
In Silico Virtual Screening of Marine Aldehyde Derivatives from Seaweeds against SARS-CoV-2



Abstract
:1. Introduction
2. Results and Discussion
2.1. Drug-Likeness Analysis of the Marine Aldehyde Derivatives
2.2. ADME/Tox Analysis of the Marine Aldehyde Derivatives
2.3. TOPKAT Analysis of Marine Aldehyde Derivatives
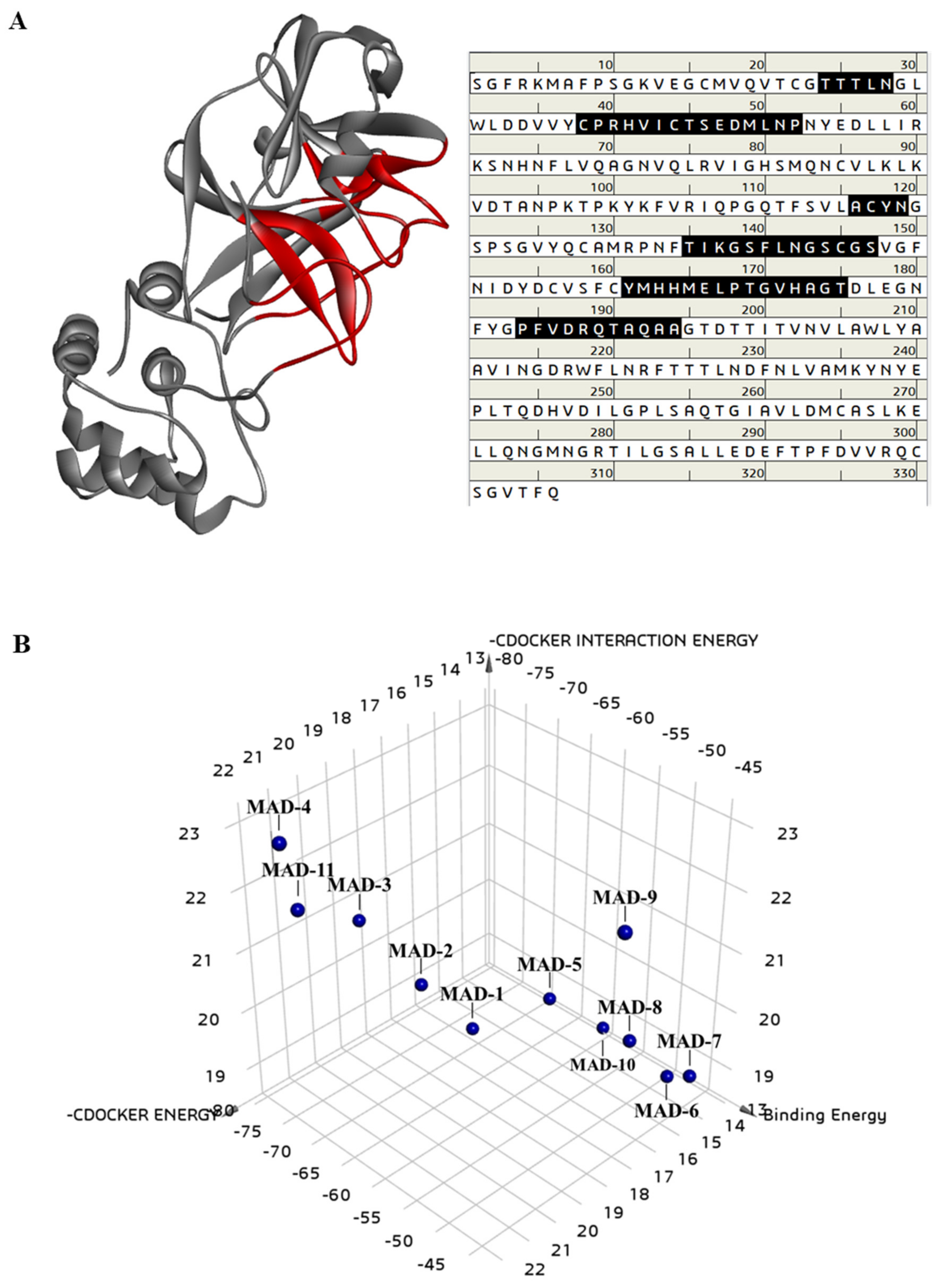
2.4. Molecular Docking Analysis of Marine Aldehyde Derivatives on 3C-like Protease
3. Materials and Methods
3.1. 3D Structure of Proteins and the Marine Aldehyde Derivatives
3.2. Drug-like Properties and ADME/Tox Predictions of the Marine Aldehyde Derivatives
3.3. TOPKAT Predictions of Marine Aldehyde Derivatives
3.4. Molecular Docking Analysis of the Marine Aldehyde Derivatives on the 3C-like Protease
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Armitage, R.; Nellums, L. Disease X: Availability bias, biotechnology and seeing beyond zoonotic risk. Public Health 2021, 190, e25. [Google Scholar] [CrossRef]
- Kuthyar, S.; Anthony, C.L.; Fashina, T.; Yeh, S.; Shantha, J.G. World health organization high priority pathogens: Ophthalmic disease findings and vision health perspectives. Pathogens 2021, 10, 442. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Zhao, P.; Praissman, J.L.; Grant, O.C.; Cai, Y.; Xiao, T.; Rosenbalm, K.E.; Aoki, K.; Kellman, B.P.; Bridger, R.; Barouch, D.H. Virus-receptor interactions of glycosylated SARS-CoV-2 spike and human ACE2 receptor. Cell Host Microbe 2020, 28, 586–601.e586. [Google Scholar] [CrossRef]
- Kandeel, M.; Al-Nazawi, M. Virtual screening and repurposing of FDA approved drugs against COVID-19 main protease. Life Sci. 2020, 251, 117627. [Google Scholar] [CrossRef]
- Vinuesa, R.; Azizpour, H.; Leite, I.; Balaam, M.; Dignum, V.; Domisch, S.; Felländer, A.; Langhans, S.D.; Tegmark, M.; Fuso Nerini, F. The role of artificial intelligence in achieving the Sustainable Development Goals. Nat. Commun. 2020, 11, 233. [Google Scholar] [CrossRef] [Green Version]
- Chassagnon, G.; Vakalopoulou, M.; Battistella, E.; Christodoulidis, S.; Hoang-Thi, T.-N.; Dangeard, S.; Deutsch, E.; Andre, F.; Guillo, E.; Halm, N. AI-driven quantification, staging and outcome prediction of COVID-19 pneumonia. Med. Image Anal. 2021, 67, 101860. [Google Scholar] [CrossRef]
- Maia, E.H.B.; Assis, L.C.; De Oliveira, T.A.; Da Silva, A.M.; Taranto, A.G. Structure-based virtual screening: From classical to artificial intelligence. Front. Chem. 2020, 8, 343. [Google Scholar] [CrossRef]
- Röhrig, U.F.; Awad, L.; Grosdidier, A.; Larrieu, P.; Stroobant, V.; Colau, D.; Cerundolo, V.; Simpson, A.J.; Vogel, P.; Van den Eynde, B.J. Rational design of indoleamine 2, 3-dioxygenase inhibitors. J. Med. Chem. 2010, 53, 1172–1189. [Google Scholar] [CrossRef]
- Glassman, P.M.; Muzykantov, V.R. Pharmacokinetic and pharmacodynamic properties of drug delivery systems. J. Pharmacol. Exp. Ther. 2019, 370, 570–580. [Google Scholar] [CrossRef]
- Tabti, K.; Elmchichi, L.; Sbai, A.; Maghat, H.; Bouachrine, M.; Lakhlifi, T.; Ghosh, A. In silico design of novel PIN1 inhibitors by combined of 3D-QSAR, molecular docking, molecular dynamic simulation and ADMET studies. J. Mol. Struct. 2022, 1253, 132291. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Zhao, W.; Li, X.; Yu, Z.; Wu, S.; Ding, L.; Liu, J. Identification of lactoferrin-derived peptides as potential inhibitors against the main protease of SARS-CoV-2. LWT 2022, 154, 112684. [Google Scholar] [CrossRef]
- Hajsafari, N.; Razaghi, Z.; Tabaian, S.H. Electrochemical study and molecular dynamics (MD) simulation of aluminum in the presence of garlic extract as a green inhibitor. J. Mol. Liq. 2021, 336, 116386. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, P.; Ye, X.; Wei, B.; Emam, M.; Zhang, H.; Wang, H. The structural diversity of marine microbial secondary metabolites based on co-culture strategy: 2009–2019. Mar. Drugs 2020, 18, 449. [Google Scholar] [CrossRef]
- Zahran, E.M.; Albohy, A.; Khalil, A.; Ibrahim, A.H.; Ahmed, H.A.; El-Hossary, E.M.; Bringmann, G.; Abdelmohsen, U.R. Bioactivity potential of marine natural products from Scleractinia-associated microbes and in silico anti-SARS-CoV-2 evaluation. Mar. Drugs 2020, 18, 645. [Google Scholar] [CrossRef]
- Kang, N.; Oh, S.; Kim, S.-Y.; Ahn, H.; Son, M.; Heo, S.-J.; Byun, K.; Jeon, Y.-J. Anti-obesity effects of Ishophloroglucin A from the brown seaweed Ishige okamurae (Yendo) via regulation of leptin signal in ob/ob mice. Algal Res. 2022, 61, 102533. [Google Scholar] [CrossRef]
- Pangestuti, R.; Haq, M.; Rahmadi, P.; Chun, B.-S. Nutritional Value and Biofunctionalities of Two Edible Green Seaweeds (Ulva lactuca and Caulerpa racemosa) from Indonesia by Subcritical Water Hydrolysis. Mar. Drugs 2021, 19, 578. [Google Scholar] [CrossRef]
- Zhu, X.; Healy, L.E.; Sevindik, O.; Sun, D.-W.; Selli, S.; Kelebek, H.; Tiwari, B.K. Impacts of novel blanching treatments combined with commercial drying methods on the physicochemical properties of Irish brown seaweed Alaria esculenta. Food Chem. 2022, 369, 130949. [Google Scholar] [CrossRef]
- Berneira, L.; da Silva, C.; Poletti, T.; Ritter, M.; dos Santos, M.; Colepicolo, P.; de Pereira, C.M.P. Evaluation of the volatile composition and fatty acid profile of seven Antarctic macroalgae. J. Appl. Phycol. 2020, 32, 3319–3329. [Google Scholar] [CrossRef]
- Cha, S.-H.; Hwang, Y.; Heo, S.-J.; Jun, H.-S. Indole-4-carboxaldehyde isolated from seaweed, Sargassum thunbergii, attenuates methylglyoxal-induced hepatic inflammation. Mar. Drugs 2019, 17, 486. [Google Scholar] [CrossRef] [Green Version]
- Han, E.J.; Fernando, I.P.S.; Kim, E.-A.; Kim, J.; Jung, K.; Kim, S.-Y.; Cha, S.-H.; Kim, K.-N.; Heo, S.-J.; Ahn, G. 5-Bromo-3, 4-dihydroxybenzaldehyde from Polysiphonia morrowii attenuate IgE/BSA-stimulated mast cell activation and passive cutaneous anaphylaxis in mice. Biochem. Pharmacol. 2020, 178, 114087. [Google Scholar] [CrossRef]
- Kang, J.-I.; Choi, Y.K.; Han, S.-C.; Nam, H.; Lee, G.; Kang, J.-H.; Koh, Y.S.; Hyun, J.W.; Yoo, E.-S.; Kang, H.-K. 5-Bromo-3, 4-dihydroxybenzaldehyde Promotes Hair Growth through Activation of Wnt/β-Catenin and Autophagy Pathways and Inhibition of TGF-β Pathways in Dermal Papilla Cells. Molecules 2022, 27, 2176. [Google Scholar] [CrossRef]
- Kang, M.-C.; Ding, Y.; Kim, J.; Kim, E.-A.; Fernando, I.S.; Heo, S.-J.; Lee, S.-H. 3-Chloro-4, 5-dihydroxybenzaldehyde inhibits adipogenesis in 3T3-L1 adipocytes by regulating expression of adipogenic transcription factors and AMPK activation. Chem. Biol. Interact. 2018, 287, 27–31. [Google Scholar] [CrossRef]
- Kim, E.-A.; Han, E.-J.; Kim, J.; Fernando, I.P.S.; Oh, J.-Y.; Kim, K.-N.; Ahn, G.; Heo, S.-J. Anti-Allergic Effect of 3, 4-Dihydroxybenzaldehyde Isolated from Polysiphonia morrowii in IgE/BSA-Stimulated Mast Cells and a Passive Cutaneous Anaphylaxis Mouse Model. Mar. Drugs 2022, 20, 133. [Google Scholar] [CrossRef]
- Wójcikowski, J.; Danek, P.J.; Basińska-Ziobroń, A.; Pukło, R.; Daniel, W.A. In vitro inhibition of human cytochrome P450 enzymes by the novel atypical antipsychotic drug asenapine: A prediction of possible drug–drug interactions. Pharmacol. Rep. 2020, 72, 612–621. [Google Scholar] [CrossRef] [Green Version]
- van Liempd, S.; Morrison, D.; Sysmans, L.; Nelis, P.; Mortishire-Smith, R. Development and validation of a higher-throughput equilibrium dialysis assay for plasma protein binding. JALA J. Assoc. Lab. Autom. 2011, 16, 56–67. [Google Scholar] [CrossRef]
- Hedvig, N.; Katarina, B.; Maurice, W.; Andrew, W. An Investigation into the Use of Computational and In Vitro Methods for Acute Systemic Toxicity Prediction; European Commission Joint Research Centre: Brussels, Belgium, 2012; p. 10. [Google Scholar]
- Kang, N.; Lee, J.-H.; Lee, W.; Ko, J.-Y.; Kim, E.-A.; Kim, J.-S.; Heu, M.-S.; Kim, G.H.; Jeon, Y.-J. Gallic acid isolated from Spirogyra sp. improves cardiovascular disease through a vasorelaxant and antihypertensive effect. Environ. Toxicol. Pharmacol. 2015, 39, 764–772. [Google Scholar] [CrossRef]
- Kang, N.; Ko, S.-C.; Kim, H.-S.; Yang, H.-W.; Ahn, G.; Lee, S.-C.; Lee, T.-G.; Lee, J.-S.; Jeon, Y.-J. Structural evidence for antihypertensive effect of an antioxidant peptide purified from the edible marine animal styela clava. J. Med. Food 2020, 23, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Kang, N.; Kim, E.-A.; Kim, J.; Lee, S.-H.; Heo, S.-J. Identifying potential antioxidant properties from the viscera of sea snails (Turbo cornutus). Mar. Drugs 2021, 19, 567. [Google Scholar] [CrossRef] [PubMed]
- Mesaik, M.A.; Murad, S.; Ismail, Z.; Abdullah, N.R.; Gill, H.K.; Yousaf, M.; Siddiqui, R.A.; Ahmad, A.; Choudhary, M.I. Biological and molecular docking studies on coagulin-H: Human IL-2 novel natural inhibitor. Mol. Immunol. 2006, 43, 1855–1863. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Robertson, D.H.; Brooks III, C.L.; Vieth, M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER—A CHARMm-based MD docking algorithm. J. Comput. Chem. 2003, 24, 1549–1562. [Google Scholar] [CrossRef]
- Wen, C.-C.; Kuo, Y.-H.; Jan, J.-T.; Liang, P.-H.; Wang, S.-Y.; Liu, H.-G.; Lee, C.-K.; Chang, S.-T.; Kuo, C.-J.; Lee, S.-S. Specific plant terpenoids and lignoids possess potent antiviral activities against severe acute respiratory syndrome coronavirus. J. Med. Chem. 2007, 50, 4087–4095. [Google Scholar] [CrossRef] [Green Version]
- Damonte, E.B.; Matulewicz, M.C.; Cerezo, A.S. Sulfated seaweed polysaccharides as antiviral agents. Curr. Med. Chem. 2004, 11, 2399–2419. [Google Scholar] [CrossRef]
- Ponce, N.M.; Pujol, C.A.; Damonte, E.B.; Flores, M.a.L.; Stortz, C.A. Fucoidans from the brown seaweed Adenocystis utricularis: Extraction methods, antiviral activity and structural studies. Carbohydr. Res. 2003, 338, 153–165. [Google Scholar] [CrossRef]
- Mandal, P.; Mateu, C.G.; Chattopadhyay, K.; Pujol, C.A.; Damonte, E.B.; Ray, B. Structural features and antiviral activity of sulphated fucans from the brown seaweed Cystoseira indica. Antivir. Chem. Chemother. 2007, 18, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Cassolato, J.E.; Noseda, M.D.; Pujol, C.A.; Pellizzari, F.M.; Damonte, E.B.; Duarte, M.E. Chemical structure and antiviral activity of the sulfated heterorhamnan isolated from the green seaweed Gayralia oxysperma. Carbohydr. Res. 2008, 343, 3085–3095. [Google Scholar] [CrossRef]
- Park, J.-Y.; Kim, J.H.; Kwon, J.M.; Kwon, H.-J.; Jeong, H.J.; Kim, Y.M.; Kim, D.; Lee, W.S.; Ryu, Y.B. Dieckol, a SARS-CoV 3CLpro inhibitor, isolated from the edible brown algae Ecklonia cava. Bioorg. Med. Chem. 2013, 21, 3730. [Google Scholar] [CrossRef]
- Kwon, H.-J.; Ryu, Y.B.; Kim, Y.-M.; Song, N.; Kim, C.Y.; Rho, M.-C.; Jeong, J.-H.; Cho, K.-O.; Lee, W.S.; Park, S.-J. In vitro antiviral activity of phlorotannins isolated from Ecklonia cava against porcine epidemic diarrhea coronavirus infection and hemagglutination. Bioorg. Med. Chem. 2013, 21, 4706–4713. [Google Scholar] [CrossRef]
- Artan, M.; Li, Y.; Karadeniz, F.; Lee, S.-H.; Kim, M.-M.; Kim, S.-K. Anti-HIV-1 activity of phloroglucinol derivative, 6, 6′-bieckol, from Ecklonia cava. Bioorg. Med. Chem. 2008, 16, 7921–7926. [Google Scholar] [CrossRef]
- Yang, H.-K.; Jung, M.-H.; Avunje, S.; Nikapitiya, C.; Kang, S.Y.; Ryu, Y.B.; Lee, W.S.; Jung, S.-J. Efficacy of algal Ecklonia cava extract against viral hemorrhagic septicemia virus (VHSV). Fish Shellfish Immunol. 2018, 72, 273–281. [Google Scholar] [CrossRef]
- Kim, K.-N.; Ko, S.-C.; Ye, B.-R.; Kim, M.-S.; Kim, J.; Ko, E.-Y.; Cho, S.-H.; Kim, D.; Heo, S.-J.; Jung, W.-K. 5-Bromo-2-hydroxy-4-methyl-benzaldehyde inhibited LPS-induced production of pro-inflammatory mediators through the inactivation of ERK, p38, and NF-κB pathways in RAW 264.7 macrophages. Chem. Biol. Interact. 2016, 258, 108–114. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Ntie-Kang, F. An in silico evaluation of the ADMET profile of the StreptomeDB database. SpringerPlus 2013, 2, 353. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| 3D structure |  |  |
| Chemical name | 4-hydroxybenzaldehyde | 3-hydroxybenzaldehyde |
| MAD no. | MAD-1 | MAD-2 |
| 3D structure |  |  |
| Chemical name | Salicylaldehyde | 3,4-dihydroxybenzaldehyde |
| MAD no. | MAD-3 | MAD-4 |
| 3D structure |  |  |
| Chemical name | indole-2-carboxaldehyde | indole-3-carboxaldehyde |
| MAD no. | MAD-5 | MAD-6 |
| 3D structure |  |  |
| Chemical name | indole-4-carboxaldehyde | indole-5-carboxaldehyde |
| MAD no. | MAD-7 | MAD-8 |
| 3D structure |  |  |
| Chemical name | indole-6-carboxaldehyde | indole-7-carboxaldehyde |
| MAD no. | MAD-9 | MAD-10 |
| 3D structure |  | |
| Chemical name | 5-bromoprotocatechualdehyde | |
| MAD no. | MAD-11 |
| Marine Aldehyde Derivatives | HBD | HBA | MW (Da) | ALogP | RB | PSA |
|---|---|---|---|---|---|---|
| MAD-1 | 1 | 2 | 122.121 | 1.347 | 1 | 37.29 |
| MAD-2 | 1 | 2 | 122.121 | 1.347 | 1 | 37.29 |
| MAD-3 | 1 | 2 | 122.121 | 1.347 | 1 | 37.29 |
| MAD-4 | 2 | 3 | 138.121 | 1.105 | 1 | 57.53 |
| MAD-5 | 1 | 2 | 145.158 | 2.174 | 1 | 32.86 |
| MAD-6 | 1 | 2 | 145.158 | 1.882 | 1 | 32.86 |
| MAD-7 | 1 | 2 | 145.158 | 1.882 | 1 | 32.86 |
| MAD-8 | 1 | 2 | 145.158 | 1.882 | 1 | 32.86 |
| MAD-9 | 1 | 2 | 145.158 | 1.882 | 1 | 32.86 |
| MAD-10 | 1 | 2 | 145.158 | 1.882 | 1 | 32.86 |
| MAD-11 | 2 | 3 | 217.017 | 1.853 | 1 | 57.53 |
| Marine Aldehyde Derivatives | AL | SL | BL | CP | HP | PP |
|---|---|---|---|---|---|---|
| MAD-1 | Good | Optimal | Medium | Non-inhibitor | Non-toxic | Binding is <90% |
| MAD-2 | Good | Optimal | Medium | Non-inhibitor | Non-toxic | Binding is <90% |
| MAD-3 | Good | Optimal | Medium | Non-inhibitor | Toxic | Binding is >90% |
| MAD-4 | Good | Optimal | Low | Non-inhibitor | Non-toxic | Binding is <90% |
| MAD-5 | Good | Good | High | Non-inhibitor | Toxic | Binding is >90% |
| MAD-6 | Good | Good | Medium | Non-inhibitor | Toxic | Binding is <90% |
| MAD-7 | Good | Good | Medium | Non-inhibitor | Toxic | Binding is <90% |
| MAD-8 | Good | Good | Medium | Non-inhibitor | Toxic | Binding is <90% |
| MAD-9 | Good | Good | Medium | Non-inhibitor | Toxic | Binding is <90% |
| MAD-10 | Good | Good | Medium | Non-inhibitor | Toxic | Binding is >90% |
| MAD-11 | Good | Optimal | Medium | Non-inhibitor | Non-toxic | Binding is <90% |
| Marine Aldehyde Derivatives | Ames Mutagenicity | Rat Oral LD50 (g/kg BW) | Rat Inhalational LC50 (mg/m3/h) |
|---|---|---|---|
| MAD-1 | Non-Mutagen | 1.13365 | 1744.04 |
| MAD-2 | Non-Mutagen | 1.31137 | 2660.83 |
| MAD-3 | Non-Mutagen | 1.0018 | 1655.42 |
| MAD-4 | Non-Mutagen | 2.67949 | 1794.97 |
| MAD-5 | Mutagen | 0.68308 | 4020.58 |
| MAD-6 | Mutagen | 0.393331 | 2431.62 |
| MAD-7 | Mutagen | 0.213938 | 2431.62 |
| MAD-8 | Mutagen | 0.535304 | 4117.71 |
| MAD-9 | Mutagen | 0.535304 | 4117.71 |
| MAD-10 | Mutagen | 0.551856 | 2431.62 |
| MAD-11 | Non-Mutagen | 1.96303 | 1975.31 |
| Asprin | Non-Mutagen | 1.57076 | 2704.1 |
| Curcumin | Non-Mutagen | 2.81353 | 1200.8 |
| Marine aldehyde derivatives | SkinIrritancy | Female RatNTP | Male Rat NTP |
| MAD-1 | Mild | Non-Carcinogen | Non-Carcinogen |
| MAD-2 | None | Non-Carcinogen | Non-Carcinogen |
| MAD-3 | None | Non-Carcinogen | Non-Carcinogen |
| MAD-4 | None | Non-Carcinogen | Non-Carcinogen |
| MAD-5 | Mild | Carcinogen | Carcinogen |
| MAD-6 | Mild | Non-Carcinogen | Carcinogen |
| MAD-7 | Mild | Non-Carcinogen | Non-Carcinogen |
| MAD-8 | Mild | Non-Carcinogen | Non-Carcinogen |
| MAD-9 | Mild | Non-Carcinogen | Non-Carcinogen |
| MAD-10 | Mild | Non-Carcinogen | Non-Carcinogen |
| MAD-11 | None | Non-Carcinogen | Carcinogen |
| Asprin | None | Non-Carcinogen | Non-Carcinogen |
| Curcumin | Mild | Carcinogen | Non-Carcinogen |
| Marine Aldehyde Derivatives | 3C-like Proteinase (6LU7) | ||
|---|---|---|---|
| –CDOCKER Energy (kcal/mol) | –CDOCKER Interaction Energy (kcal/mol) | Binding Energy (kcal/mol) | |
| MAD-1 | 16.3341 | 18.8237 | −69.5871 |
| MAD-2 | 17.2844 | 19.576 | −74.1383 |
| MAD-3 | 18.0852 | 20.4885 | −80.9339 |
| MAD-4 | 22.4808 | 23.2915 | −71.9725 |
| MAD-5 | 14.1641 | 19.0705 | −65.4523 |
| MAD-6 | 13.4791 | 18.5471 | −49.2269 |
| MAD-7 | 13.4622 | 18.7735 | −46.0088 |
| MAD-8 | 14.7365 | 19.4259 | −50.8909 |
| MAD-9 | 17.7453 | 22.5439 | −42.0406 |
| MAD-10 | 12.8445 | 18.4353 | −61.5275 |
| MAD-11 | 21.4484 | 21.8194 | −74.9887 |
| Curcumin | 35.4411 | 45.3384 | −63.3906 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, N.; Heo, S.-Y.; Cha, S.-H.; Ahn, G.; Heo, S.-J. In Silico Virtual Screening of Marine Aldehyde Derivatives from Seaweeds against SARS-CoV-2. Mar. Drugs 2022, 20, 399. https://doi.org/10.3390/md20060399
Kang N, Heo S-Y, Cha S-H, Ahn G, Heo S-J. In Silico Virtual Screening of Marine Aldehyde Derivatives from Seaweeds against SARS-CoV-2. Marine Drugs. 2022; 20(6):399. https://doi.org/10.3390/md20060399
Chicago/Turabian StyleKang, Nalae, Seong-Yeong Heo, Seon-Heui Cha, Ginnae Ahn, and Soo-Jin Heo. 2022. "In Silico Virtual Screening of Marine Aldehyde Derivatives from Seaweeds against SARS-CoV-2" Marine Drugs 20, no. 6: 399. https://doi.org/10.3390/md20060399
APA StyleKang, N., Heo, S.-Y., Cha, S.-H., Ahn, G., & Heo, S.-J. (2022). In Silico Virtual Screening of Marine Aldehyde Derivatives from Seaweeds against SARS-CoV-2. Marine Drugs, 20(6), 399. https://doi.org/10.3390/md20060399