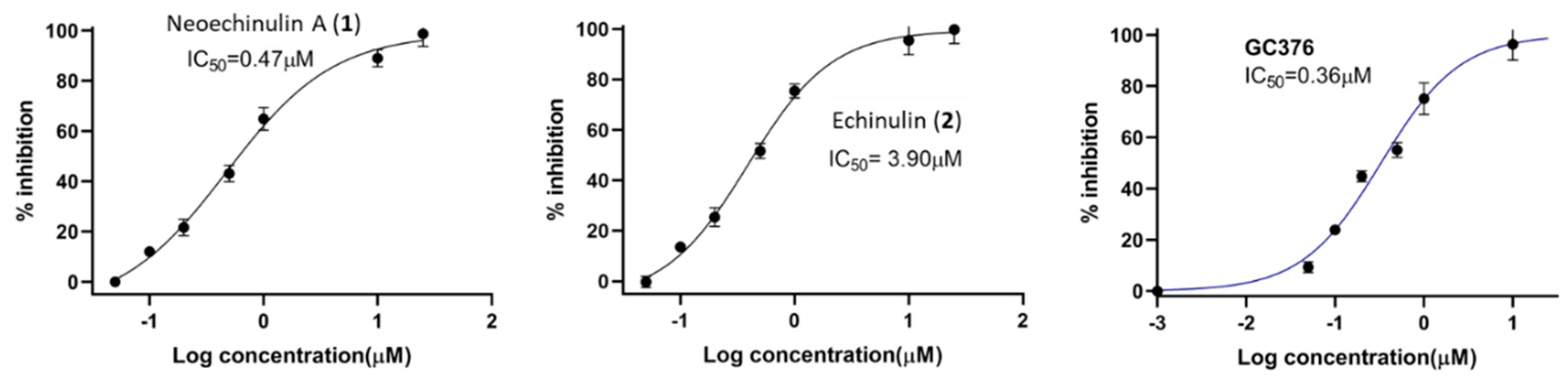
2.2. In Vitro Screening against SARS-CoV-2 Mpro
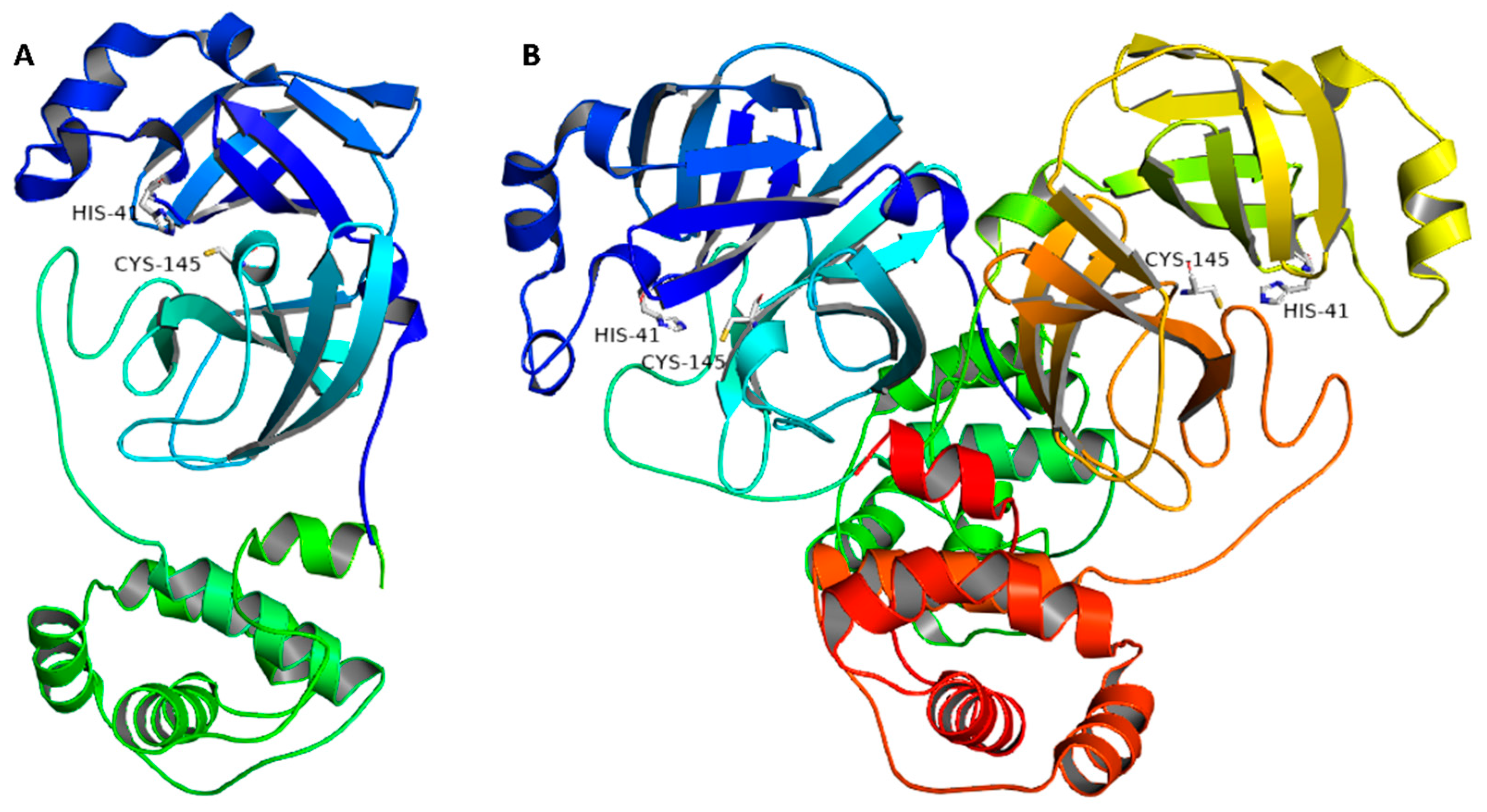
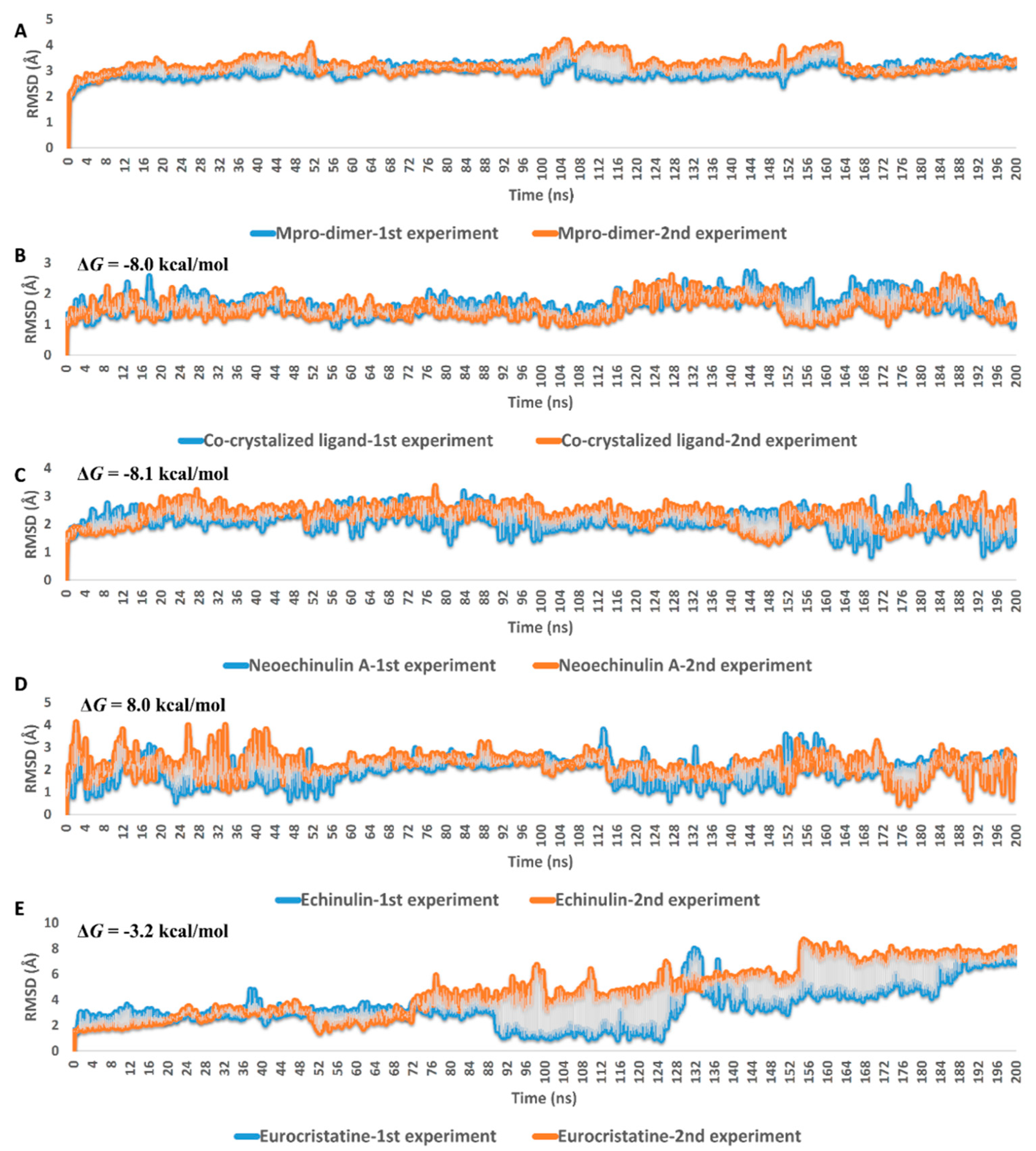
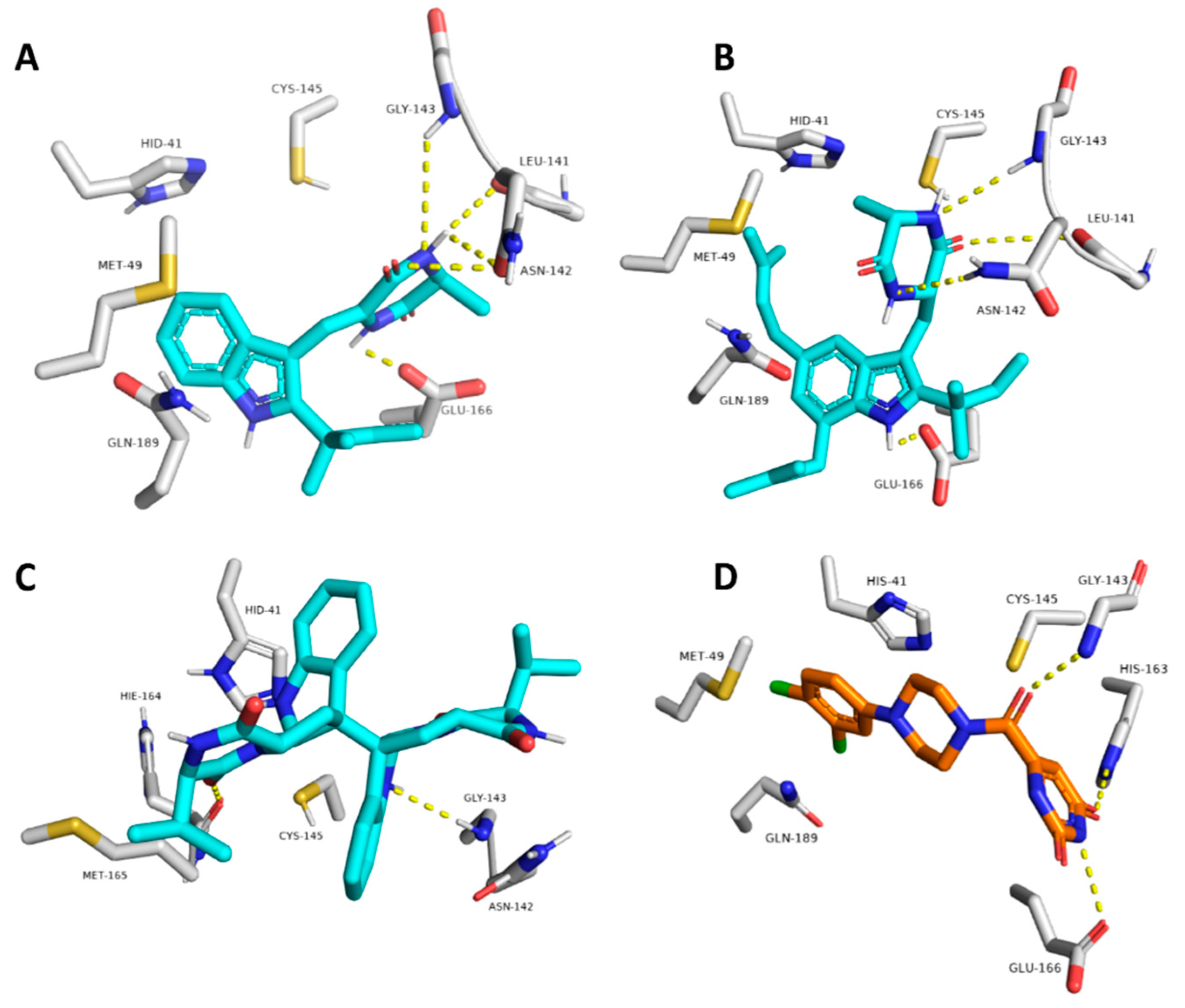
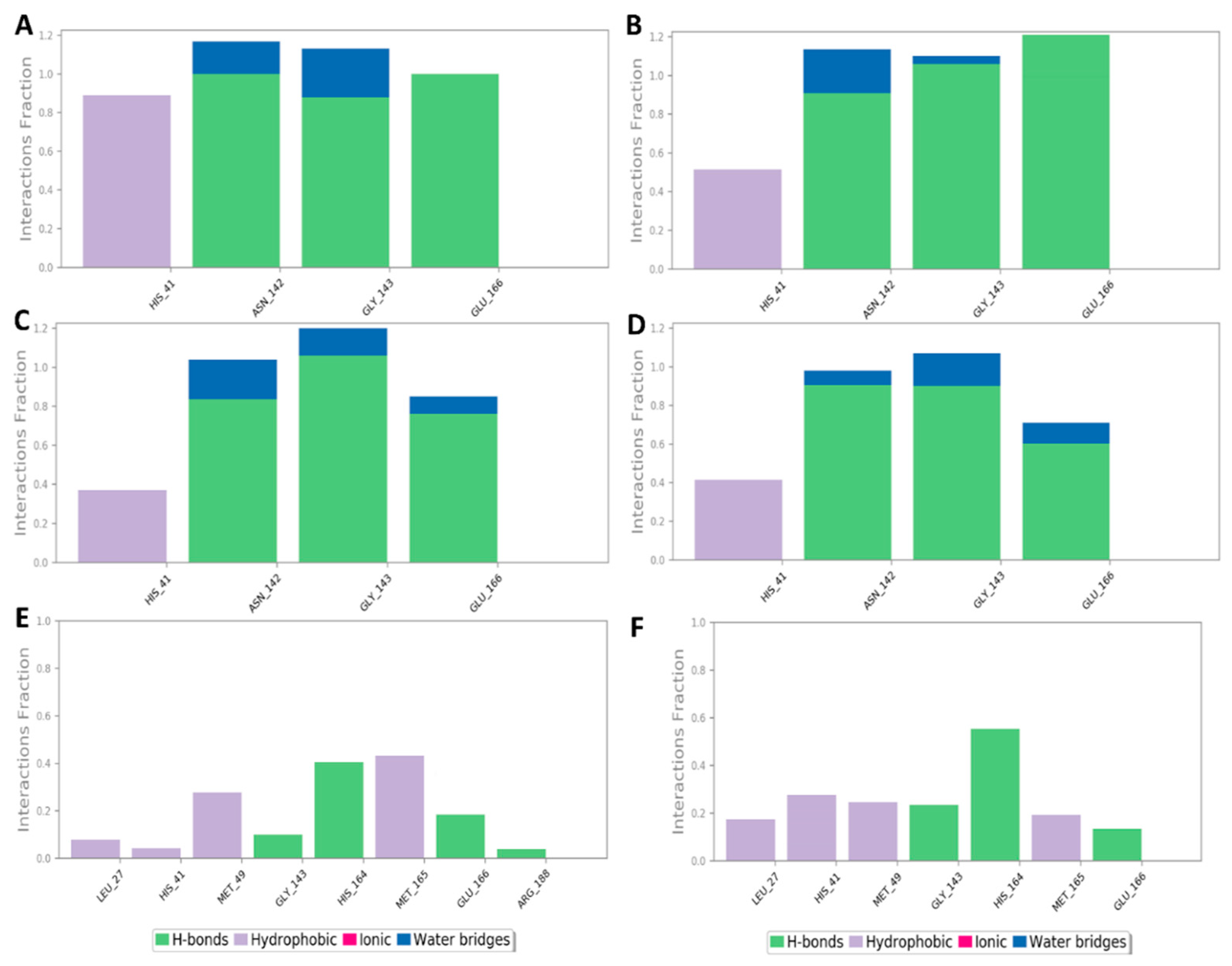
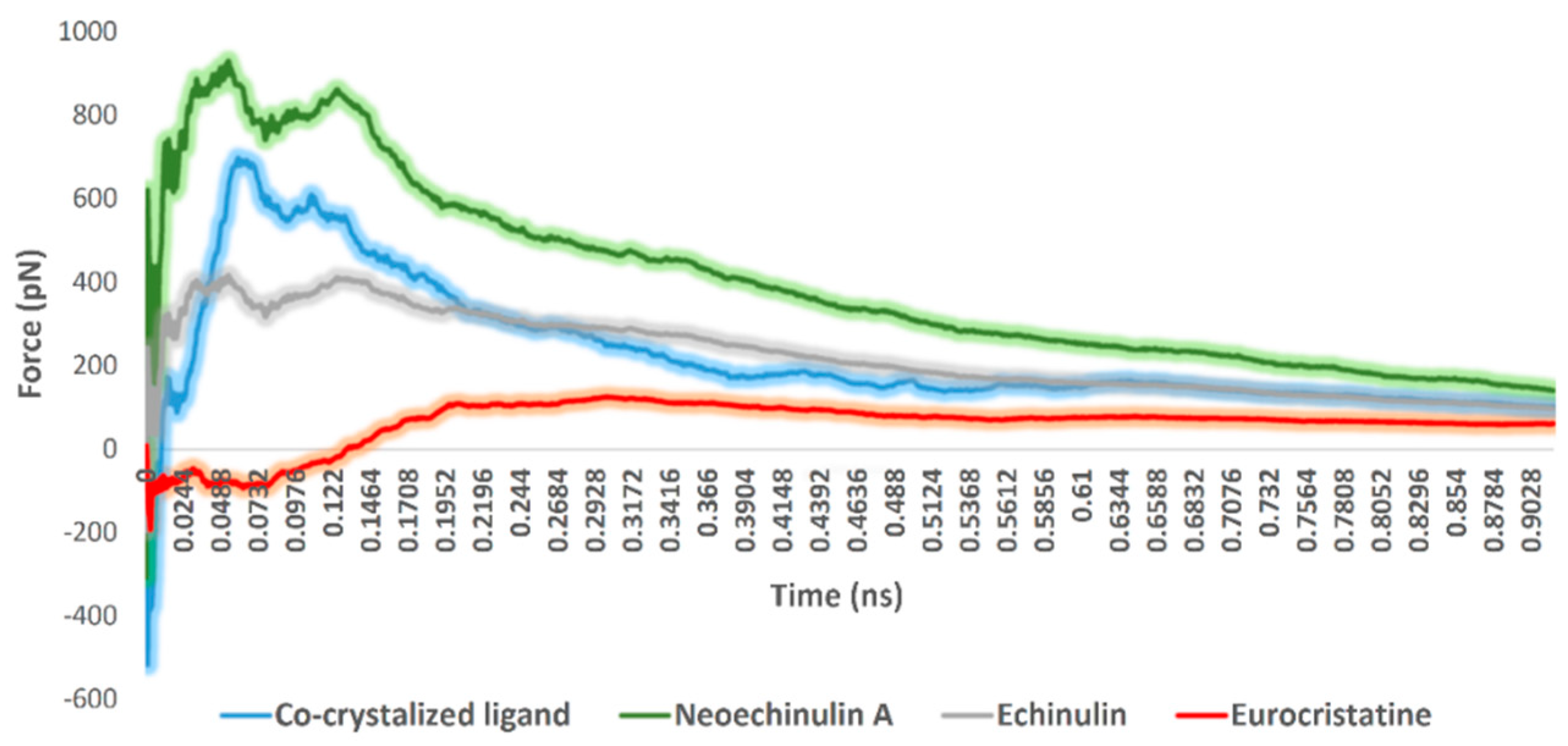
2.3. Docking and Molecular Dynamics Simulations
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Instruments
4.3. Fungal Isolation and Identification
4.4. Small- and Large-Scale Fermentation and Extraction
4.5. Isolation of Fungal Metabolites
4.6. In Vitro Assay against SARS-CoV-2 Main Protease
4.7. Docking and Molecular Dynamics Simulations
- Step1 (docking step): the compounds under investigation were docked into the Mpro active site using Autodock Vina [39]. The resulting top-ranked pose was almost identical to that of the co-crystallized one with an RMSD of 1.1 Å (Figure S12). The generated binding poses were then visually investigated using Pymol software [13,39].
- Step 2 (ΔGbind estimation step): Top-scoring poses resulted from the docking step were subjected to molecular dynamic simulations (MDS)-based binding free energy estimation (ΔGbind) using the Free Energy Perturbation (FEP) method [40]. MDS experiments carried out to calculate ΔGbind were performed by NAMD software [41], and all input files required for simulation by NAMD were prepared by using the online website Charmm-GUI (https://charmm-gui.org/?doc=input/afes.abinding, accessed on 15 February 2022) [40,41].
- Step 4 (SMD step): steered molecular dynamics (SMD), was carried out to determine the relative binding affinity of the investigated compounds. All SMD experiments were carried out by NAMD software as described previously [41].
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Available online: https://covid19.who.int/ (accessed on 15 February 2022).
- Available online: https://news.un.org/en/story/2021/04/1089392 (accessed on 15 February 2022).
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Ng, Y.L.; Salim, C.K.; Chu, J.J.H. Drug repurposing for COVID-19: Approaches, challenges and promising candidates. Pharmacol. Ther. 2021, 228, 107930. [Google Scholar] [CrossRef] [PubMed]
- Mercorelli, B.; Palù, G.; Loregian, A. Drug repurposing for viral infectious diseases: How far are we? Trends Microbiol. 2018, 26, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.fda.gov/consumers/consumer-updates/know-your-treatment-options-covid-19 (accessed on 15 February 2022).
- Available online: https://www.ema.europa.eu/en/human-regulatory/overview/public-health-threats/coronavirus-disease-covid-19/treatments-vaccines/covid-19-treatments (accessed on 15 February 2022).
- Jayk Bernal, A.; Gomes da Silva, M.M.; Musungaie, D.B.; Kovalchuk, E.; Gonzalez, A.; Delos Reyes, V.; Martín-Quirós, A.; Caraco, Y.; Williams-Diaz, A.; Brown, M.L.; et al. Molnupiravir for Oral Treatment of COVID-19 in Nonhospitalized Patients. N. Engl. J. Med. 2022, 386, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.R.; Allerton, C.M.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Mahase, E. COVID-19: Pfizer’s paxlovid is 89% effective in patients at risk of serious illness, company reports. Br. Med. J. 2021, 375, n2713. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Caly, L.; Druce, J.D.; Catton, M.G.; Jans, D.A.; Wagstaff, K.M. The FDA-approved drug ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antivir. Res. 2020, 178, 104787. [Google Scholar] [CrossRef]
- Thissera, B.; Sayed, A.M.; Hassan, M.H.A.; Abdelwahab, S.F.; Amaeze, N.; Semler, V.T.; Alenezi, F.N.; Yaseen, M.; Alhadrami, H.A.; Belbahri, L.; et al. Bioguided Isolation of Cyclopenin Analogues as Potential SARS-CoV-2 Mpro Inhibitors from Penicillium citrinum TDPEF34. Biomolecules 2021, 11, 1366. [Google Scholar] [CrossRef] [PubMed]
- Salih, A.E.; Thissera, B.; Yaseen, M.; Hassane, A.S.; El-Seedi, H.R.; Sayed, A.M.; Rateb, M.E. Marine sulfated polysaccharides as promising antiviral agents: A comprehensive report and modeling study focusing on SARS CoV-2. Mar. Drugs 2021, 19, 406. [Google Scholar] [CrossRef]
- Alhadrami, H.A.; Sayed, A.M.; Al-Khatabi, H.; Alhakamy, N.A.; Rateb, M.E. Scaffold Hopping of α-Rubromycin Enables Direct Access to FDA-Approved Cromoglicic Acid as a SARS-CoV-2 MPro Inhibitor. Pharmaceuticals 2021, 14, 541. [Google Scholar] [CrossRef]
- Alhadrami, H.A.; Sayed, A.M.; Hassan, H.M.; Youssif, K.A.; Gaber, Y.; Moatasim, Y.; Kutkat, O.; Mostafa, A.; Ali, M.A.; Rateb, M.E.; et al. Cnicin as an Anti-SARS-CoV-2: An Integrated in Silico and in Vitro Approach for the Rapid Identification of Potential COVID-19 Therapeutics. Antibiotics 2021, 10, 542. [Google Scholar] [CrossRef] [PubMed]
- Alhadrami, H.A.; Sayed, A.M.; Sharif, A.M.; Azhar, E.I.; Rateb, M.E. Olive-Derived Triterpenes Suppress SARS-CoV-2 Main Protease: A Promising Scaffold for Future Therapeutics. Molecules 2021, 26, 2654. [Google Scholar] [CrossRef]
- Orfali, R.; Rateb, M.E.; Hassan, H.M.; Alonazi, M.; Gomaa, M.R.; Mahrous, N.; GabAllah, M.; Kandeil, A.; Perveen, S.; Abdelmohsen, U.R.; et al. Sinapic Acid Suppresses SARS CoV-2 Replication by Targeting Its Envelope Protein. Antibiotics 2021, 10, 420. [Google Scholar] [CrossRef]
- Reher, R.; Kim, H.W.; Zhang, C.; Mao, H.H.; Wang, M.; Nothias, L.F.; Caraballo-Rodriguez, A.M.; Glukhov, E.; Teke, B.; Leao, T.; et al. A convolutional neural network-based approach for the rapid annotation of molecularly diverse natural products. J. Am. Chem. Soc. 2020, 142, 4114–4120. [Google Scholar] [CrossRef]
- van Santen, J.A.; Poynton, E.F.; Iskakova, D.; McMann, E.; Alsup, T.A.; Clark, T.N.; Fergusson, C.H.; Fewer, D.P.; Hughes, A.H.; McCadden, C.A.; et al. The Natural Products Atlas 2.0: A database of microbially-derived natural products. Nucleic Acids Res. 2021, 50, D1317–D1323. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Feng, C.; Wang, S.Y.; Zhang, D.M.; Li, X.H.; Zhang, C.X. New Indole Diketopiperazine Alkaloids from Soft Coral-Associated Epiphytic Fungus Aspergillus sp. EGF 15-0-3. Chem. Biodivers. 2020, 17, e2000106. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Li, Y.; Zhang, X.; Li, Q.; Liu, X.; Huang, Y.; Tang, T.; Zheng, S.; Wang, W.; Tang, J. A new prenylated indole diketopiperazine alkaloid from Eurotium cristatum. Molecules 2014, 19, 17839–17847. [Google Scholar] [CrossRef]
- Gomes, N.M.; Dethoup, T.; Singburaudom, N.; Gales, L.; Silva, A.M.; Kijjoa, A. Eurocristatine, a new diketopiperazine dimer from the marine sponge-associated fungus Eurotium cristatum. Phytochem. Lett. 2012, 5, 717–720. [Google Scholar] [CrossRef]
- Chen, M.; Shao, C.L.; Wang, K.L.; Xu, Y.; She, Z.G.; Wang, C.Y. Dihydroisocoumarin derivatives with antifouling activities from a gorgonian derived Eurotium sp. fungus. Tetrahedron 2014, 70, 9132–9138. [Google Scholar] [CrossRef]
- Clyde, A.; Galanie, S.; Kneller, D.W.; Ma, H.; Babuji, Y.; Blaiszik, B.; Brace, A.; Brettin, T.; Chard, K.; Chard, R.; et al. High Throughput Virtual Screening and Validation of a SARS-CoV-2 Main Protease Non-Covalent Inhibitor. J. Chem. Inf. Model. 2022, 62, 116–128. [Google Scholar] [CrossRef]
- Ullrich, S.; Nitsche, C. The SARS-CoV-2 main protease as drug target. Bioorganic Med. Chem. Lett. 2020, 30, 127377. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- El-Gendy, B.E.D.M.; Rateb, M.E. Antibacterial activity of diketopiperazines isolated from a marine fungus using t-butoxycarbonyl group as a simple tool for purification. Bioorg. Med. Chem. Lett. 2015, 25, 3125–3128. [Google Scholar] [CrossRef] [PubMed]
- Wakefield, J.; Hassan, H.M.; Jaspars, M.; Ebel, R.; Rateb, M.E. Dual induction of new microbial secondary metabolites by fungal bacterial co-cultivation. Front. Microbiol. 2017, 8, 1284. [Google Scholar] [CrossRef] [Green Version]
- Yagi, R.; Doi, M. Isolation of an antioxidative substance produced by Aspergillus repens. Biosci. Biotechnol. Biochem. 1999, 63, 932–933. [Google Scholar] [CrossRef]
- Kamisuki, S.; Himeno, N.; Tsurukawa, Y.; Kusayanagi, T.; Takeno, M.; Kamakura, T.; Kuramochi, K.; Sugawara, F. Identification of proteins that bind to the neuroprotective agent neoechinulin A. Biosci. Biotechnol. Biochem. 2018, 82, 442–448. [Google Scholar] [CrossRef]
- Kim, K.S.; Cui, X.; Lee, D.S.; Sohn, J.H.; Yim, J.H.; Kim, Y.C.; Oh, H. Anti-inflammatory effect of neoechinulin a from the marine fungus Eurotium sp. SF-5989 through the suppression of NF-кB and p38 MAPK pathways in lipopolysaccharide-stimulated RAW264. 7 macrophages. Molecules 2013, 18, 13245–13259. [Google Scholar] [CrossRef] [Green Version]
- Sasaki-Hamada, S.; Hoshi, M.; Niwa, Y.; Ueda, Y.; Kokaji, A.; Kamisuki, S.; Kuramochi, K.; Sugawara, F.; Oka, J.I. Neoechinulin A induced memory improvements and antidepressant-like effects in mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 71, 155–161. [Google Scholar] [CrossRef]
- Chen, X.; Si, L.; Liu, D.; Proksch, P.; Zhang, L.; Zhou, D.; Lin, W. Neoechinulin B and its analogues as potential entry inhibitors of influenza viruses, targeting viral hemagglutinin. Eur. J. Med. Chem. 2015, 93, 182–195. [Google Scholar] [CrossRef]
- Muratov, E.N.; Amaro, R.; Andrade, C.H.; Brown, N.; Ekins, S.; Fourches, D.; Tropsha, A. A critical overview of computational approaches employed for COVID-19 drug discovery. Chem. Soc. Rev. 2021, 50, 9121–9151. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, T.S.; Okimoto, N.; Koyama, Y.M.; Hirano, Y.; Morimoto, G.; Ohno, Y.; Taiji, M. Drug binding dynamics of the dimeric SARS-CoV-2 main protease, determined by molecular dynamics simulation. Sci. Rep. 2020, 10, 1698. [Google Scholar] [CrossRef]
- Padhi, A.K.; Rath, S.L.; Tripathi, T. Accelerating COVID-19 research using molecular dynamics simulation. J. Phys. Chem. B 2021, 125, 9078–9091. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeliger, D.; de Groot, B.L. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput. Aided Mol. Des. 2010, 24, 417–422. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Oshima, H.; Zhang, H.; Kern, N.R.; Re, S.; Lee, J.; Rous, B.; Sugita, Y.; Jiang, W.; Im, W. CHARMM-GUI free energy calculator for absolute and relative ligand solvation and binding free energy simulations. J. Chem. Theory Comput. 2020, 16, 7207–7218. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [Green Version]
- Release, S. 3: Desmond molecular dynamics system, DE shaw research, New York, NY, 2017. In Maestro-Desmond Interoperability Tools; Schrödinger: New York, NY, USA, 2017. [Google Scholar]
- Schrodinger, LLC. Maestro, Version 9.0; Schrodinger, LLC.: New York, NY, USA, 2009. [Google Scholar]
- Sayed, A.M.; Alhadrami, H.A.; El-Gendy, A.O.; Shamikh, Y.I.; Belbahri, L.; Hassan, H.M.; Abdelmohsen, U.R.; Rateb, M.E. Microbial natural products as potential inhibitors of SARS-CoV-2 main protease (Mpro). Microorganisms 2020, 8, 970. [Google Scholar] [CrossRef]
- Amaro, R.E.; Baudry, J.; Chodera, J.; Demir, Ö.; McCammon, J.A.; Miao, Y.; Smith, J.C. Ensemble docking in drug discovery. Biophys. J. 2018, 114, 2271–2278. [Google Scholar] [CrossRef] [Green Version]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the SC’06: Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; IEEE: New York, NY, USA, 2006; p. 43. [Google Scholar]
- Ngo, S.T.; Tam, N.M.; Quan, P.M.; Nguyen, T.H. Benchmark of Popular Free Energy Approaches Revealing the Inhibitors Binding to SARS-CoV-2 Mpro. J. Chem. Inf. Model. 2021, 61, 2302–2312. [Google Scholar] [CrossRef]
- Tutone, M.; Perricone, U.; Almerico, A.M. Conf-VLKA: A structure-based revisitation of the Virtual Lock-and-key Approach. J. Mol. Graph. Model. 2017, 71, 50–57. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alhadrami, H.A.; Burgio, G.; Thissera, B.; Orfali, R.; Jiffri, S.E.; Yaseen, M.; Sayed, A.M.; Rateb, M.E. Neoechinulin A as a Promising SARS-CoV-2 Mpro Inhibitor: In Vitro and In Silico Study Showing the Ability of Simulations in Discerning Active from Inactive Enzyme Inhibitors. Mar. Drugs 2022, 20, 163. https://doi.org/10.3390/md20030163
Alhadrami HA, Burgio G, Thissera B, Orfali R, Jiffri SE, Yaseen M, Sayed AM, Rateb ME. Neoechinulin A as a Promising SARS-CoV-2 Mpro Inhibitor: In Vitro and In Silico Study Showing the Ability of Simulations in Discerning Active from Inactive Enzyme Inhibitors. Marine Drugs. 2022; 20(3):163. https://doi.org/10.3390/md20030163
Chicago/Turabian StyleAlhadrami, Hani A., Gaia Burgio, Bathini Thissera, Raha Orfali, Suzan E. Jiffri, Mohammed Yaseen, Ahmed M. Sayed, and Mostafa E. Rateb. 2022. "Neoechinulin A as a Promising SARS-CoV-2 Mpro Inhibitor: In Vitro and In Silico Study Showing the Ability of Simulations in Discerning Active from Inactive Enzyme Inhibitors" Marine Drugs 20, no. 3: 163. https://doi.org/10.3390/md20030163
APA StyleAlhadrami, H. A., Burgio, G., Thissera, B., Orfali, R., Jiffri, S. E., Yaseen, M., Sayed, A. M., & Rateb, M. E. (2022). Neoechinulin A as a Promising SARS-CoV-2 Mpro Inhibitor: In Vitro and In Silico Study Showing the Ability of Simulations in Discerning Active from Inactive Enzyme Inhibitors. Marine Drugs, 20(3), 163. https://doi.org/10.3390/md20030163