
Homo/Hetero-Dimers of Aromatic Bisabolane Sesquiterpenoids with Neuroprotective Activity from the Fungus Aspergillus versicolor A18 from South China Sea

 and
and
Abstract
:1. Introduction
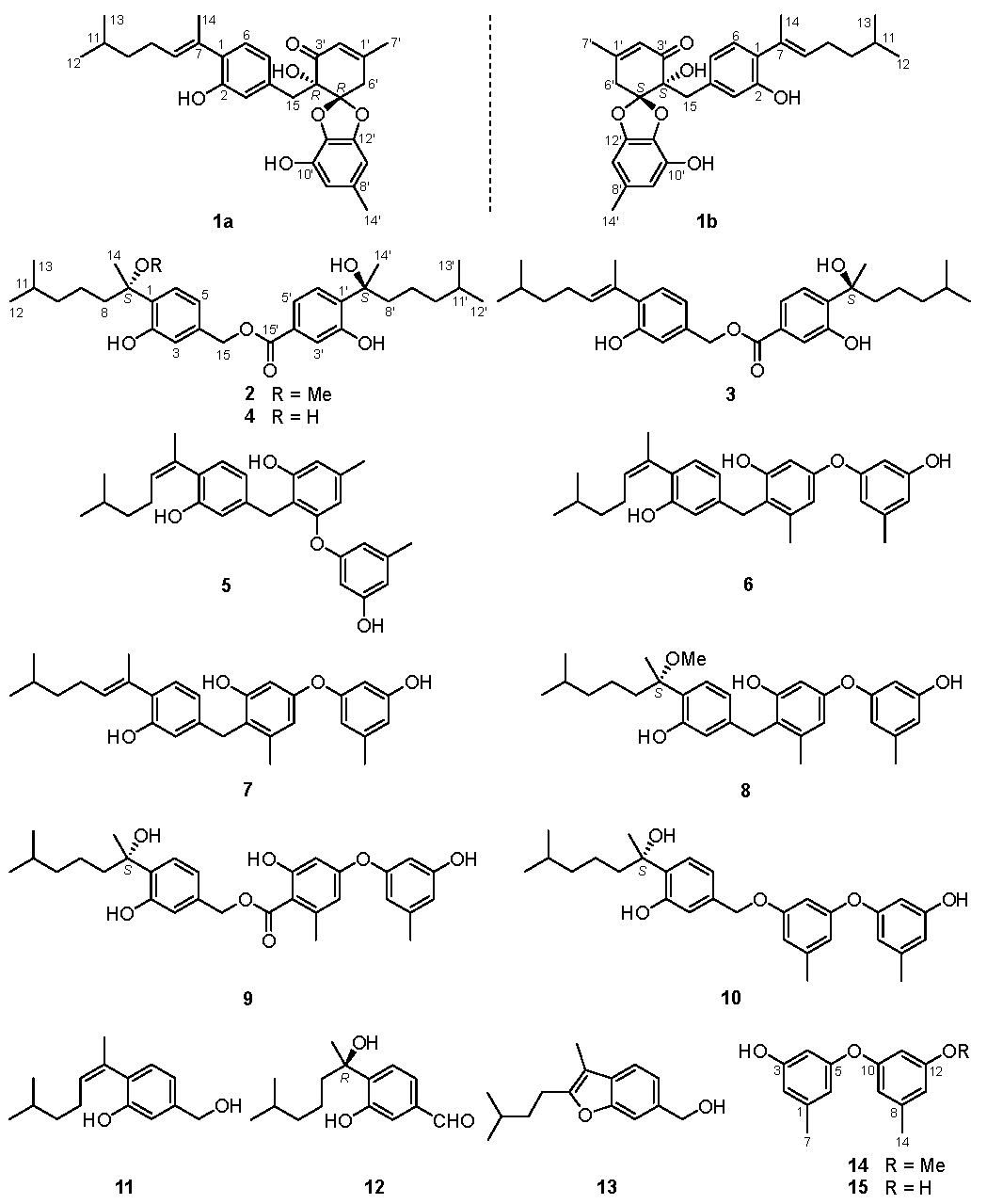
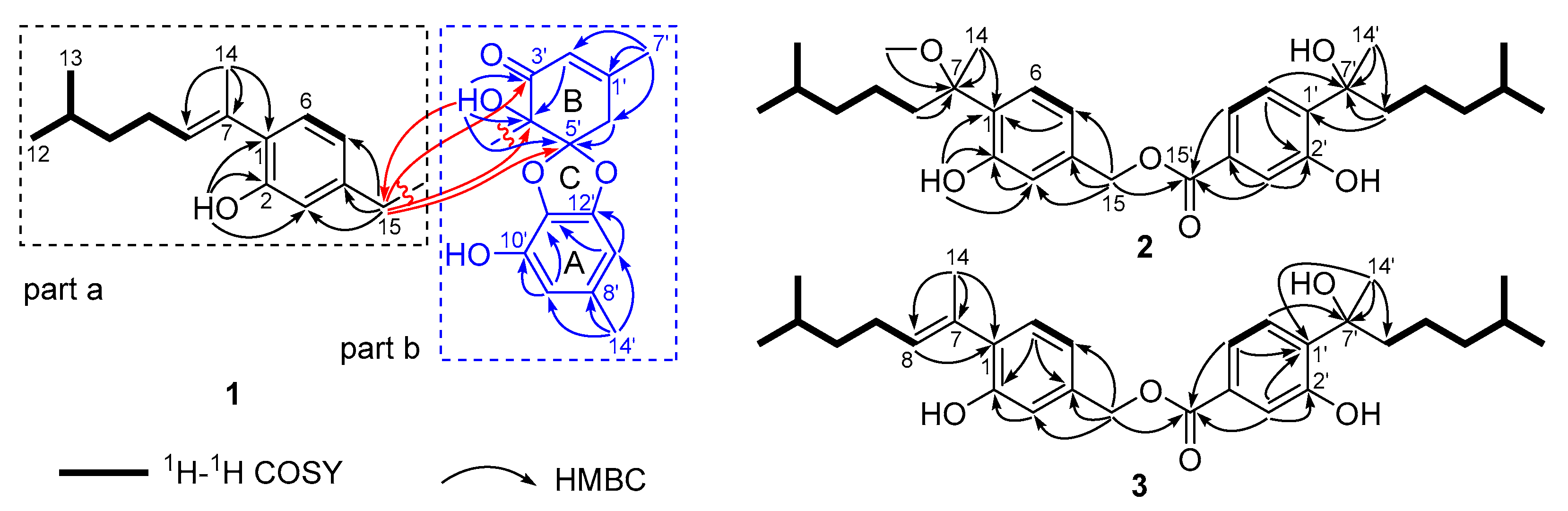
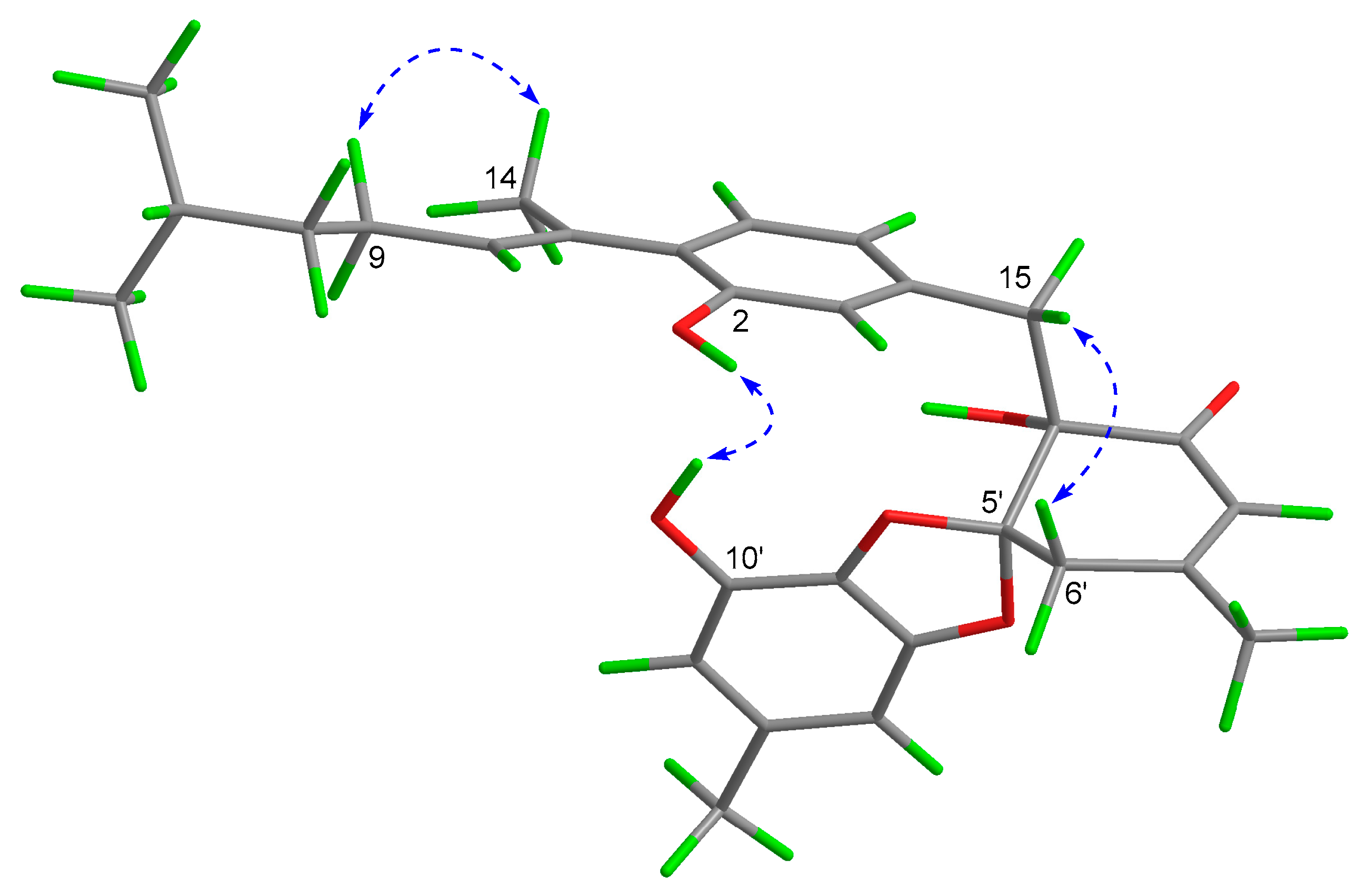
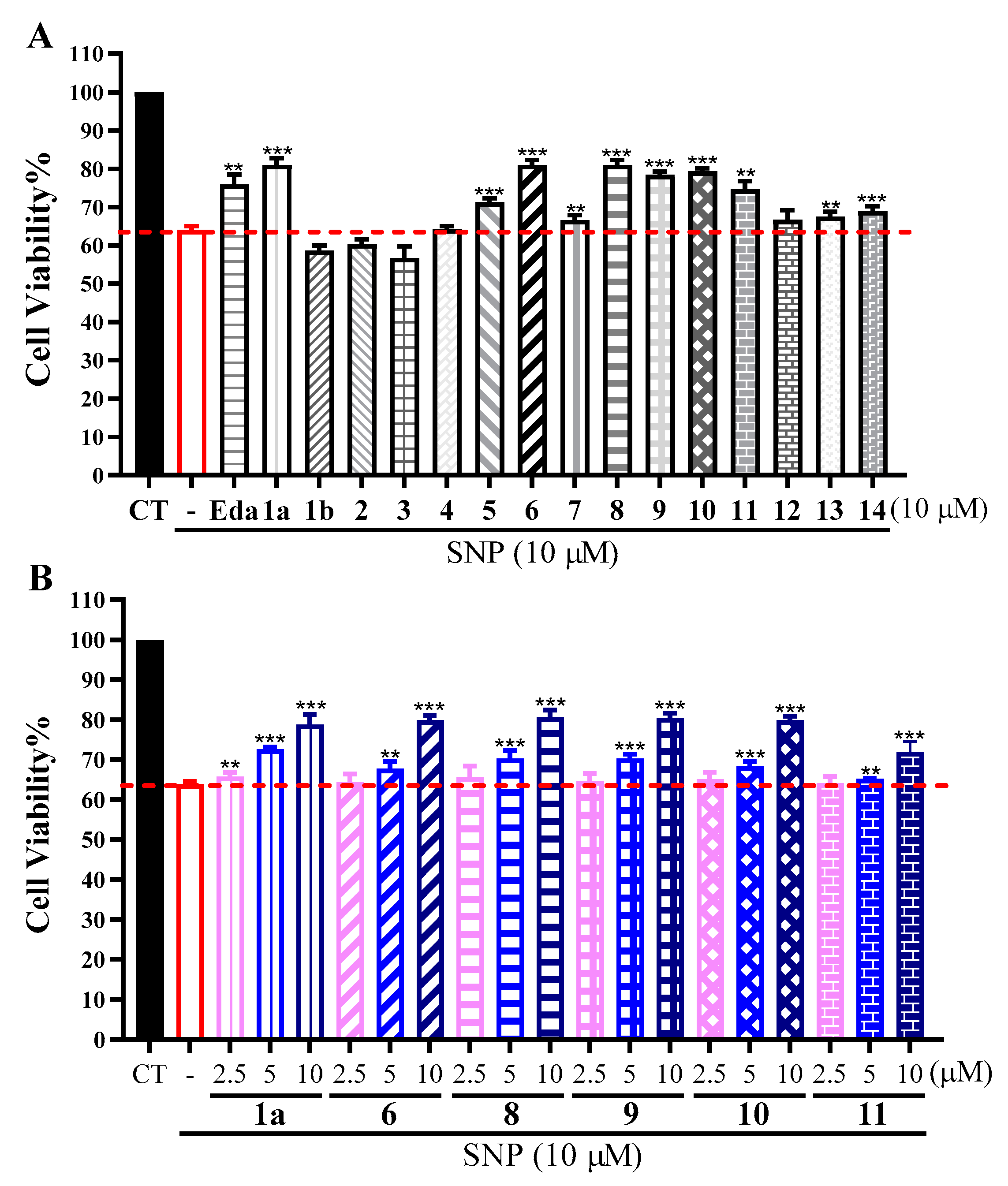
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Fermentation and Extraction
3.4. Isolation and Purification
3.5. ECD Calculation for Assigning the Absolute Configurations of 1a and 1b
3.6. Neuroprotective Bioassays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sun, C.; Zhang, Z.; Ren, Z.; Yu, L.; Zhou, H.; Han, Y.; Mudassir, S.; Che, Q.; Zhang, G.; Li, D.; et al. Antibacterial Cyclic Tripeptides from Antarctica-Sponge-Derived Fungus Aspergillus insulicola HDN151418. Mar. Drugs 2020, 18, 532. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.-L.; Shao, C.-L.; Chen, J.-F.; Guo, Z.-Y.; Fu, X.-M.; Chen, Y.-Y.; Li, R.; de Voogd, N.J.; She, Z.-G.; Lin, Y.-C.; et al. New Bisabolane Sesquiterpenoids from a Marine-Derived Fungus Aspergillus sp. Isolated from The Sponge Xestospongia testudinaria. Bioorg. Med. Chem. Lett. 2012, 22, 1326–1329. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-D.; Li, X.-M.; Xu, G.-M.; Zhang, P.; Wang, B.-G. Antimicrobial Phenolic Bisabolanes and Related Derivatives from Penicillium aculeatum SD-321, a Deep Sea Sediment-Derived Fungus. J. Nat. Prod. 2015, 78, 844–849. [Google Scholar] [CrossRef] [PubMed]
- Prompanya, C.; Dethoup, T.; Bessa, L.J.; Pinto, M.M.M.; Gales, L.; Costa, P.M.; Silva, A.M.S.; Kijjoa, A. New Isocoumarin Derivatives and Meroterpenoids from the Marine Sponge-Associated Fungus Aspergillus similanensis sp. nov. KUFA 0013. Mar. Drugs 2014, 12, 5160–5173. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Salim, A.A.; Capon, R.J. Millmerranones A−F: A Meroterpene Cyclic Carbonate and Related Metabolites from the Australian Fungus Aspergillus sp. CMB-MRF324. Org. Lett. 2021, 23, 8424–8428. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Su, J.-C.; Hu, J.-S.; He, X.-X.; Lin, S.-J.; Zhang, D.-M.; Ye, W.-C.; Chen, M.-F.; Lin, H.-W.; Zhang, C.-X. Probing Indole Diketopiperazine-Based Hybrids as Environmental Induced Products from Aspergillus sp. EGF 15-0-3. Org. Lett. 2022, 24, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Ye, G.; Huang, C.; Li, J.; Chen, T.; Tang, J.; Liu, W.; Long, Y. Isolation, Structural Characterization and Antidiabetic Activity of New Diketopiperazine Alkaloids from Mangrove Endophytic Fungus Aspergillus sp. 16-5c. Mar. Drugs 2021, 19, 402. [Google Scholar] [CrossRef]
- Qin, X.; Huang, J.; Zhou, D.; Zhang, W.; Zhang, Y.; Li, J.; Yang, R.; Huang, X. Polyketide Derivatives, Guhypoxylonols A–D from a Mangrove Endophytic Fungus Aspergillus sp. GXNU-Y45 That Inhibit Nitric Oxide Production. Mar. Drugs 2022, 20, 5. [Google Scholar] [CrossRef]
- Shu, H.-Z.; Peng, C.; Bu, L.; Guo, L.; Liu, F.; Xiong, L. Bisabolane-Type Sesquiterpenoids: Structural Diversity and Biological Activity. Phytochemistry 2021, 192, 112927. [Google Scholar] [CrossRef]
- Zhao, Z.-M.; Sun, Z.-H.; Chen, M.-H.; Liao, Q.; Tan, M.; Zhang, X.-W.; Zhu, H.-D.; Pi, R.-B.; Yin, S. Neuroprotective Polyhydroxypregnane Glycosides from Cynanchum otophyllum. Steroids 2013, 78, 1015–1020. [Google Scholar] [CrossRef]
- Tang, G.-H.; Chen, Z.-W.; Lin, T.-T.; Tan, M.; Gao, X.-Y.; Bao, J.-M.; Cheng, Z.-B.; Sun, Z.-H.; Huang, G.; Yin, S. Neolignans from Aristolochia fordiana Prevent Oxidative Stress-Induced Neuronal Death through Maintaining the Nrf2/HO-1 Pathway in HT22 Cells. J. Nat. Prod. 2015, 78, 1894–1903. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.-L.; Tang, G.-H.; Guo, Y.-Q.; Xu, Y.-K.; Huang, Z.-S.; Yin, S. Mulberry Diels-Alder Type Adducts from Morus alba as Multi-Targeted Agents for Alzheimer’s Disease. Phytochemistry 2019, 157, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.-Y.; Pan, Y.-H.; Zhang, J.-S.; Yin, S.; Tang, G.-H. Prenylated Xanthones from the Pericarps of Garcinia mangostana. Acta Sci. Nat. Univ. Sunyatseni 2020, 59, 21–32. [Google Scholar]
- Wang, J.; Lu, Z.; Liu, P.; Wang, Y.; Li, J.; Hong, K.; Zhu, W. Cytotoxic Polyphenols from the Fungus Penicillium expansum 091006 Endogenous with the Mangrove Plant Excoecaria agallocha. Planta Med. 2012, 78, 1861–1866. [Google Scholar]
- Lu, Z.; Zhu, H.; Fu, P.; Wang, Y.; Zhang, Z.; Lin, H.; Liu, P.; Zhuang, Y.; Hong, K.; Zhu, W. Cytotoxic Polyphenols from the Marine-Derived Fungus Penicillium expansum. J. Nat. Prod. 2010, 73, 911–914. [Google Scholar] [CrossRef]
- Liu, L.; Liu, R.; Buddha, B.B.; Bao, L.; Han, J.; Wang, L.; Liu, H. New Phenolic Bisabolane Sesquiterpenoid Derivatives with Cytotoxicity from Aspergillus tennesseensis. J. Antibiot. 2018, 71, 538–542. [Google Scholar] [CrossRef]
- Sumarah, M.W.; Kesting, J.R.; Sorensen, D.; Miller, J.D. Antifungal Metabolites from Fungal Endophytes of Pinus strobus. Phytochemistry 2011, 72, 1833–1837. [Google Scholar] [CrossRef]
- Nukina, M.; Sato, Y.; Ikeda, M.; Sassa, T. Sydonol, and New Fungal Morphogenic Substance Produced by an Unidentified Aspergillus sp. Agric. Biol. Chem. 1981, 45, 789–790. [Google Scholar] [CrossRef]
- Wu, J.-S.; Yao, G.-S.; Shi, X.-H.; Rehman, U.R.; Xu, Y.; Fu, X.-M.; Zhang, X.-L.; Liu, Y.; Wang, C.-Y. Epigenetic Agents Trigger the Production of Bioactive Nucleoside Derivatives and Bisabolane Sesquiterpenes from the Marine-Derived Fungus Aspergillus versicolor. Front. Microbiol. 2020, 11, 85. [Google Scholar] [CrossRef] [Green Version]
- Lan, W.-J.; Liu, W.; Liang, W.-L.; Xu, Z.; Le, X.; Xu, J.; Lam, C.-K.; Yang, D.-P.; Li, H.-J.; Wang, L.-Y. Pseudaboydins A and B: Novel Isobenzofuranone Derivatives from Marine Fungus Pseudallescheria boydii Associated with Starfish Acanthaster planci. Mar. Drugs 2014, 12, 4188–4199. [Google Scholar] [CrossRef] [Green Version]
- Julianti, E.; Abrian, I.A.; Wibowo, M.S.; Azhari, M.; Tsurayya, N.; Izzati, F.; Juanssilfero, A.B.; Bayu, A.; Rahmawati, S.I.; Putra, M.Y. Secondary Metabolites from Marine-Derived Fungi and Actinobacteria as Potential Sources of Novel Colorectal Cancer Drugs. Mar. Drugs 2022, 20, 67. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Fan, C.-Q.; Chen, S.; Zhou, J.; Zhou, J.-F.; Lu, Y.-N.; Ma, L.-Y.; Tian, X.-Q. Microbial Diversity and Activity of Some Fungi in the Surface Seawater of Northern South China Sea. Mar. Fish. 2022. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 a | 2 b | 3 a | |||
|---|---|---|---|---|---|---|
| δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | |
| 1 | 129.9, C | 128.0, C | 131.1, C | |||
| 2 | 151.6, C | 156.1, C | 152.2, C | |||
| 3 | 117.4, CH | 6.64, s | 116.4, CH | 6.92, s | 115.0, CH | 6.98, s |
| 4 | 134.3, C | 137.3, C | 136.2, C | |||
| 5 | 122.4, CH | 6.62, d (7.7) | 119.1, CH | 6.89, d (7.9) | 119.9, CH | 6.93, d (7.8) |
| 6 | 127.9, CH | 6.93, d (7.7) | 127.7, CH | 7.00, d (7.9) | 128.6, CH | 7.07, d (7.8) |
| 7 | 131.7, C | 82.9, C | 131.6, C | |||
| 8 | 132.0, CH | 5.53, t (7.0) | 40.2, CH2 | 1.82, m 1.93, m | 132.3, CH | 5.56, t (7.1) |
| 9 | 26.5, CH2 | 2.20, q (8.0) | 21.8, CH2 | 1.33, m | 26.5, CH2 | 2.22, q (7.5) |
| 10 | 38.7, CH2 | 1.32, q (7.5) | 39.1, CH2 | 1.14, m | 38.7, CH2 | 1.33, m |
| 11 | 27.9, CH | 1.60, m | 27.9, CH | 1.50, m | 27.9, CH | 1.60, m |
| 12 | 22.6, CH3 | 0.91, d (6.6) | 22.7, CH3 | 0.83, d (6.6) | 22.7, CH3 | 0.93, d (6.6) |
| 13 | 22.6, CH3 | 0.91, d (6.6) | 22.8, CH3 | 0.84, d (6.6) | 22.7, CH3 | 0.93, d (6.6) |
| 14 | 18.0, CH3 | 1.94, s | 22.4, CH3 | 1.58, s | 18.0, CH3 | 1.98, s |
| 15 | 40.3, CH2 | 2.94, d (13.4) 3.31, d (13.4) | 66.2, CH2 | 5.27, s | 66.4, CH2 | 5.27, s |
| 1′ | 157.1, C | 134.5, C | 134.5, C | |||
| 2′ | 124.0, CH | 6.10, s | 156.3, C | 156.3, C | ||
| 3′ | 197.6, C | 119.2, CH | 7.56, s | 119.0, CH | 7.55, s | |
| 4′ | 81.8, C | 130.7, C | 130.7, C | |||
| 5′ | 118.9, C | 120.8, CH | 7.52, d (8.2) | 120.8, CH | 7.52, d (8.2) | |
| 6′ | 42.0, CH2 | 3.01, d (19.1) 3.14, d (19.0) | 126.4, CH | 7.05, d (8.2) | 126.4, CH | 7.04, d (8.1) |
| 7′ | 24.3, CH3 | 2.10, s | 79.2, C | 79.2, C | ||
| 8′ | 132.2, C | 43.1, CH2 | 1.81, m 1.90, m | 43.1, CH2 | 1.79, m 1.91, m | |
| 9′ | 102.4, CH | 6.33, s | 21.8, CH2 | 1.29, m | 21.8, CH2 | 1.28, m |
| 10′ | 148.6, C | 39.3, CH2 | 1.14, m | 39.1, CH2 | 1.14, m | |
| 11′ | 132.2, C | 27.9, CH | 1.50, m | 27.9, CH | 1.49, m | |
| 12′ | 138.1, C | 22.7, CH3 | 0.83, d (6.6) | 22.7, CH3 | 0.82, d (6.6) | |
| 13′ | 111.3, CH | 6.26, s | 22.7, CH3 | 0.83, d (6.6) | 22.7, CH3 | 0.83, d (6.6) |
| 14′ | 21.5, CH3 | 2.23, s | 29.3, CH3 | 1.66, s | 29.3, CH3 | 1.66, s |
| 15′ | 166.3, C | 166.3, C | ||||
| 2-OH | 5.51, s | 8.88, s | ||||
| 2′-OH | 9.30, s | 9.30, s | ||||
| 4′-OH | 3.52, s | |||||
| 10′-OH | 4.78, s | |||||
| 7-OMe | 50.6, CH3 | 3.22, s | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weng, H.-Z.; Zhu, J.-Y.; Yuan, F.-Y.; Tang, Z.-Y.; Tian, X.-Q.; Chen, Y.; Fan, C.-Q.; Tang, G.-H.; Yin, S. Homo/Hetero-Dimers of Aromatic Bisabolane Sesquiterpenoids with Neuroprotective Activity from the Fungus Aspergillus versicolor A18 from South China Sea. Mar. Drugs 2022, 20, 322. https://doi.org/10.3390/md20050322
Weng H-Z, Zhu J-Y, Yuan F-Y, Tang Z-Y, Tian X-Q, Chen Y, Fan C-Q, Tang G-H, Yin S. Homo/Hetero-Dimers of Aromatic Bisabolane Sesquiterpenoids with Neuroprotective Activity from the Fungus Aspergillus versicolor A18 from South China Sea. Marine Drugs. 2022; 20(5):322. https://doi.org/10.3390/md20050322
Chicago/Turabian StyleWeng, Han-Zhuang, Jun-Yu Zhu, Fang-Yu Yuan, Zhuo-Ya Tang, Xiao-Qing Tian, Ye Chen, Cheng-Qi Fan, Gui-Hua Tang, and Sheng Yin. 2022. "Homo/Hetero-Dimers of Aromatic Bisabolane Sesquiterpenoids with Neuroprotective Activity from the Fungus Aspergillus versicolor A18 from South China Sea" Marine Drugs 20, no. 5: 322. https://doi.org/10.3390/md20050322
APA StyleWeng, H. -Z., Zhu, J. -Y., Yuan, F. -Y., Tang, Z. -Y., Tian, X. -Q., Chen, Y., Fan, C. -Q., Tang, G. -H., & Yin, S. (2022). Homo/Hetero-Dimers of Aromatic Bisabolane Sesquiterpenoids with Neuroprotective Activity from the Fungus Aspergillus versicolor A18 from South China Sea. Marine Drugs, 20(5), 322. https://doi.org/10.3390/md20050322